
Discovering Genomic Regions Associated with Reproductive Traits and Frame Score in Mexican Simmental and Simbrah Cattle Using Individual SNP and Haplotype Markers

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phenotypic Data
- Frame score (FS): Hip height converted to frame score represents a linear measurement employed by cattle producers to assess the potential lean-to-fat ratio of an individual animal within a performance-oriented program. Calculated using the reference tables provided by the Beef Improvement Federation [15].
- Scrotal circumference (SC): For yearling bulls, scrotal circumference measurement entails encircling the broadest section of the scrotum with a scrotal tape while the testicles are in a fully extended state.
- Heifer fertility (HF): Represents the probability that the daughters of a sire have their first calving at the age of 3 years or earlier.
- Stayability (STAY): Indicates the probability that the daughters of a sire, having had a calf before 3 years of age, will have at least a second calf before 6 years of age.
2.2. Genotypic Data
2.3. Association Analyses
2.3.1. Single-Marker GWAS
2.3.2. Bayesian GWAS
2.4. Candidate Gene Annotations and Previously Reported QTL
3. Results
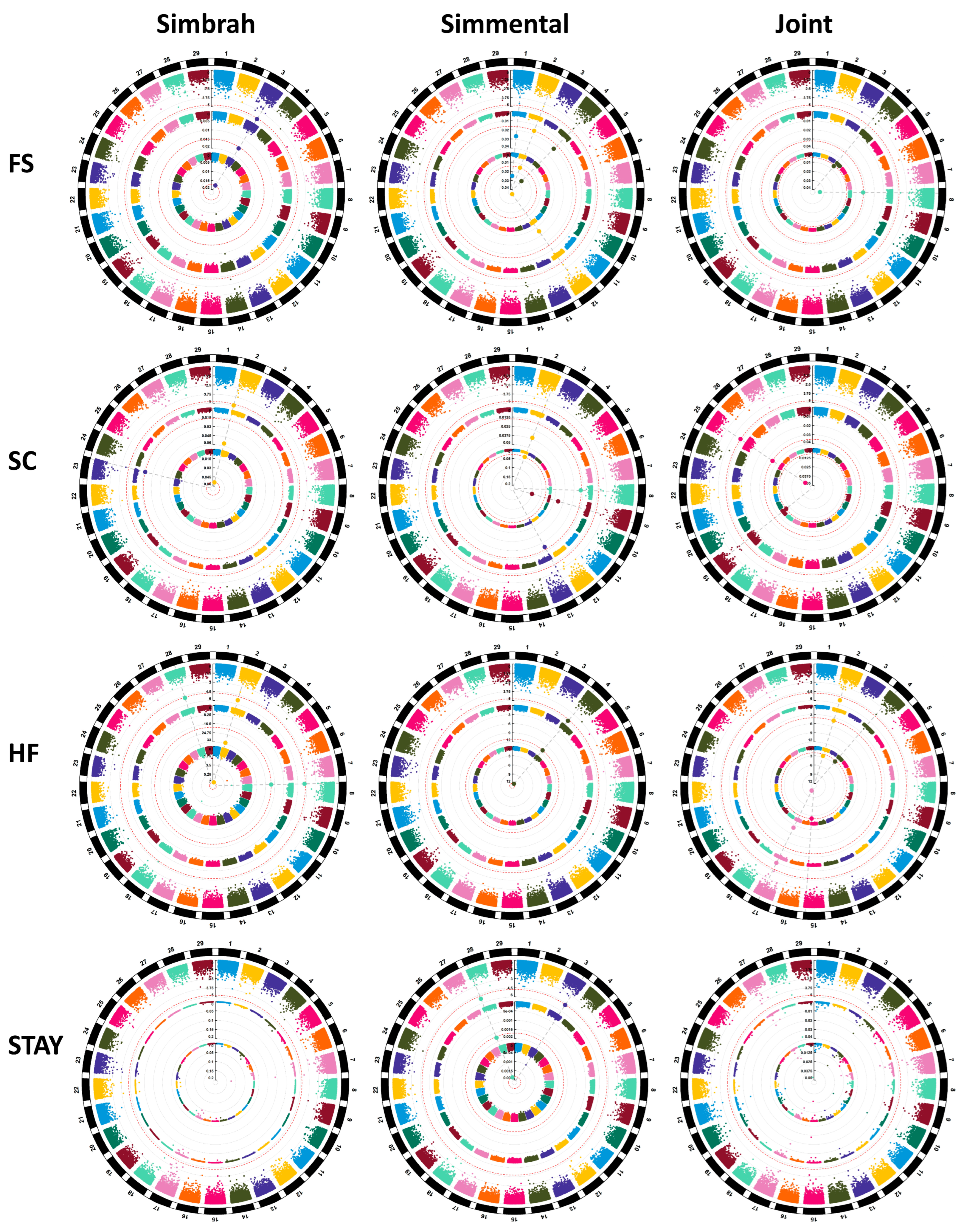
3.1. Frame Score
3.2. Scrotal Circumference
3.3. Heifer Fertility
3.4. Stayability
4. Discussion
4.1. Frame Score
4.2. Scrotal Circumference
4.3. Heifer Fertility
4.4. Stayability
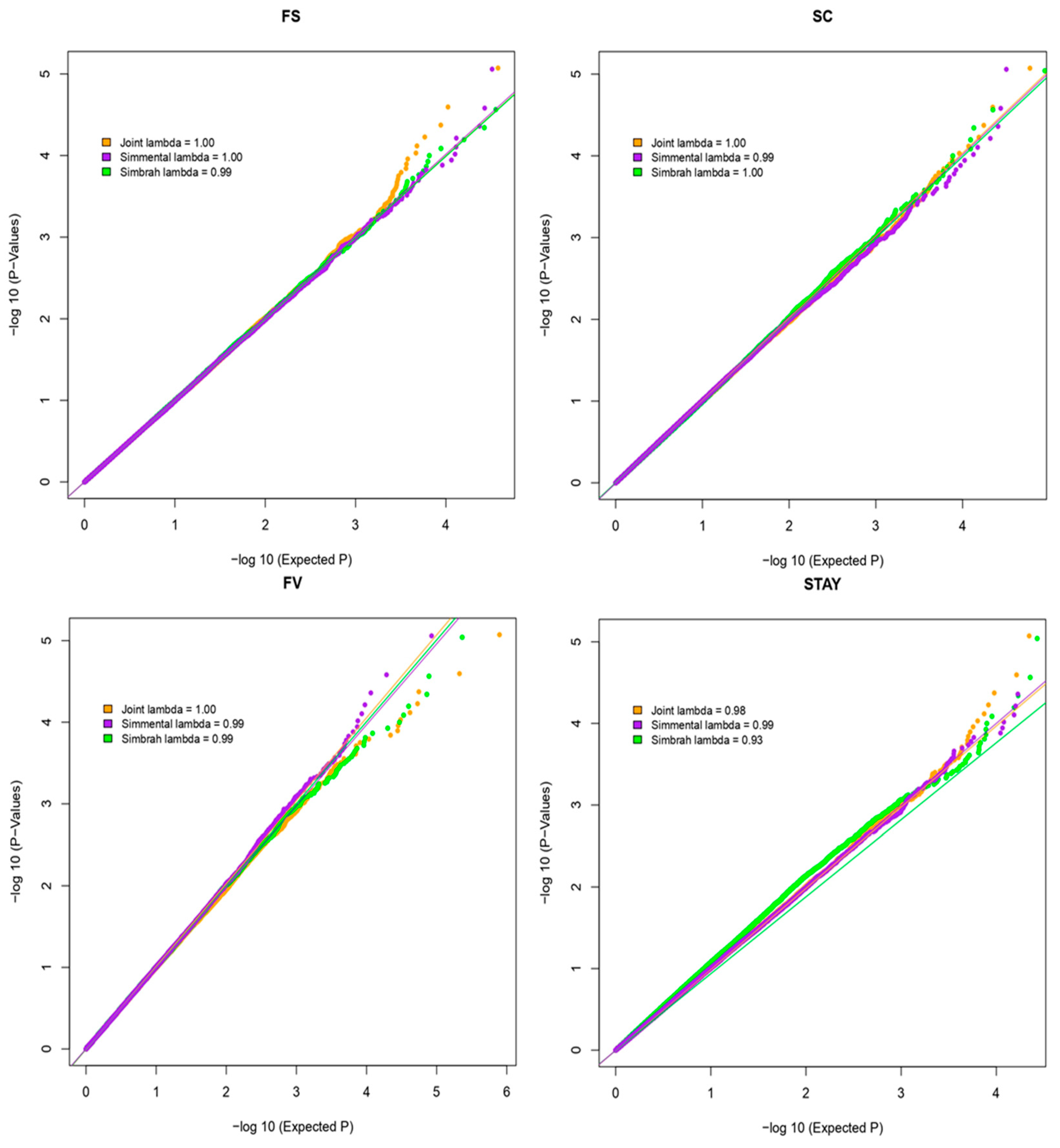
4.5. Single-Marker GWAS and Two Bayesian GWAS
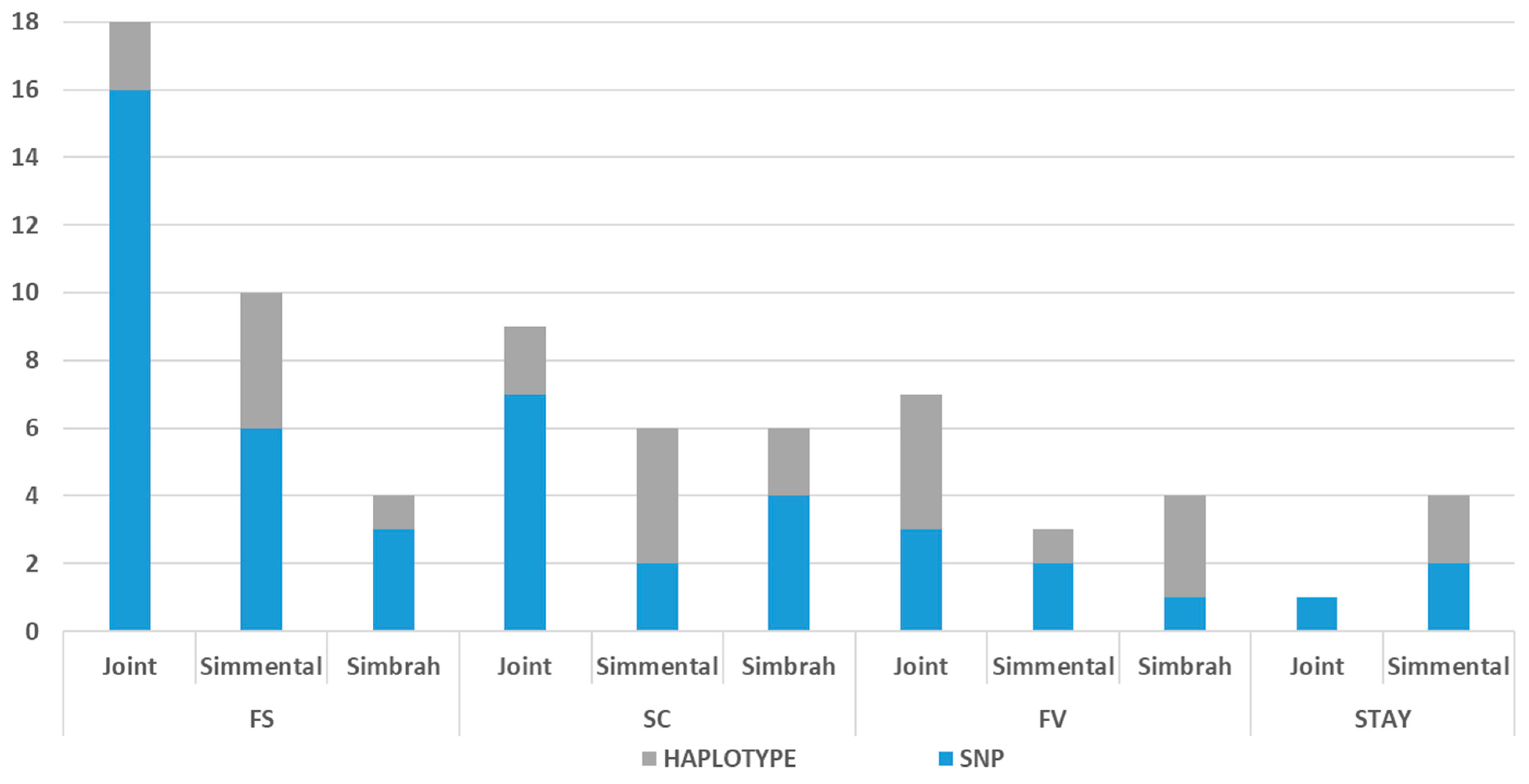
4.6. SNP- and Haplotype-Based Association Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baeza Rodríguez, J.J.; Vega Murillo, V.E.; Ríos Utrera, Á.; Martínez Velázquez, G.; Arechavaleta Velasco, M.E.; Montaño Bermúdez, M. Prototipo de Evaluación Genética Nacional Para Fertilidad de Vaquillas Simmental-Simbrah y Charolais-Charbray. Rev. Mex. Ciencias Pecu. 2017, 8, 249. [Google Scholar] [CrossRef]
- Burns, B.M.; Fordyce, G.; Holroyd, R.G. A Review of Factors That Impact on the Capacity of Beef Cattle Females to Conceive, Maintain a Pregnancy and Wean a Calf—Implications for Reproductive Efficiency in Northern Australia. Anim. Reprod. Sci. 2010, 122, 1–22. [Google Scholar] [CrossRef]
- Berry, D.P.; Wall, E.; Pryce, J.E. Genetics and Genomics of Reproductive Performance in Dairy and Beef Cattle. Animal 2014, 8, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Kijas, J.W.; Townley, D.; Dalrymple, B.P.; Heaton, M.P.; Maddox, J.F.; McGrath, A.; Wilson, P.; Ingersoll, R.G.; McCulloch, R.; McWilliam, S.; et al. A Genome Wide Survey of SNP Variation Reveals the Genetic Structure of Sheep Breeds. PLoS ONE 2009, 4, e4668. [Google Scholar] [CrossRef]
- Hamazaki, K.; Iwata, H. RAINBOW: Haplotype-Based Genome-Wide Association Study Using a Novel SNP-Set Method. PLoS Comput. Biol. 2020, 16, e1007663. [Google Scholar] [CrossRef]
- Mokry, F.B.; Buzanskas, M.E.; de Alvarenga Mudadu, M.; do Amaral Grossi, D.; Higa, R.H.; Ventura, R.V.; de Lima, A.O.; Sargolzaei, M.; Meirelles, S.L.C.; Schenkel, F.S.; et al. Linkage Disequilibrium and Haplotype Block Structure in a Composite Beef Cattle Breed. BMC Genom. 2014, 15, S6. [Google Scholar] [CrossRef]
- Melo, T.P.; Fortes, M.R.S.; Bresolin, T.; Mota, L.F.M.; Albuquerque, L.G.; Carvalheiro, R. Multitrait Meta-Analysis Identified Genomic Regions Associated with Sexual Precocity in Tropical Beef Cattle1. J. Anim. Sci. 2018, 96, 4087–4099. [Google Scholar] [CrossRef]
- McClure, M.C.; Morsci, N.S.; Schnabel, R.D.; Kim, J.W.; Yao, P.; Rolf, M.M.; McKay, S.D.; Gregg, S.J.; Chapple, R.H.; Northcutt, S.L.; et al. A Genome Scan for Quantitative Trait Loci Influencing Carcass, Post-Natal Growth and Reproductive Traits in Commercial Angus Cattle. Anim. Genet. 2010, 41, 597–607. [Google Scholar] [CrossRef]
- Sweett, H.; Fonseca, P.A.S.; Suárez-Vega, A.; Livernois, A.; Miglior, F.; Cánovas, A. Genome-Wide Association Study to Identify Genomic Regions and Positional Candidate Genes Associated with Male Fertility in Beef Cattle. Sci. Rep. 2020, 10, 20102. [Google Scholar] [CrossRef]
- Barreto Amaral Teixeira, D.; Alves Fernandes Júnior, G.; Beraldo dos Santos Silva, D.; Bermal Costa, R.; Takada, L.; Gustavo Mansan Gordo, D.; Bresolin, T.; Carvalheiro, R.; Baldi, F.; Galvão de Albuquerque, L. Genomic Analysis of Stayability in Nellore Cattle. PLoS ONE 2017, 12, e0179076. [Google Scholar] [CrossRef]
- Speidel, S.E.; Buckley, B.A.; Boldt, R.J.; Enns, R.M.; Lee, J.; Spangler, M.L.; Thomas, M.G. Genome-Wide Association Study of Stayability and Heifer Pregnancy in Red Angus Cattle. J. Anim. Sci. 2018, 96, 846–853. [Google Scholar] [CrossRef]
- McDaneld, T.G.; Kuehn, L.A.; Thomas, M.G.; Snelling, W.M.; Smith, T.P.L.; Pollak, E.J.; Cole, J.B.; Keele, J.W. Genomewide Association Study of Reproductive Efficiency in Female Cattle. J. Anim. Sci. 2014, 92, 1945–1957. [Google Scholar] [CrossRef] [PubMed]
- Akanno, E.C.; Chen, L.; Abo-Ismail, M.K.; Crowley, J.J.; Wang, Z.; Li, C.; Basarab, J.A.; MacNeil, M.D.; Plastow, G.S. Genome-Wide Association Scan for Heterotic Quantitative Trait Loci in Multi-Breed and Crossbred Beef Cattle. Genet. Sel. Evol. 2018, 50, 48. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, I.; Boichard, D.; Lund, M.S. Sequence Variants Selected from a Multi-Breed GWAS Can Improve the Reliability of Genomic Predictions in Dairy Cattle. Genet. Sel. Evol. 2016, 48, 83. [Google Scholar] [CrossRef]
- Beef Improvement Federation Guidelines for Uniform Beef Improvement Programs. Available online: https://guidelines.beefimprovement.org/index.php/Guidelines_for_Uniform_Beef_Improvement_Programs (accessed on 11 July 2023).
- Garrick, D.J.; Taylor, J.F.; Fernando, R.L. Deregressing Estimated Breeding Values and Weighting Information for Genomic Regression Analyses. Genet. Sel. Evol. 2009, 41, 55. [Google Scholar] [CrossRef]
- Calderón-Chagoya, R.; Vega-Murillo, V.E.; García-Ruiz, A.; Ríos-Utrera, Á.; Martínez-Velázquez, G.; Montaño-Bermúdez, M. Genome and Chromosome Wide Association Studies for Growth Traits in Simmental and Simbrah Cattle. Anim. Biosci. 2022, 36, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.; Daly, M.; et al. PLINK (1.07). PLINK: A Toolset for Whole-Genome Association and Population-Based Linkage Analysis. Am. J. Hum. Genet. 2007, 81, 1–293. Available online: http://pngu.mgh.harvard.edu/purcell/plink/ (accessed on 1 January 2022). [CrossRef]
- Wen, Y.-J.; Zhang, H.; Ni, Y.-L.; Huang, B.; Zhang, J.; Feng, J.-Y.; Wang, S.-B.; Dunwell, J.M.; Zhang, Y.-M.; Wu, R. Methodological Implementation of Mixed Linear Models in Multi-Locus Genome-Wide Association Studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Lander, E.; Kruglyak, L. Genetic Dissection of Complex Traits: Guidelines for Interpreting and Reporting Linkage Results. Nat. Genet. 1995, 11, 241–247. [Google Scholar] [CrossRef]
- Guo, Y.; Huang, Y.; Hou, L.; Ma, J.; Chen, C.; Ai, H.; Huang, L.; Ren, J. Genome-Wide Detection of Genetic Markers Associated with Growth and Fatness in Four Pig Populations Using Four Approaches. Genet. Sel. Evol. 2017, 49, 1–11. [Google Scholar] [CrossRef]
- Pérez, P.; de los Campos, G. Genome-Wide Regression and Prediction with the BGLR Statistical Package. Genetics 2014, 198, 483–495. [Google Scholar] [CrossRef]
- Habier, D.; Fernando, R.L.; Kizilkaya, K.; Garrick, D.J. Extension of the Bayesian Alphabet for Genomic Selection. BMC Bioinform. 2011, 12, 186. [Google Scholar] [CrossRef] [PubMed]
- Meuwissen, T.H.E.; Hayes, B.J.; Goddard, M.E. Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 2001, 157, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Allen, J.E.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Bennett, R.; et al. Ensembl 2022. Nucleic Acids Res. 2022, 50, D988–D995. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-L.; Park, C.A.; Reecy, J.M. Bringing the Animal QTLdb and CorrDB into the Future: Meeting New Challenges and Providing Updated Services. Nucleic Acids Res. 2022, 50, D956–D961. [Google Scholar] [CrossRef] [PubMed]
- Ehret, G.B. Genome-Wide Association Studies: Contribution of Genomics to Understanding Blood Pressure and Essential Hypertension. Curr. Hypertens. Rep. 2010, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, M.S.; Heyen, D.W.; Weller, J.I.; Ron, M.; Sonstegard, T.S.; Van Tassell, C.P.; Lewin, H.A. Detection of Quantitative Trait Loci Influencing Conformation Traits and Calving Ease in Holstein-Friesian Cattle. J. Dairy Sci. 2005, 88, 4111–4119. [Google Scholar] [CrossRef]
- Ríos Utrera, Á.; Martínez Velázquez, G.; Calderón Chagoya, R.; Montaño Bermúdez, M.; Vega Murillo, V.E. Logros, Retos y Perspectivas de La Investigación En Mejoramiento Genético de Bovinos Productores de Carne En El INIFAP. Rev. Mex. Ciencias Pecu. 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-Wide Association Analysis of Thirty One Production, Health, Reproduction and Body Conformation Traits in Contemporary U.S. Holstein Cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef]
- Snelling, W.M.; Allan, M.F.; Keele, J.W.; Kuehn, L.A.; McDaneld, T.; Smith, T.P.L.; Sonstegard, T.S.; Thallman, R.M.; Bennett, G.L. Genome-Wide Association Study of Growth in Crossbred Beef Cattle12. J. Anim. Sci. 2010, 88, 837–848. [Google Scholar] [CrossRef]
- Setoguchi, K.; Watanabe, T.; Weikard, R.; Albrecht, E.; Kühn, C.; Kinoshita, A.; Sugimoto, Y.; Takasuga, A. The SNP c.1326T>G in the Non-SMC Condensin I Complex, Subunit G (NCAPG) Gene Encoding a p.Ile442Met Variant Is Associated with an Increase in Body Frame Size at Puberty in Cattle. Anim. Genet. 2011, 42, 650–655. [Google Scholar] [CrossRef]
- Imumorin, I.G.; Kim, E.-H.; Lee, Y.-M.; De Koning, D.-J.; van Arendonk, J.A.; De Donato, M.; Taylor, J.F.; Kim, J.-J. Genome Scan for Parent-of-Origin QTL Effects on Bovine Growth and Carcass Traits. Front. Genet. 2011, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-J.; Farnir, F.; Savell, J.; Taylor, J.F. Detection of Quantitative Trait Loci for Growth and Beef Carcass Fatness Traits in a Cross between Bos Taurus (Angus) and Bos Indicus (Brahman) Cattle1. J. Anim. Sci. 2003, 81, 1933–1942. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, B.W.; Byla, B.M.; Gregory, K.E. Mapping Quantitative Trait Loci for Bovine Ovulation Rate. Mamm. Genome 2000, 11, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Alijani, S.; Rafat, S.A.; Abdollahi-Arpanahi, R. Genome-Wide Association Study and Pathway Analysis for Female Fertility Traits in Iranian Holstein Cattle. Ann. Anim. Sci. 2020, 20, 825–851. [Google Scholar] [CrossRef]
- Muncie, S.A.; Cassady, J.P.; Ashwell, M.S. Refinement of Quantitative Trait Loci on Bovine Chromosome 18 Affecting Health and Reproduction in US Holsteins. Anim. Genet. 2006, 37, 273–275. [Google Scholar] [CrossRef]
- Hawken, R.J.; Zhang, Y.D.; Fortes, M.R.S.; Collis, E.; Barris, W.C.; Corbet, N.J.; Williams, P.J.; Fordyce, G.; Holroyd, R.G.; Walkley, J.R.W.; et al. Genome-Wide Association Studies of Female Reproduction in Tropically Adapted Beef Cattle1. J. Anim. Sci. 2012, 90, 1398–1410. [Google Scholar] [CrossRef]
- Freebern, E.; Santos, D.J.A.; Fang, L.; Jiang, J.; Parker Gaddis, K.L.; Liu, G.E.; Vanraden, P.M.; Maltecca, C.; Cole, J.B.; Ma, L. GWAS and Fine-Mapping of Livability and Six Disease Traits in Holstein Cattle. BMC Genom. 2020, 21, 1–11. [Google Scholar] [CrossRef]
- Jiang, J.; Ma, L.; Prakapenka, D.; VanRaden, P.M.; Cole, J.B.; Da, Y. A Large-Scale Genome-Wide Association Study in U.S. Holstein Cattle. Front. Genet. 2019, 10, 442321. [Google Scholar] [CrossRef]
- Lu, B.; Poirier, C.; Gaspar, T.; Gratzke, C.; Harrison, W.; Busija, D.; Matzuk, M.M.; Andersson, K.-E.; Overbeek, P.A.; Bishop, C.E. A Mutation in the Inner Mitochondrial Membrane Peptidase 2-Like Gene (Immp2l) Affects Mitochondrial Function and Impairs Fertility in Mice1. Biol. Reprod. 2008, 78, 601–610. [Google Scholar] [CrossRef]
- Kang, Z.; Zhang, S.; He, L.; Zhu, H.; Wang, Z.; Yan, H.; Huang, Y.; Dang, R.; Lei, C.; Chen, H.; et al. A 14-Bp Functional Deletion within the CMTM2 Gene Is Significantly Associated with Litter Size in Goat. Theriogenology 2019, 139, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Höglund, J.K.; Guldbrandtsen, B.; Su, G.; Thomsen, B.; Lund, M.S. Genome Scan Detects Quantitative Trait Loci Affecting Female Fertility Traits in Danish and Swedish Holstein Cattle. J. Dairy Sci. 2009, 92, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Yasothai, R. Importance of Energy on Reproduction in Dairy Cattle. Int. J. Sci. Environ. Technol. 2014, 3, 2020–2023. [Google Scholar]
- Boichard, D.; Grohs, C.; Bourgeois, F.; Cerqueira, F.; Faugeras, R.; Neau, A.; Rupp, R.; Amigues, Y.; Boscher, M.Y.; Levéziel, H. Detection of Genes Influencing Economic Traits in Three French Dairy Cattle Breeds. Genet. Sel. Evol. 2003, 35, 77. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.C.; Pérez, J.E.; Herrera, N.; Martínez, R.; Bejarano, D.; Rocha, J.F. Research Article Genomic Association Study for Age at First Calving and Calving Interval in Romosinuano and Costeño Con Cuernos Cattle. Genet. Mol. Res. 2019, 18, 1–13. [Google Scholar] [CrossRef]
- Zhao, Y.; Pu, Y.; Liang, B.; Bai, T.; Liu, Y.; Jiang, L.; Ma, Y. A Study Using Single-locus and Multi-locus Genome-wide Association Study to Identify Genes Associated with Teat Number in Hu Sheep. Anim. Genet. 2022, 53, 203–211. [Google Scholar] [CrossRef]
- Baker, M. Statisticians Issue Warning over Misuse of P Values. Nature 2016, 531, 151. [Google Scholar] [CrossRef]
- Raymond, B.; Bouwman, A.C.; Wientjes, Y.C.J.; Schrooten, C.; Houwing-Duistermaat, J.; Veerkamp, R.F. Genomic Prediction for Numerically Small Breeds, Using Models with Pre-Selected and Differentially Weighted Markers. Genet. Sel. Evol. 2018, 50, 49. [Google Scholar] [CrossRef]
- Legarra, A.; Ricard, A.; Varona, L. GWAS by GBLUP: Single and Multimarker EMMAX and Bayes Factors, with an Example in Detection of a Major Gene for Horse Gait. G3 Genes Genomes Genet. 2018, 8, 2301–2308. [Google Scholar] [CrossRef]
- Banerjee, S.; Zeng, L.; Schunkert, H.; Söding, J. Bayesian Multiple Logistic Regression for Case-Control GWAS. PLoS Genet. 2018, 14, e1007856. [Google Scholar] [CrossRef]
- Contreras-Soto, R.I.; Mora, F.; De Oliveira, M.A.R.; Higashi, W.; Scapim, C.A.; Schuster, I. A Genome-Wide Association Study for Agronomic Traits in Soybean Using SNP Markers and SNP-Based Haplotype Analysis. PLoS ONE 2017, 12, e0171105. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Onteru, S.K.; Du, Z.Q.; Garrick, D.J.; Stalder, K.J.; Rothschild, M.F. Genome-Wide Association Study Identifies Loci for Body Composition and Structural Soundness Traits in Pigs. PLoS ONE 2011, 6, e14726. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; O’Connor, T.D.; Akey, J.M. Genetic Architecture of Quantitative Traits and Complex Diseases. Curr. Opin. Genet. Dev. 2013, 23, 678–683. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calderón-Chagoya, R.; Vega-Murillo, V.E.; García-Ruiz, A.; Ríos-Utrera, Á.; Martínez-Velázquez, G.; Montaño-Bermúdez, M. Discovering Genomic Regions Associated with Reproductive Traits and Frame Score in Mexican Simmental and Simbrah Cattle Using Individual SNP and Haplotype Markers. Genes 2023, 14, 2004. https://doi.org/10.3390/genes14112004
Calderón-Chagoya R, Vega-Murillo VE, García-Ruiz A, Ríos-Utrera Á, Martínez-Velázquez G, Montaño-Bermúdez M. Discovering Genomic Regions Associated with Reproductive Traits and Frame Score in Mexican Simmental and Simbrah Cattle Using Individual SNP and Haplotype Markers. Genes. 2023; 14(11):2004. https://doi.org/10.3390/genes14112004
Chicago/Turabian StyleCalderón-Chagoya, René, Vicente Eliezer Vega-Murillo, Adriana García-Ruiz, Ángel Ríos-Utrera, Guillermo Martínez-Velázquez, and Moisés Montaño-Bermúdez. 2023. "Discovering Genomic Regions Associated with Reproductive Traits and Frame Score in Mexican Simmental and Simbrah Cattle Using Individual SNP and Haplotype Markers" Genes 14, no. 11: 2004. https://doi.org/10.3390/genes14112004