
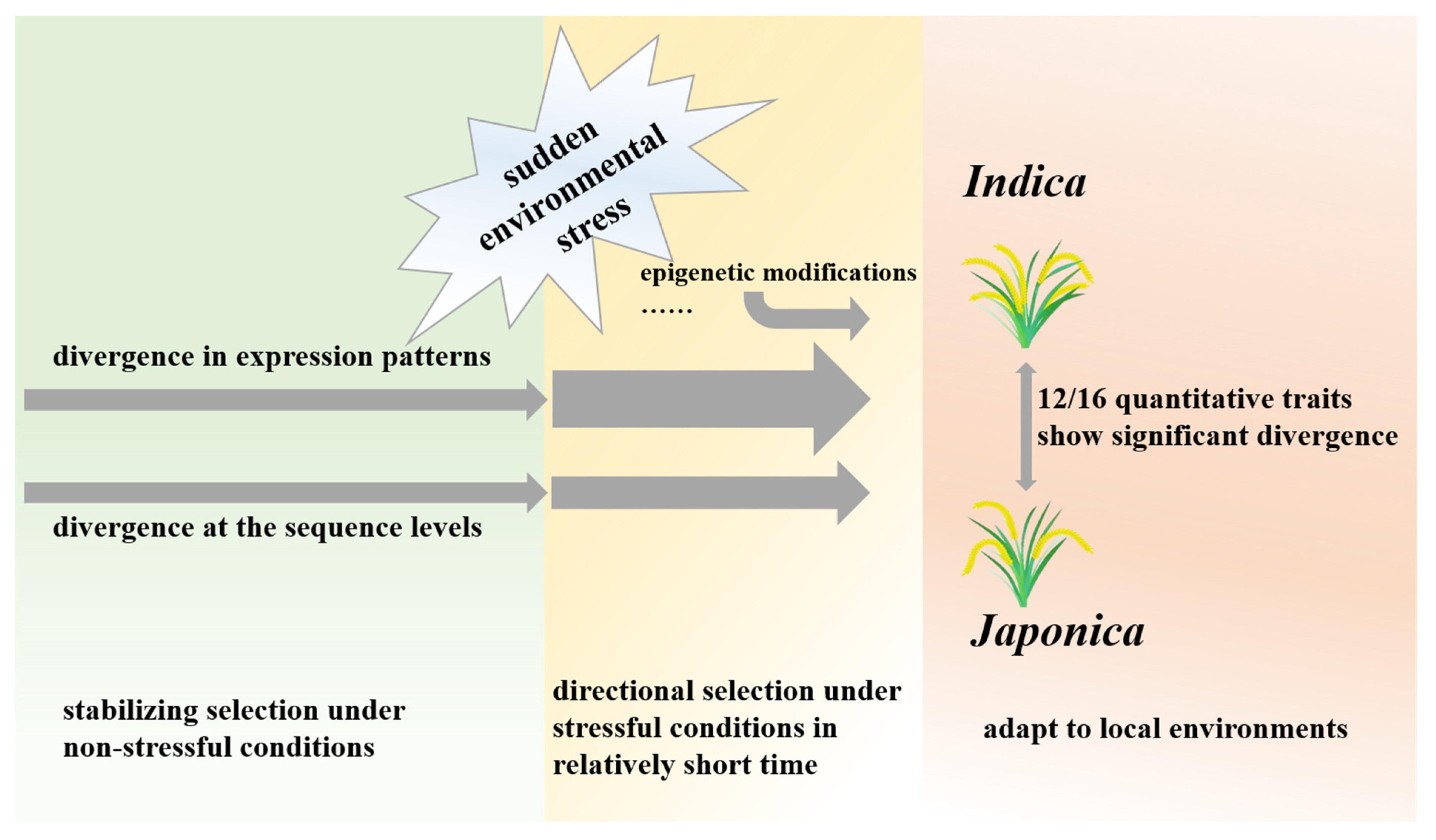
Differentiation of Morphological Traits and Genome-Wide Expression Patterns between Rice Subspecies Indica and Japonica

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection for Phenotypic Observation and RNA-Seq
2.2. Whole-Genome Resequencing Data
2.3. Phylogeny Based on SNPs from Resequencing Data
2.4. Phylogeny Based on RNA-Seq Data
2.5. Identifying Differentially Expressed Genes (DEGs) between Indica and Japonica Rice
3. Results
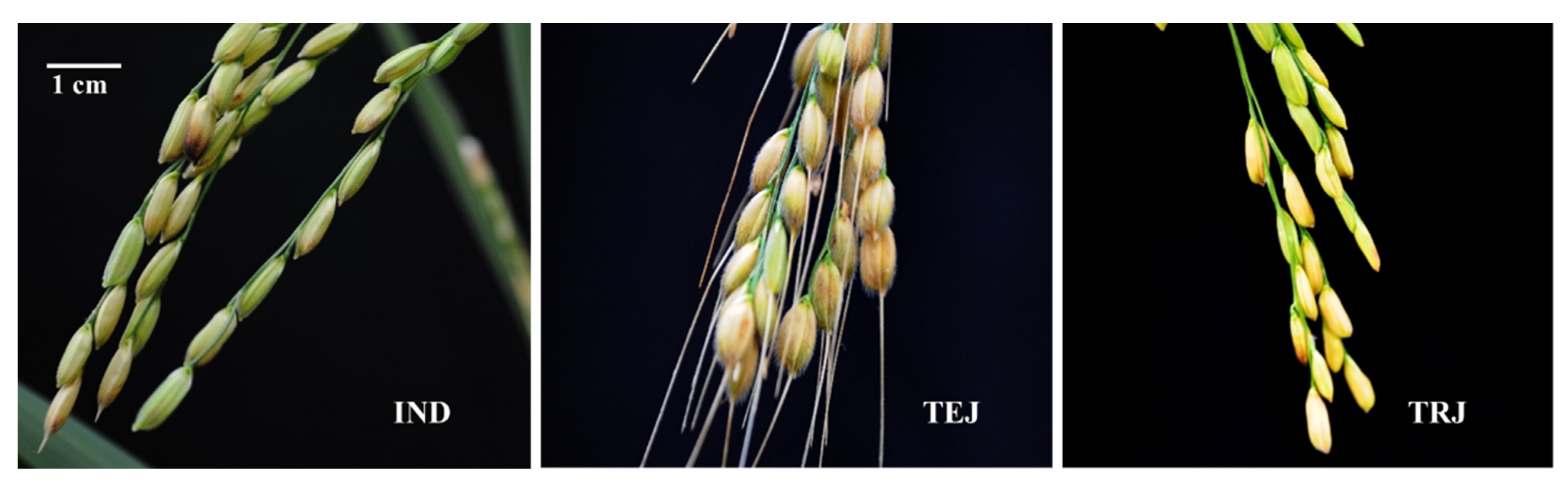
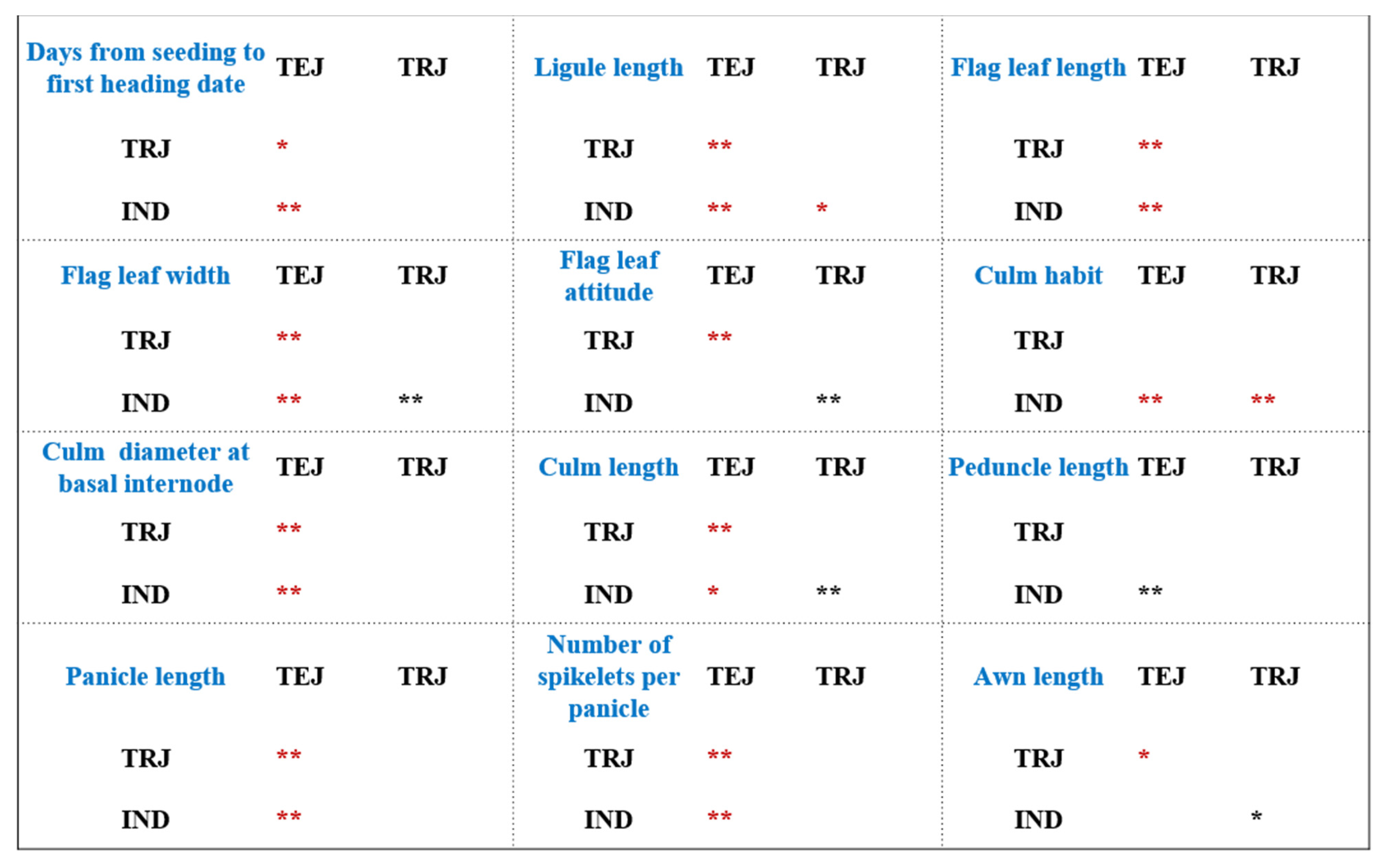
3.1. Morphological Variation between Two Subspecies of Rice
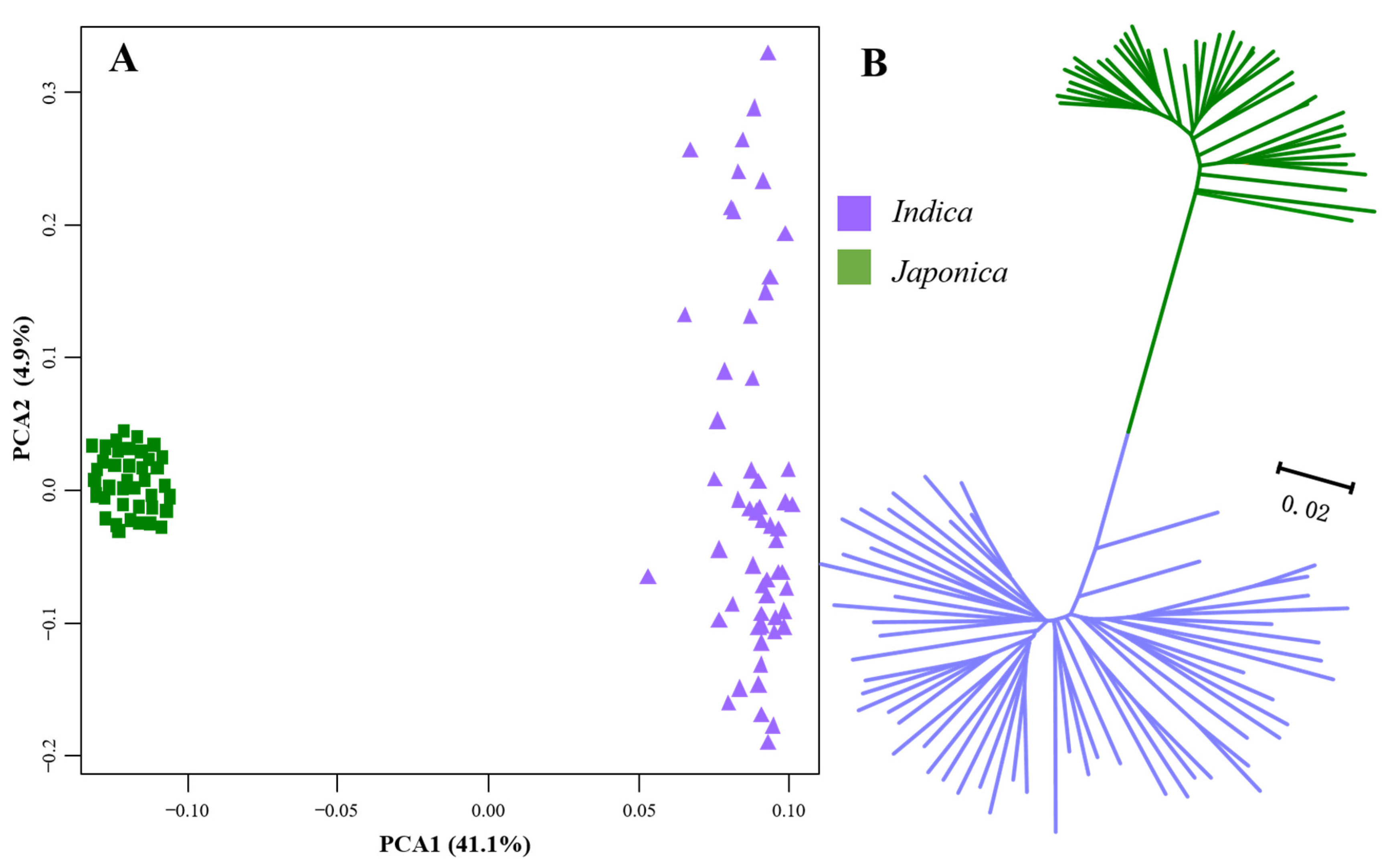
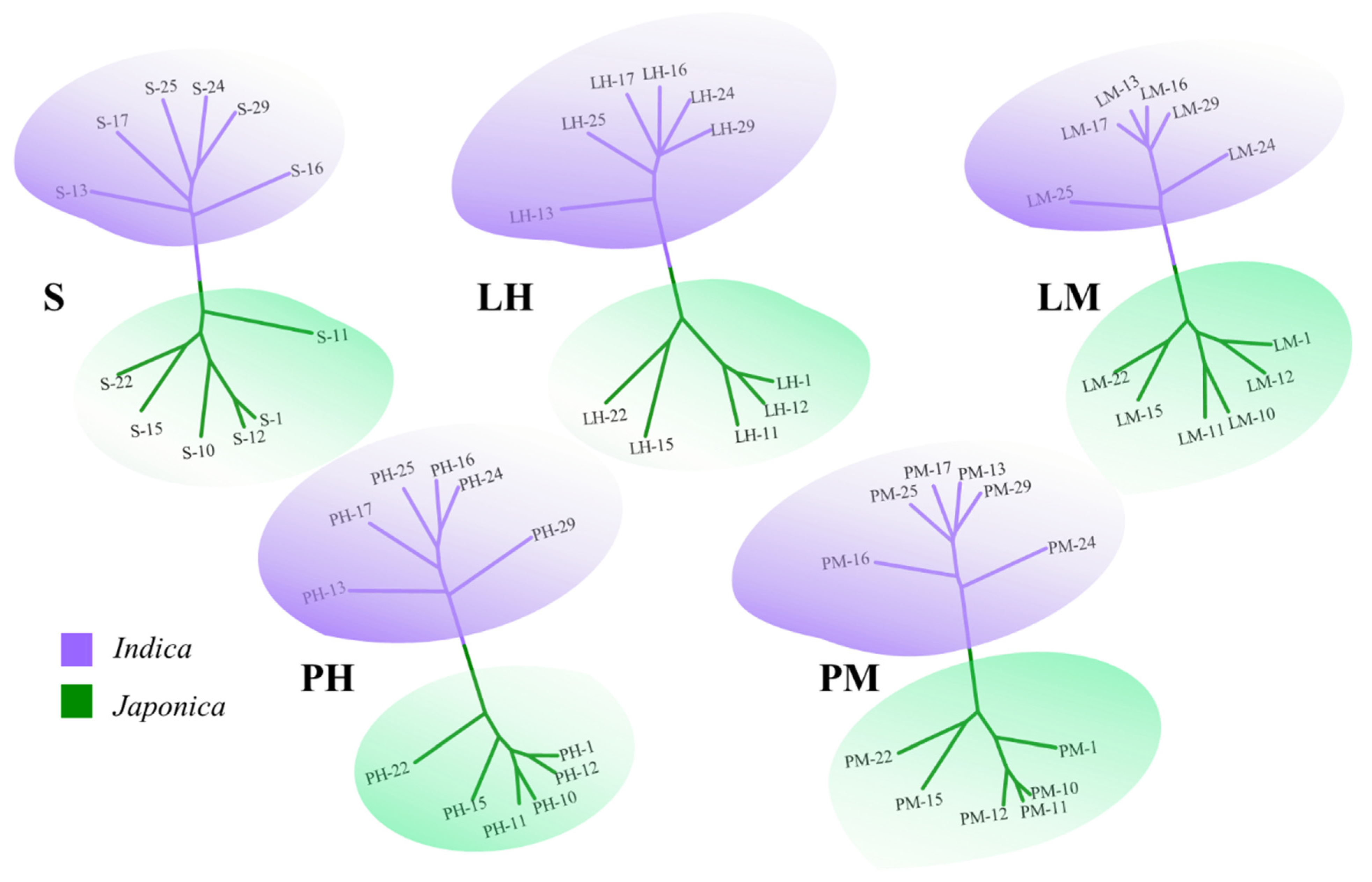
3.2. Divergence at Genome-Wide DNA Sequence Level and Expression Level between the Two Subspecies of Rice
4. Discussion
4.1. Significant Divergence in Morphological Traits between the Two Subspecies of Rice
4.2. Divergence between Indica and Japonica Rice at Both DNA Sequence and Expression Levels
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- King, M.C.; Wilson, A.C. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar] [CrossRef]
- Wilson, A.C.; Maxson, L.R.; Sarich, V.M. Two types of molecular evolution evidence from studies of interspecific hybridization. Proc. Natl. Acad. Sci. USA 1974, 71, 2843–2847. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Doebley, J.; Stec, A.; Hubbard, L. The evolution of apical dominance in maize. Nature 1997, 386, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Konishi, S.; Izawa, T.; Lin, S.Y.; Ebana, K.; Fukuta, Y.; Sasaki, T.; Yano, M. An SNP caused loss of seed shattering during rice domestication. Science 2006, 312, 1392–1396. [Google Scholar] [CrossRef]
- Li, C.B.; Zhou, A.L.; Sang, T. Rice domestication by reducing shattering. Science 2006, 311, 1936–1939. [Google Scholar] [CrossRef]
- Purugganan, M.D.; Fuller, D.Q. The nature of selection during plant domestication. Nature 2009, 457, 843–848. [Google Scholar] [CrossRef]
- Romero, I.G.; Ruvinsky, I.; Gilad, Y. Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 2012, 13, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Groen, S.C.; Calic, I.; Joly-Lopez, Z.; Platts, A.E.; Choi, J.Y.; Natividad, M.; Dorph, K.; Mauck, W.M.; Bracken, B.; Cabral, C.L.U.; et al. The strength and pattern of natural selection on gene expression in rice. Nature 2020, 578, 572–576. [Google Scholar] [CrossRef]
- Khush, G.S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef]
- Sang, T.; Ge, S. Genetics and phylogenetics of rice domestication. Curr. Opin. Genet. Dev. 2007, 17, 533–538. [Google Scholar] [CrossRef]
- Wang, C.H.; Zheng, X.M.; Xu, Q.; Yuan, X.P.; Huang, L.; Zhou, H.F.; Wei, X.H.; Ge, S. Genetic diversity and classification of Oryza sativa with emphasis on Chinese rice germplasm. Heredity 2014, 112, 489–496. [Google Scholar] [CrossRef]
- Vaughan, D.A.; Lu, B.R.; Tomooka, N. The evolving story of rice evolution. Plant Sci. 2008, 174, 394–408. [Google Scholar] [CrossRef]
- Kovach, M.J.; Sweeney, M.T.; McCouch, S.R. New insights into the history of rice domestication. Trends Genet. 2007, 23, 578–587. [Google Scholar] [CrossRef] [PubMed]
- Bioversity International; International Rice Research Institute; WARDA. Descriptors for Wild and Cultivated Rice (Oryza spp.); Bioversity International: Rome, Italy, 2007. [Google Scholar]
- Wang, M.X.; Jing, C.Y.; Wang, X.H.; Cai, Z.; Zhou, L.; Geng, M.F.; Han, J.D.; Guo, J.; Zhang, F.M.; Ge, S. Genomic landscape of parallel domestication of upland rice and its implications. J. Syst. Evol. 2021, 59, 229–239. [Google Scholar] [CrossRef]
- Li, J.Y.; Wang, J.; Zeigler, R.S. The 3000 rice genomes project: New opportunities and challenges for future rice research. GigaScience 2014, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. Differential expression analysis of complex RNA-seq experiments using edgeR. In Statistical Analysis of Next Generation Sequence Data; Datta, S., Nettleton, D., Eds.; Springer: New York, NY, USA, 2014; pp. 25–50. [Google Scholar]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.6; University of Washington: Seattle, WA, USA, 2005; Available online: https://phylipweb.github.io/phylip/ (accessed on 1 January 2021).
- Stebbins, G. Variation and Evolution in Plants; Columbia University Press: New York, NY, USA, 1950. [Google Scholar]
- Futuyma, D.J. Evolution, 3rd ed.; Sinauer Associates: Sunderland, MA, USA, 2013. [Google Scholar]
- Wu, W.X.; Zheng, X.M.; Lu, G.W.; Zhong, Z.Z.; Gao, H.; Chen, L.P.; Wu, C.Y.; Wang, H.J.; Wang, Q.; Zhou, K.N.; et al. Association of functional nucleotide polymorphisms at DTH2 with the northward expansion of rice cultivation in Asia. Proc. Natl. Acad. Sci. USA 2013, 110, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Lee, S.; Kim, T.H.; Lee, J.H.; Park, J.; Lee, J.; Lee, J.Y.; Cho, L.H.; Choi, J.Y.; Lee, W.; et al. Natural variations at the Stay-Green gene promoter control lifespan and yield in rice cultivars. Nat. Commun. 2020, 11, 2819. [Google Scholar] [CrossRef] [PubMed]
- Garris, A.J.; Tai, T.H.; Coburn, J.; Kresovich, S.; McCouch, S. Genetic structure and diversity in Oryza sativa L. Genetics 2005, 169, 1631–1638. [Google Scholar] [CrossRef]
- Bolnick, D.I.; Barrett, R.D.H.; Oke, K.B.; Rennison, D.J.; Stuart, Y.E. (Non)Parallel evolution. Annu. Rev. Ecol. Evol. Syst. 2018, 49, 303–330. [Google Scholar] [CrossRef]
- Shibata, Y.; Sheffield, N.C.; Fedrigo, O.; Babbitt, C.C.; Wortham, M.; Tewari, A.K.; London, D.; Song, L.; Lee, B.K.; Iyer, V.R.; et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012, 8, e1002789. [Google Scholar] [CrossRef]
- Guo, J.; Liu, R.; Huang, L.; Zheng, X.M.; Liu, P.L.; Du, Y.S.; Cai, Z.; Zhou, L.; Wei, X.H.; Zhang, F.M.; et al. Widespread and adaptive alterations in genome-wide gene expression associated with ecological divergence of two Oryza species. Mol. Biol. Evol. 2016, 33, 62–78. [Google Scholar] [CrossRef]
- Wolf, J.B.W.; Bayer, T.; Haubold, B.; Schilhabel, M.; Rosenstiel, P.; Tautz, D. Nucleotide divergence vs. gene expression differentiation: Comparative transcriptome sequencing in natural isolates from the carrion crow and its hybrid zone with the hooded crow. Mol. Ecol. 2010, 19, 162–175. [Google Scholar] [CrossRef]
- Martinez-Fernandez, M.; Bernatchez, L.; Rolan-Alvarez, E.; Quesada, H. Insights into the role of differential gene expression on the ecological adaptation of the snail Littorina saxatilis. BMC Evol. Biol. 2010, 10, 356. [Google Scholar] [CrossRef]
- Nosil, P. Ecological Speciation; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Seymour, D.K.; Koenig, D.; Hagmann, J.; Becker, C.; Weigel, D. Evolution of DNA methylation patterns in the Brassicaceae is driven by differences in genome organization. PLoS Genet. 2014, 10, e1004785. [Google Scholar] [CrossRef]
- Yu, X.L.; Wei, P.; Zhao, S.Q.; Chen, Z.Y.F.; Li, X.Z.; Zhang, W.C.; Liu, C.L.; Yang, Y.J.; Li, X.Y.; Liu, X. Population transcriptomics uncover the relative roles of positive selection and differential expression in Batrachium bungei adaptation to the Qinghai–Tibetan plateau. Plant Cell Rep. 2023, 42, 879–893. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.C.; Yao, C.T.; Liao, B.Y.; Weng, M.P.; Dong, F.; Hsu, Y.C.; Hung, C.M. Weak gene–gene interaction facilitates the evolution of gene expression plasticity. BMC Biol. 2023, 21, 57. [Google Scholar] [CrossRef] [PubMed]
- Jing, C.Y.; Zhang, F.M.; Wang, X.H.; Wang, M.X.; Zhou, L.; Cai, Z.; Han, J.D.; Geng, M.F.; Yu, W.H.; Jiao, Z.H.; et al. Multiple domestications of Asian rice. Nat. Plants 2023, 9, 1221–1235. [Google Scholar] [CrossRef] [PubMed]
- Pickersgill, B. Parallel vs. convergent evolution in domestication and diversification of crops in the Americas. Front. Ecol. Evol. 2018, 6, 56. [Google Scholar] [CrossRef]
- Martinez-Ainsworth, N.E.; Tenaillon, M.I. Superheroes and masterminds of plant domestication. Comptes Rendus Biol. 2016, 339, 268–273. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Accession No. | Sample Origin | Biological Repetition |
|---|---|---|---|
| TEJ 1 ** | 680 | Zhejiang | 8 |
| TEJ 2 | IRGC2545 | Japan | 6 |
| TEJ 10 ** | 20708 | Shanxi | 8 |
| TEJ 11 ** | 20721 | Liaoning | 6 |
| TEJ 12 ** | 20876 | Jiangsu | 7 |
| TEJ 14 | 21773 | Yunnan | 4 |
| TEJ 27 * | 49357 | Guangdong | 8 |
| TEJ 28 | IRGC55457 | Korea | 9 |
| TEJ 30 | Nipponbare | Japan | 8 |
| TRJ 3 | IRGC3782 | Philippines | 2 |
| TRJ 15 ** | 22128 | Guizhou | 5 |
| TRJ 22 ** | 32447 | Yunnan | 6 |
| TRJ 26 | IRGC43675 | Indonesia | 8 |
| IND 7 | IRGC9147 | Indonesia | 8 |
| IND 13 ** | 21327 | Hubei | 7 |
| IND 16 ** | 22233 | Guizhou | 7 |
| IND 17 ** | 24380 | Yunnan | 6 |
| IND 19 | IRGC27748 | Thailand | 8 |
| IND 24 ** | 36467 | Guangxi | 4 |
| IND 25 ** | 41949 | Hainan | 5 |
| IND 29 ** | 60993 | Jiangxi | 8 |
| Total | 138 |
| Quantitative Traits | CV (Standard Deviation/Mean) | Mean ± Standard Deviation (SD) | ||||
|---|---|---|---|---|---|---|
| Japonica | Indica | Japonica | Indica | |||
| TEJ | TRJ | TEJ | TRJ | |||
| Days from seeding to first heading date (d) | 0.20 | 0.13 | 0.26 | 107.68 ± 21.20 a | 122.24 ± 15.66 b | 130.36 ± 33.59 b |
| Ligule length (cm) | 0.46 | 0.41 | 0.33 | 1.06 ± 0.49 a | 1.81 ± 0.74 b | 2.32 ± 0.75 c |
| Flag leaf length (cm) | 0.35 | 0.29 | 0.41 | 21.40 ± 7.53 a | 31.71 ± 9.19 b | 30.87 ± 12.63 b |
| Flag leaf width (cm) | 0.28 | 0.22 | 0.17 | 1.02 ± 0.28 a | 1.55 ± 0.34 b | 1.27 ± 0.22 c |
| Flag leaf attitude (°) | 0.63 | 0.54 | 0.58 | 21.89 ± 13.75 a | 71.00 ± 38.37 b | 26.19 ± 15.17 a |
| Culm habit (°) | 0.65 | 0.94 | 0.55 | 14.57 ± 9.42 a | 11.33 ± 10.60 a | 21.36 ± 11.78 b |
| Culm diameter at basal internode (cm) | 0.37 | 0.49 | 0.46 | 0.60 ± 0.23 a | 0.87 ± 0.42 b | 0.92 ± 0.42 b |
| Culm length (cm) | 0.33 | 0.16 | 0.22 | 91.59 ± 30.37 a | 133.53 ± 21.26 b | 103.82 ± 23.22 c |
| Attitude of flowering panicle branches (°) | 6.78 | — | 5.44 | 1.09 ± 7.37 | 0.00 ± 0.00 | 0.57 ± 3.09 |
| Attitude of seed setting panicle branches (°) | 3.71 | 1.92 | 2.41 | 2.17 ± 8.07 | 4.67 ± 8.96 | 4.50 ± 10.85 |
| Anther length (cm) | 0.27 | 0.25 | 0.27 | 0.15 ± 0.04 | 0.16 ± 0.04 | 0.16 ± 0.04 |
| Peduncle length (cm) | 0.87 | 0.73 | 1.38 | 5.48 ± 4.79 a | 4.58 ± 3.34 ab | 2.76 ± 3.80 b |
| Panicle length (cm) | 0.32 | 0.25 | 0.15 | 17.54 ± 5.55 a | 25.92 ± 6.39 b | 23.39 ± 3.40 b |
| Number of spikelets per panicle | 0.63 | 0.44 | 0.36 | 67.07 ± 42.20 a | 138.38 ± 60.37 b | 122.47 ± 44.63 b |
| Awn length (cm) | 2.34 | 1.34 | 2.03 | 0.40 ± 0.92 a | 1.12 ± 1.50 b | 0.27 ± 0.55 a |
| Number of panicles per plant | 0.49 | 0.47 | 0.52 | 5.93 ± 2.93 | 4.40 ± 2.06 | 5.53 ± 2.87 |
| Tissue | EEGs | DEGs | DEGs Total (% of EEGs) | |
|---|---|---|---|---|
| Up-Regulated | Down-Regulated | |||
| S | 23,477 | 2320 | 1006 | 3326 (14.2%) |
| LH | 22,917 | 1727 | 919 | 2646 (11.5%) |
| PH | 26,406 | 1880 | 1036 | 2916 (11.0%) |
| LM | 23,377 | 2052 | 1561 | 3613 (15.5%) |
| PM | 25,260 | 1699 | 799 | 2498 (9.9%) |
| Mean | 24,287 | 1936 | 1064 | 3000 (12.4%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Huang, L.; Kou, Y.; Li, D.; Hu, W.; Fan, D.; Cheng, S.; Yang, Y.; Zhang, Z. Differentiation of Morphological Traits and Genome-Wide Expression Patterns between Rice Subspecies Indica and Japonica. Genes 2023, 14, 1971. https://doi.org/10.3390/genes14101971
Wang M, Huang L, Kou Y, Li D, Hu W, Fan D, Cheng S, Yang Y, Zhang Z. Differentiation of Morphological Traits and Genome-Wide Expression Patterns between Rice Subspecies Indica and Japonica. Genes. 2023; 14(10):1971. https://doi.org/10.3390/genes14101971
Chicago/Turabian StyleWang, Meixia, Lei Huang, Yixuan Kou, Danqi Li, Wan Hu, Dengmei Fan, Shanmei Cheng, Yi Yang, and Zhiyong Zhang. 2023. "Differentiation of Morphological Traits and Genome-Wide Expression Patterns between Rice Subspecies Indica and Japonica" Genes 14, no. 10: 1971. https://doi.org/10.3390/genes14101971