

STS and PUDP Deletion Identified by Targeted Panel Sequencing with CNV Analysis in X-Linked Ichthyosis: A Case Report and Literature Review


Abstract
:1. Introduction
2. Materials and Methods
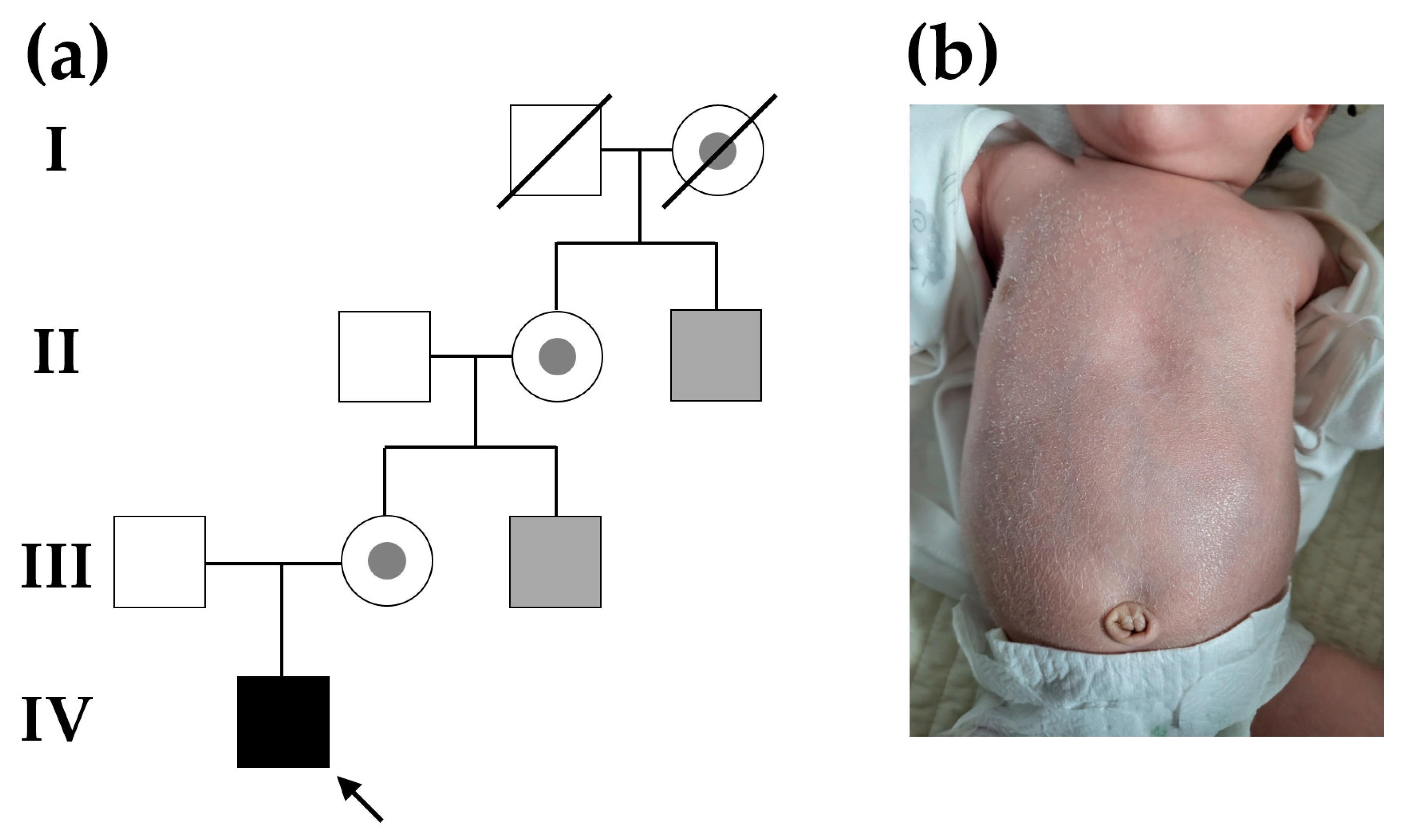
2.1. Patient
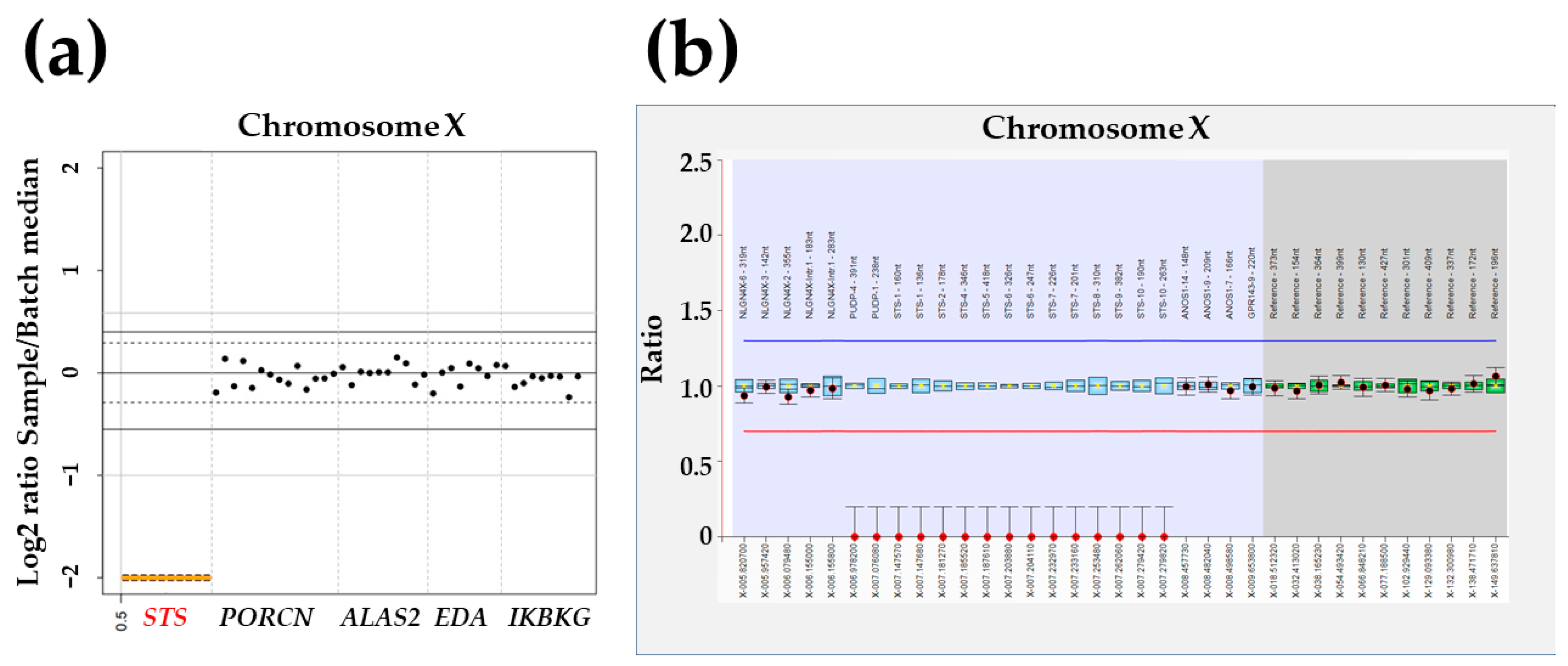
2.2. Targeted Panel Sequencing
2.3. Multiplex Ligation-Dependent Probe Amplification
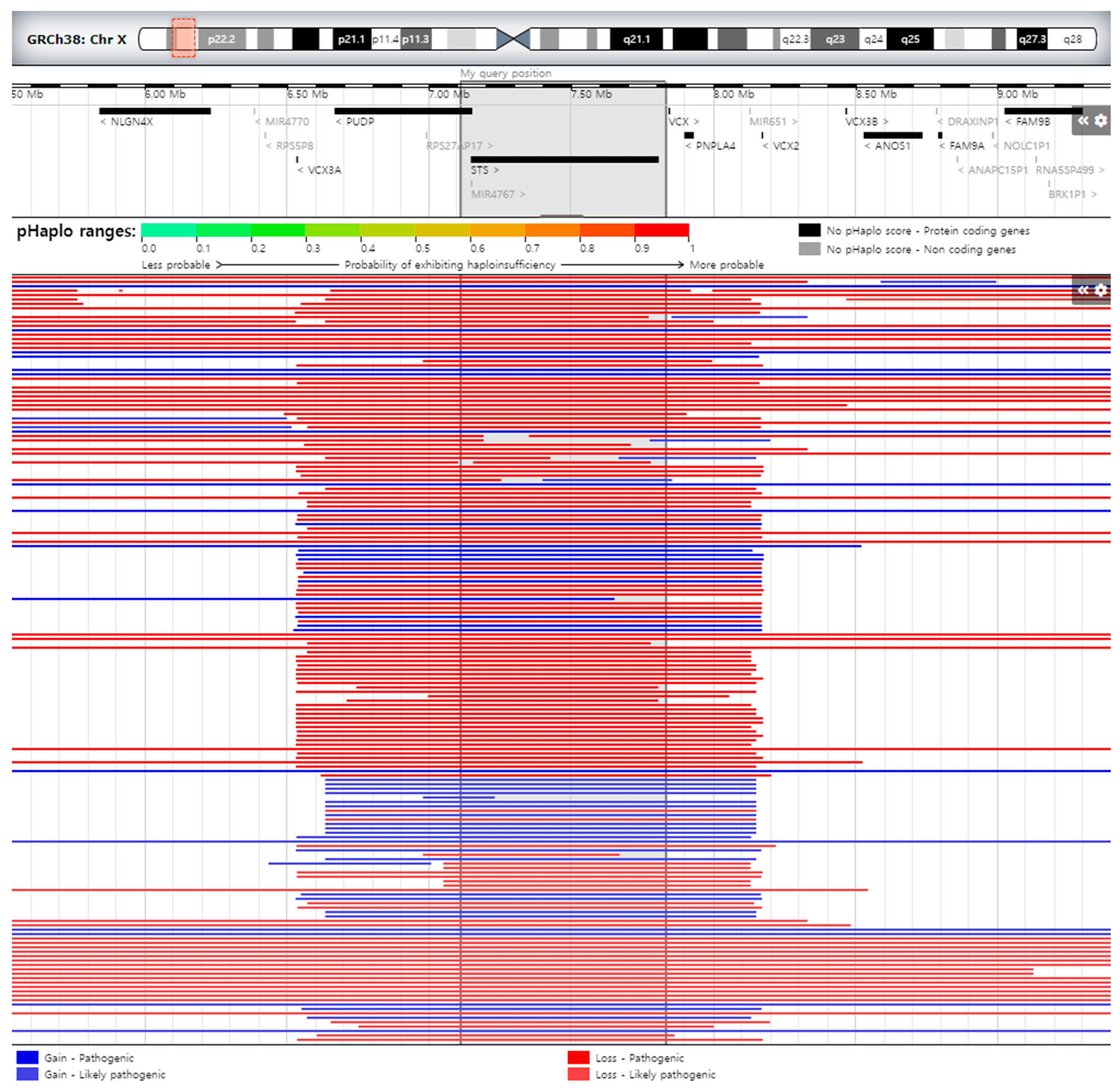
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Craiglow, B.G. Ichthyosis in the newborn. Semin. Perinatol. 2013, 37, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Traupe, H. Revealing the mysteries of X-linked recessive ichthyosis. Br. J. Dermatol. 2018, 179, 821–822. [Google Scholar] [CrossRef] [PubMed]
- Takeichi, T.; Akiyama, M. Inherited ichthyosis: Non-syndromic forms. J. Dermatol. 2016, 43, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.H.; Kim, S.Y.; Kim, J.K. A case of 9.7 Mb terminal Xp deletion including OA1 locus associated with contiguous gene syndrome. J. Korean Med. Sci. 2012, 27, 1273–1277. [Google Scholar] [CrossRef]
- Lee, N.R.; Yoon, N.Y.; Jung, M.; Kim, J.-Y.; Seo, S.J.; Wang, H.-y.; Lee, H.; Sohn, Y.B.; Choi, E.H. Skin Barrier Function Is Not Impaired and Kallikrein 7 Gene Polymorphism Is Frequently Observed in Korean X-linked Ichthyosis Patients Diagnosed by Fluorescence in Situ Hybridization and Array Comparative Genomic Hybridization. J. Korean Med. Sci. 2016, 31, 1307–1318. [Google Scholar] [CrossRef]
- Yun, J.M.; Na, K.S.; Kim, M.S.; Kim, H.S.; Hwang, H.B. Two Cases of Pre-descemet Corneal Dystrophy Associated with X-linked Ichthyosis: A Case Report by Genetic Analysis. J. Korean Ophthalmol. Soc. 2017, 58, 993–997. [Google Scholar] [CrossRef]
- Park, S.J.; Park, K.H.; Bae, M.H.; Kim, Y.M. Developmental Delay in Children with X-Linked Ichthyosis: A Case Series. Ann. Child. Neurol. 2021, 29, 186–189. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Pugh, T.J.; Amr, S.S.; Bowser, M.J.; Gowrisankar, S.; Hynes, E.; Mahanta, L.M.; Rehm, H.L.; Funke, B.; Lebo, M.S. VisCap: Inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet. Med. 2016, 18, 712–719. [Google Scholar] [CrossRef]
- Liu, P.; Erez, A.; Nagamani, S.C.; Bi, W.; Carvalho, C.M.; Simmons, A.D.; Wiszniewska, J.; Fang, P.; Eng, P.A.; Cooper, M.L.; et al. Copy number gain at Xp22.31 includes complex duplication rearrangements and recurrent triplications. Hum. Mol. Genet. 2011, 20, 1975–1988. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Daoud, H.; Luco, S.M.; Li, R.; Bareke, E.; Beaulieu, C.; Jarinova, O.; Carson, N.; Nikkel, S.M.; Graham, G.E.; Richer, J.; et al. Next-generation sequencing for diagnosis of rare diseases in the neonatal intensive care unit. Cmaj 2016, 188, E254–E260. [Google Scholar] [CrossRef]
- Lonardo, F.; Parenti, G.; Luquetti, D.V.; Annunziata, I.; Della Monica, M.; Perone, L.; De Gregori, M.; Zuffardi, O.; Brunetti-Pierri, N.; Andria, G.; et al. Contiguous gene syndrome due to an interstitial deletion in Xp22.3 in a boy with ichthyosis, chondrodysplasia punctata, mental retardation and ADHD. Eur. J. Med. Genet. 2007, 50, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Mao, J.; Wang, X.; Duan, L.; Song, Y.; Lian, X.; Zheng, J.; Liu, Z.; Nie, M.; Wu, X. Novel Microdeletion in the X Chromosome Leads to Kallmann Syndrome, Ichthyosis, Obesity, and Strabismus. Front. Genet. 2020, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhou, H.; Zhou, L.; Gong, Y.; Lin, J.; Chen, Y. Clinical features and genetic analysis of two Chinese families with X-linked ichthyosis. J. Int. Med. Res. 2020, 48, 300060520962292. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, H.; Lin, N.; He, S.; An, G.; Wang, Y.; Chen, M.; Chen, L.; Lin, Y.; Xu, L. X-linked ichthyosis: Molecular findings in four pedigrees with inconspicuous clinical manifestations. J. Clin. Lab. Anal. 2020, 34, e23201. [Google Scholar] [CrossRef]
- Boere, P.M.; Bonnet, C.; Frausto, R.F.; Fung, S.S.M.; Aldave, A.J. Multimodal Imaging of Pre-Descemet Corneal Dystrophy Associated with X-Linked Ichthyosis and Deletion of the STS Gene. Cornea 2020, 39, 1442–1445. [Google Scholar] [CrossRef]
- Williams, D.; Onyia, O.; Chung, D.D.; Kirakosyan, A.; Hovakimyan, A.; Payne, C.; Moshirfar, M.; Aldave, A.J. Identification of a novel partial deletion of STS associated with pre-Descemet corneal dystrophy and X-linked ichthyosis. Mol. Vis. 2023, 29, 25–30. [Google Scholar]
- Brcic, L.; Underwood, J.F.; Kendall, K.M.; Caseras, X.; Kirov, G.; Davies, W. Medical and neurobehavioural phenotypes in carriers of X-linked ichthyosis-associated genetic deletions in the UK Biobank. J. Med. Genet. 2020, 57, 692–698. [Google Scholar] [CrossRef]
- Ben Khelifa, H.; Soyah, N.; Ben-Abdallah-Bouhjar, I.; Gritly, R.; Sanlaville, D.; Elghezal, H.; Saad, A.; Mougou-Zerelli, S. Xp22.3 interstitial deletion: A recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene 2013, 527, 578–583. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.; Zhang, L.; Kong, W.; Xie, H.; Wang, J.; Wu, Y.; Wu, X.; Liu, X.; Zhang, Y.; et al. Large De Novo Microdeletion in Epilepsy with Intellectual and Developmental Disabilities, with a Systems Biology Analysis. Adv. Neurobiol. 2018, 21, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Labonne, J.D.J.; Driessen, T.M.; Harris, M.E.; Kong, I.K.; Brakta, S.; Theisen, J.; Sangare, M.; Layman, L.C.; Kim, C.H.; Lim, J.; et al. Comparative Genomic Mapping Implicates LRRK2 for Intellectual Disability and Autism at 12q12, and HDHD1, as Well as PNPLA4, for X-Linked Intellectual Disability at Xp22.31. J. Clin. Med. 2020, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Myers, K.A.; Simard-Tremblay, E.; Saint-Martin, C. X-Linked Familial Focal Epilepsy Associated With Xp22.31 Deletion. Pediatr. Neurol. 2020, 108, 113–116. [Google Scholar] [CrossRef]
- Schierz, I.A.M.; Giuffrè, M.; Cimador, M.; D’Alessandro, M.M.; Serra, G.; Favata, F.; Antona, V.; Piro, E.; Corsello, G. Hypertrophic pyloric stenosis masked by kidney failure in a male infant with a contiguous gene deletion syndrome at Xp22.31 involving the steroid sulfatase gene: Case report. Ital. J. Pediatr. 2022, 48, 19. [Google Scholar] [CrossRef]
- Diociaiuti, A.; Angioni, A.; Pisaneschi, E.; Alesi, V.; Zambruno, G.; Novelli, A.; El Hachem, M. X-linked ichthyosis: Clinical and molecular findings in 35 Italian patients. Exp. Dermatol. 2019, 28, 1156–1163. [Google Scholar] [CrossRef]
- Hu, H.; Huang, Y.; Hou, R.; Xu, H.; Liu, Y.; Liao, X.; Xu, J.; Jiang, L.; Wang, D. Xp22.31 copy number variations in 87 fetuses: Refined genotype-phenotype correlations by prenatal and postnatal follow-up. BMC Med. Genom. 2023, 16, 69. [Google Scholar] [CrossRef] [PubMed]
- Souche, E.; Beltran, S.; Brosens, E.; Belmont, J.W.; Fossum, M.; Riess, O.; Gilissen, C.; Ardeshirdavani, A.; Houge, G.; van Gijn, M.; et al. Recommendations for whole genome sequencing in diagnostics for rare diseases. Eur. J. Hum. Genet. 2022, 30, 1017–1021. [Google Scholar] [CrossRef]
- de Castro, M.J.; González-Vioque, E.; Barbosa-Gouveia, S.; Salguero, E.; Rite, S.; López-Suárez, O.; Pérez-Muñuzuri, A.; Couce, M.L. Rapid Phenotype-Driven Gene Sequencing with the NeoSeq Panel: A Diagnostic Tool for Critically Ill Newborns with Suspected Genetic Disease. J. Clin. Med. 2020, 9, 2362. [Google Scholar] [CrossRef]
- Hosomi, N.; Oiso, N.; Fukai, K.; Hanada, K.; Fujita, H.; Ishii, M. Deletion of distal promoter of VCXA in a patient with X-linked ichthyosis associated with borderline mental retardation. J. Dermatol. Sci. 2007, 45, 31–36. [Google Scholar] [CrossRef]
- Abdel-Hamed, M.F.; Hussein, H.A.; Helmy, N.A.; Elsaie, M.L. The detection of steroid sulfatase gene deletion (STS) in Egyptian males with X-linked ichthyosis. J. Drugs Dermatol. 2010, 9, 1192–1196. [Google Scholar]
- Ramesh, R.; Chen, H.; Kukula, A.; Wakeling, E.L.; Rustin, M.H.; McLean, W.H. Exacerbation of X-linked ichthyosis phenotype in a female by inheritance of filaggrin and steroid sulfatase mutations. J. Dermatol. Sci. 2011, 64, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xi, Q.; Liu, X.; Yue, F.; Zhang, H.; Sun, M.; Liu, R. Prenatal Diagnosis and Molecular Cytogenetic Characterization of Copy Number Variations on 4p15.2p16.3, Xp22.31, and 12p11.1q11 in a Fetus with Ultrasound Anomalies: A Case Report and Literature Review. Biomed. Res. Int. 2020, 2020, 1761738. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Zhang, J.; Yang, L.; Bai, J.; Fan, P. Detection of Significant Copy Number Variations From Multiple Samples in Next-Generation Sequencing Data. IEEE Trans. Nanobiosci. 2018, 17, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Royer-Bertrand, B.; Cisarova, K.; Niel-Butschi, F.; Mittaz-Crettol, L.; Fodstad, H.; Superti-Furga, A. CNV Detection from Exome Sequencing Data in Routine Diagnostics of Rare Genetic Disorders: Opportunities and Limitations. Genes 2021, 12, 1427. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| AAGAB, ABCA12, ALAS2, ALOX12B, ALOXE3, ANAPC1, APCDD1, AQP5, CARD14, CAST, CDH3, CDSN, CERS3, COL17A1, COL7A1, CSTA, CTSC, CYP4F22, DSC2, DSG1, DSG4, DSP, DST, EDA, EDAR, EDARADD, ENPP1, EXPH5, FECH, FERMT1, GJA1, GJB2, GJB3, GJB4, GJB6, HOXC13, HR, IKBKG, ITGA3, ITGA6, ITGB4, JUP, KDSR, KLHL24, KRT1, KRT10, KRT14, KRT16, KRT17, KRT5, KRT6A, KRT6B, KRT6C, KRT74, KRT85, KRT9, LAMA3, LAMB3, LAMC2, LIPH, LPAR6, LSS, MSX1, NECTIN1, NECTIN4, NFKB2, NFKBIA, NIPAL4, PKP1, PLEC, PNPLA1, PORCN, RHBDF2, RMRP, RSPO1, SDR9C7, SERPINB7, SERPINB8, SLC27A4, SLURP1, SNAP29, ST14, STS, SULT2B1, TAT, TGM1, TGM5, TP63, TRPV3, TSPEAR, WNT10A, ALDH3A2, C3orf52, SMARCAD1, SNRPE |
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | Case 7 | |
|---|---|---|---|---|---|---|---|
| Reference | [4] | [6] | [6] | [7] | [7] | [7] | This study |
| Method | CMA | MLPA | MLPA | CMA | CMA | CMA | MPS,MLPA |
| Deletion size | 9.7 Mb | 0.720 Mb? | 0.720 Mb? | 1.686 Mb | 1.654 Mb | 1.670 Mb | 0.719 Mb |
| Deletion range | Xpter to 9,786,297 | 7,108,996 to 7,828,312? | 7,108,996 to 7,828,312? | 6,455,151 to 8,141,076 | 6,473,896 to 8,127,579 | 6,458,939 to 8,143,509 | 7,108,996 to 7,828,312 |
| Age at Dx | 13.5 yr | 19 yr | 19 yr | 7 yr | 17 mo | 3 yr | 1 yr |
| Sex | Male | Male | Male | Male | Male | Female | Male |
| Symptoms | |||||||
| Skin | |||||||
| Dry skin | P | P | P | P | P | P | P |
| Ichthyosis | P | P | P | P | P | P | P |
| Eye | |||||||
| Myopia | P | N | N | P | N | N | N |
| Strabismus | P | N | N | N | N | P | N |
| Macular hypoplasia | P | N | N | N | N | N | N |
| Hypopigmented fundus | P | N | N | N | N | N | N |
| Pre-dCD | N | P | P | N | N | N | N |
| Neurological | |||||||
| Cognitive impairment | P | N | N | P | P | P | N |
| Delayed GMD | P | N | N | P | P | P | N |
| GDD | P | N | N | P | P | P | N |
| Epilepsy | N | N | N | P | N | N | N |
| IAH | N | N | N | P | N | P | N |
| Face | |||||||
| Dysmorphism | P | N | N | N | N | N | N |
| Cleft lip | P | N | N | N | N | N | N |
| Bone & growth | |||||||
| Short stature | P | N | N | N | N | N | N |
| Skeletal malformation | P | N | N | N | N | N | N |
| Scoliosis | N | N | N | P | N | N | N |
| Others | |||||||
| CoA | N | N | N | P | N | N | N |
| IUGR | N | N | N | P | P | N | N |
| Premature birth | N | N | N | P | N | N | N |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Cho, Y.G.; Kim, J.K.; Kim, H.H. STS and PUDP Deletion Identified by Targeted Panel Sequencing with CNV Analysis in X-Linked Ichthyosis: A Case Report and Literature Review. Genes 2023, 14, 1925. https://doi.org/10.3390/genes14101925
Park J, Cho YG, Kim JK, Kim HH. STS and PUDP Deletion Identified by Targeted Panel Sequencing with CNV Analysis in X-Linked Ichthyosis: A Case Report and Literature Review. Genes. 2023; 14(10):1925. https://doi.org/10.3390/genes14101925
Chicago/Turabian StylePark, Joonhong, Yong Gon Cho, Jin Kyu Kim, and Hyun Ho Kim. 2023. "STS and PUDP Deletion Identified by Targeted Panel Sequencing with CNV Analysis in X-Linked Ichthyosis: A Case Report and Literature Review" Genes 14, no. 10: 1925. https://doi.org/10.3390/genes14101925