
The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases
by
 , ,
, ,
Marco Salluzzo
1,
Clara Vianello
2,†,
Sandra Abdullatef
3,
Roberto Rimondini
2,
Giovanni Piccoli
3 and
Lucia Carboni
1,* 1
Department of Pharmacy and Biotechnology, Alma Mater Studiorum University of Bologna, 40126 Bologna, Italy
2
Department of Medical and Surgical Sciences, Alma Mater Studiorum University of Bologna, 40126 Bologna, Italy
3
Department of Cellular, Computational and Integrative Biology, University of Trento, 38123 Trento, Italy
*
Author to whom correspondence should be addressed.
†
Present address: Department for Life Quality Studies, Alma Mater Studiorum University of Bologna, 47921 Rimini, Italy.
Genes 2023, 14(10), 1886; https://doi.org/10.3390/genes14101886
Submission received: 1 September 2023
/
Revised: 26 September 2023
/
Accepted: 27 September 2023
/
Published: 28 September 2023
(This article belongs to the Special Issue Study on Genotypes and Phenotypes of Neurodegenerative Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:In the brain, cell adhesion molecules (CAMs) are critical for neurite outgrowth, axonal fasciculation, neuronal survival and migration, and synapse formation and maintenance. Among CAMs, the IgLON family comprises five members: Opioid Binding Protein/Cell Adhesion Molecule Like (OPCML or OBCAM), Limbic System Associated Membrane Protein (LSAMP), neurotrimin (NTM), Neuronal Growth Regulator 1 (NEGR1), and IgLON5. IgLONs exhibit three N-terminal C2 immunoglobulin domains; several glycosylation sites; and a glycosylphosphatidylinositol anchoring to the membrane. Interactions as homo- or heterodimers in cis and in trans, as well as binding to other molecules, appear critical for their functions. Shedding by metalloproteases generates soluble factors interacting with cellular receptors and activating signal transduction. The aim of this review was to analyse the available data implicating a role for IgLONs in neuropsychiatric disorders. Starting from the identification of a pathological role for antibodies against IgLON5 in an autoimmune neurodegenerative disease with a poorly understood mechanism of action, accumulating evidence links IgLONs to neuropsychiatric disorders, albeit with still undefined mechanisms which will require future thorough investigations.
1. Introduction
Cell adhesion molecules (CAMs) placed on the cell surface perform critical functions in a number of biological processes requiring contact between cells or with the extracellular matrix, such as cell recognition, adhesion, migration, and differentiation. The principal CAM groups are integrins, selectins, cadherins, and the immunoglobulin superfamily. In the developing brain, CAMs are crucial for the correct assembly of synaptic connections as well as the interactions with supporting glial cells. In the mature brain, complex functions depend on the correct performance of CAMs in establishing contacts between neuronal bodies, axonal interactions with myelinating glial cells, axon fasciculation, and connections to non-nervous cells [1,2]. Integrins are type I transmembrane proteins consisting of a large multidomain extracellular portion, a single-pass transmembrane region, and a short cytoplasmic component. The integrin family encompasses heterodimers of α and β subunits, which can combine to form several different integrins exhibiting overlapping but non-redundant functions, with specific ligand and signalling preferences depending on the α and β subunit combinations [3,4]. Selectins consist of an N-terminal carbohydrate-recognition domain allowing for the binding to glycoconjugates, an epidermal growth factor-like domain, a series of short consensus repeats, a transmembrane region, and a short C-terminal intracellular tail. Three selectin family members exist, P-selectin, L-selectin, and E-selectin, expressed in platelet-endothelial cells, leukocytes, and endothelial cells, respectively [5,6]. In the brain, selectins expressed by endothelial cells are involved in inflammatory responses, in damage after ischemic events, and in autoimmune diseases [6,7,8]. Cadherins are a large number of calcium-dependent adhesion proteins. Their structure comprises a calcium-binding extracellular domain consisting of several cadherin repeats of about 100 amino acids, a transmembrane domain, and a cytoplasmic domain which interacts with signalling molecules. Cadherins can form lateral dimers (cis-dimers) as well as trans-dimers with cadherins expressed by other cells [9,10].
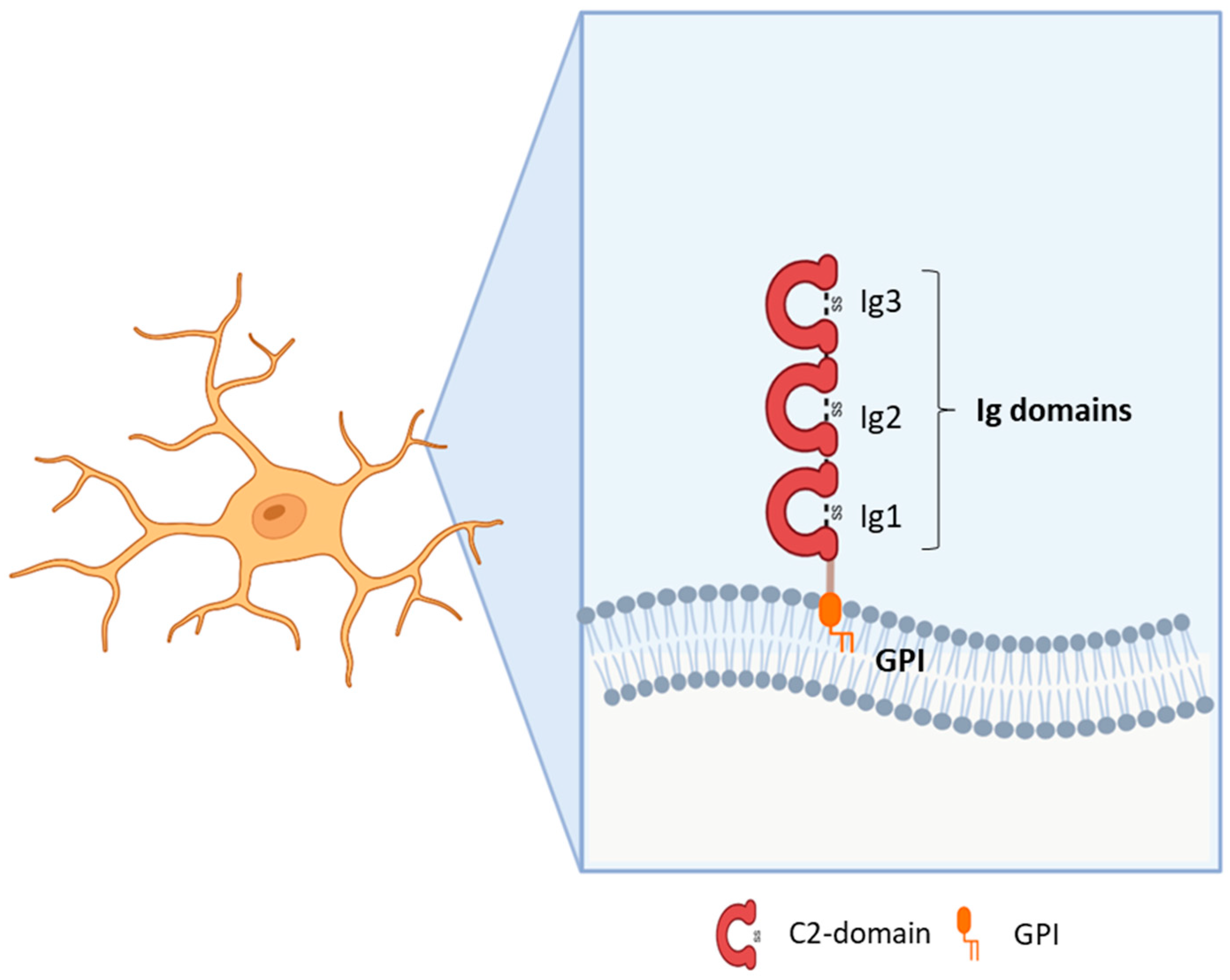
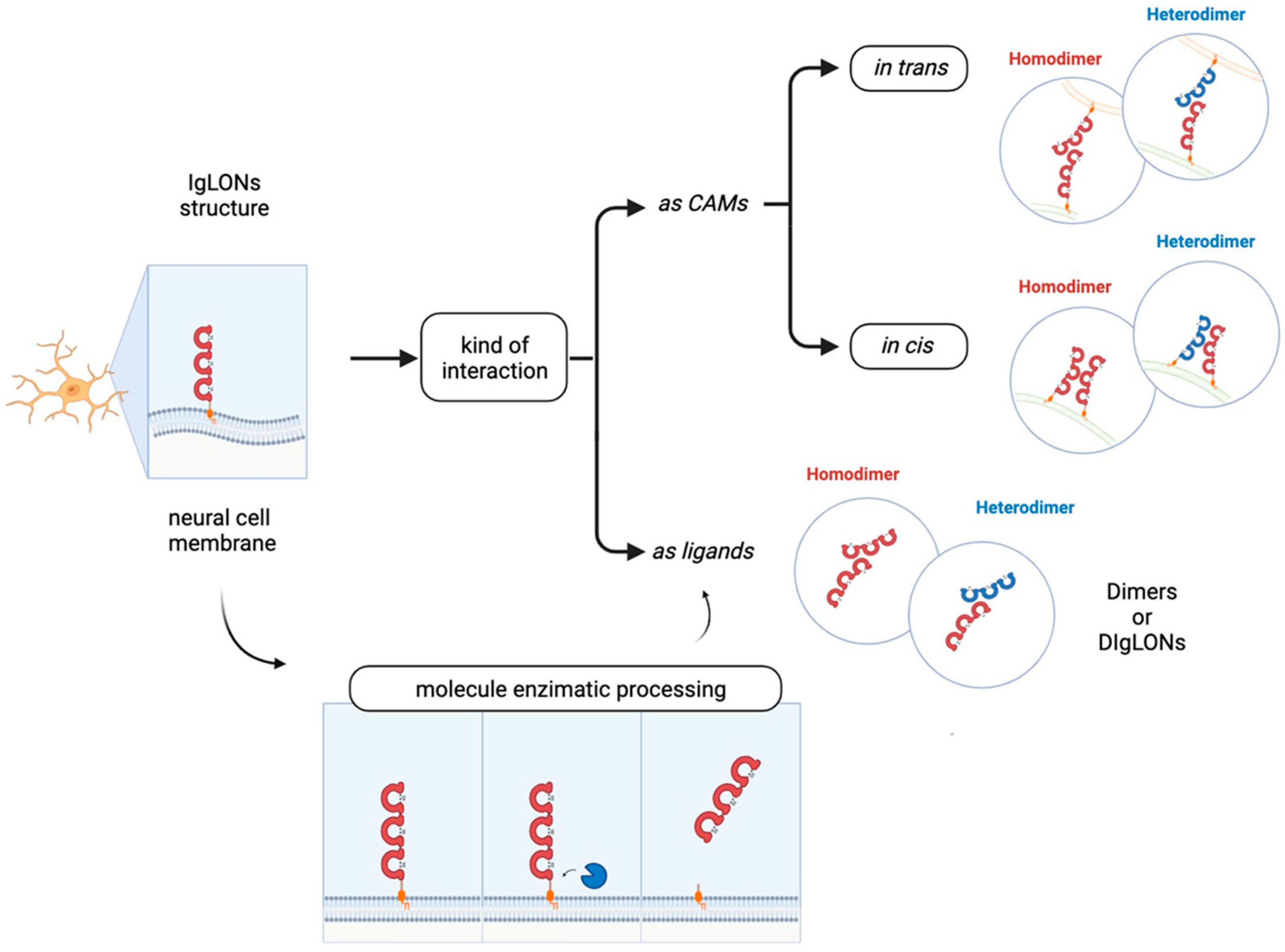
Immunoglobulin (Ig)-like molecules are an ancient and diverse family of proteins performing a variety of functions, such as immune and signalling molecules and CAMs. Four different subtypes of Ig-like domains exist which are named constant 1, constant 2, variable, and intermediate (C1, C2, V, and I) for their resemblance to immunoglobulin domains. The presence of at least one Ig-like domain is a requirement of this class [11,12]. Generally, the core of all Ig domains encompasses two β-sheets facing each other and an intra-chain disulfide bridge which provides stability to the structure [13]. In addition, variable numbers of fibronectin type III domains or other protein modules characterise the sub-families within this large group. Ig-like CAMs generally include type I transmembrane proteins with a large N-terminal domain, a transmembrane portion, and a cytoplasmic domain [1,14]. However, some CAMs are anchored to the membrane through a glycosylphosphatidylinositol (GPI) segment [15]. Some proteins demonstrate homophilic binding specificity, whereas others have heterophilic specificity, thus interacting with other Ig-like CAMs or with surface proteins [1,16]. Ig domains play critical roles in mediating homophilic and heterophilic interactions in trans, namely between CAMs on adjacent cells or CAMs localised to the extracellular environment, as well as in cis, i.e., with proteins located in the plasma membrane of the same cell [17,18]. The first Ig-like CAMs identified in the central nervous system (CNS) were neural cell adhesion molecule (NCAM) and L1, which, along with their isoforms, are recognised for their major functions in axon outgrowth and fasciculation, neuronal survival and migration, synapse formation, and synaptic plasticity [11,14,19]. Subsequent research led to the identification of additional sub-families, such as the nectins, MAM domain–containing GPI anchors, IGSF9, IGSF21, contactins, and IgLONs [11,20]. The IgLON subfamily, which is the focus of this review, includes five members sharing the presence of three C2 immunoglobulin domains in the N-terminus and a GPI-anchoring to the membrane (Figure 1). The first family members identified were Limbic System Associated Membrane Protein (LSAMP, initially named LAMP) [21], Opioid Binding Protein/Cell Adhesion Molecule Like (OPCML, also known as OBCAM) [22], and neurotrimin (NTM) [23]. Indeed, the name IgLON stands for Ig family containing LAMP, OBCAM, NTM. Subsequently, another family member was identified initially as KILON/neurotractin, and later re-named Neuronal Growth Regulator 1 (NEGR1) [24,25]. Lastly, antibodies against the IgLON member IgLON5 (IgLON Family Member 5) have been associated with autoimmune encephalitis [26]. Since the discovery of this family, its prominent roles in brain development, axon fasciculation, neurite extension, and synapse formation and maintenance have been identified [21,22,23,24,25]. Although the role in synapse regulation is well-established, further research is needed to characterise the molecular mechanisms underlying the functional outcomes [17,27,28,29,30]. A meaningful contribution to the comprehension of IgLONs’ physiological functions derived from the analysis of its molecular evolution, originating from an ancestor around the emergence of Arthropods [31]. Functional analysis allowed for the identification of common as well as specific sequences for interacting partner recognition, post-translational modifications, metalloproteinase cleavage sites, and signal transduction pathways [31].
Available evidence supports the involvement of CAMs in the onset of neuropsychiatric disorders. In Alzheimer’s disease, a role for CAMs has been suggested by genome-wide association studies (GWAS) as well as by the identification of altered levels in diseased brains [32,33]. Molecules belonging to several CAM families have been implicated in pathological manifestations through different mechanisms, involving amyloid-β metabolism, cell plasticity, and neuroinflammation [33,34]. The detection of an increase in the enzymes involved in GPI-anchoring in Alzheimer’s disease brains provided further support to a relevant role of these sub-families [35]. Moreover, genetic studies have associated CAMs with a number of neuropsychiatric disorders, including CRASH syndrome, MASA syndrome, X-linked mental retardation, intellectual disability, autism spectrum disorder, schizophrenia, addiction, and bipolar disorder [15,19]. The roles of CAMs in axon growth, guidance, and fasciculation; in target recognition; and in synapse formation and maintenance might contribute to the circuit alterations characterising neuropsychiatric disorders. The main proposed mechanisms include the ability to target the formation of specific circuitries responsible for specialised brain functions through localised expression. Additionally, CAMs may impact the occurrence of neuropsychiatric diseases by modifying the balance between excitatory and inhibitory signals, thereby affecting local neural circuit activity. This influence extends to the modulation of neuromodulatory systems like monoaminergic circuits and the alteration of synaptic strength and composition [15]. Focussing more specifically on Ig-like CAMs, mutations in the genes belonging to the best-studied NCAM and L1 families, as well as changes in their expression patterns or post-translational modifications, have been associated with psychiatric and neurodegenerative disorders, including the L1 syndrome, Alzheimer’s disease, schizophrenia, and bipolar disorder [36]. When further restricting the focus to GPI-anchored sub-families, their role as functional receptors controlling neurite outgrowth, synapse formation, synapse plasticity, and learning behaviours has been implicated in the pathophysiological mechanisms associated with neurodegenerative and psychiatric disorders [17]. Since a wealth of data has been recently accumulating to implicate the IgLONs, the aim of the present review is to analyse and discuss the findings indicating possible connections between this family and neurological and psychiatric disorders.
2. IgLON5 (IgLON Family Member 5)
Anti-IgLON5 disease (ORPHA:420789) is a rare disease of the CNS. Since its discovery in 2014, 60 cases have been reported worldwide [37,38,39]. The clinical manifestation of the disease is highly heterogeneous, encompassing unique sleep and movement disorders, bulbar dysfunction, and cognitive impairment [26]. Disease onset is around the age of 60 years with progressive symptoms that can culminate in life-threatening respiratory problems [37]. Post-mortem analysis has revealed a tauopathy restricted to neurons in the brainstem, tegmentum, hypothalamus, and hippocampus [40]. The mortality rate associated with the anti-IgLON5 syndrome is considerably high, contributing to an overall mortality rate of 34%, with no discernible link between mortality and treatment response [38]. While respiratory complications are the primary cause of death, the additional neurological symptoms (cognitive decline, sleep disturbances) significantly diminish the quality of life for patients and their families.
The role of the IgLON5 protein and the underlying mechanism of disease associated with anti-IgLON5 antibodies remain poorly understood. IgLON5 is widely expressed in the CNS, with the highest levels reached in the olfactory bulb, cortical plate, and hippocampus in mice and humans. Comprising a chain of 336 amino acids, IgLON5 shares significant structural resemblance with other proteins such as OPCML (50% similarity), NTM (48–49% similarity), LSAMP (46–47% similarity), and NEGR1 (41% similarity) (Figure 1). IgLON5 is involved in forming both homomeric and heteromeric interactions with other IgLON family members [41] (Figure 2). However, the functional implications stemming from these interactions remain poorly characterised.
The detection of anti-IgLON5 antibodies is crucial for the diagnosing of anti-IgLON5 disease. These antibodies are detectable in both serum and cerebrospinal fluid (CSF), typically coinciding with the initial diagnosis timeframe. The question of whether anti-IgLON5 antibodies directly cause neuronal dysfunction and degeneration or instead appear as a consequence of the neurodegenerative process remain unresolved. Among anti-IgLON5 antibodies, the non-complement fixing IgG4 subclass predominates over IgG1. In vitro experiments have shown that IgG4 antibodies lead to the internalization of IgLON5 in cultures of hippocampal neurons [42]. Upon exposure to anti-IgLON5 antibodies, cultured rat or human neurons showed increased neurodegenerative features such as neuronal blebbing and fragmentation [43,44]. Prolonged exposure to anti-IgLON5 IgG prompts tau hyperphosphorylation and cellular death [44].
Similar to Alzheimer’s disease, the tau filaments evident in individuals with anti-IgLON5 syndrome encompass both 3R-tau and 4R-tau isoforms. However, this accumulation manifests in a distinct spatial pattern, indicative of anti-IgLON5 disease constituting a novel form of tauopathy. Recent autoptic studies have described anti-IgLON5 cases lacking hyperphosphorylated tau deposits [45,46]. These findings raise the possibility that tauopathy might arise subsequently in the disease progression, evolving as a prolonged outcome of the antibody-related effects. Thus, earlier events still depending on anti-IgLON5 antibodies may play a role in the aetiology of the disease, in addition to the proposition that a genetic predisposition to autoimmunity might exert influence, exemplified by the robust correlation with the exceptionally rare HLA-DRB1*1001 and HLA-DQB1*0501 alleles [40].
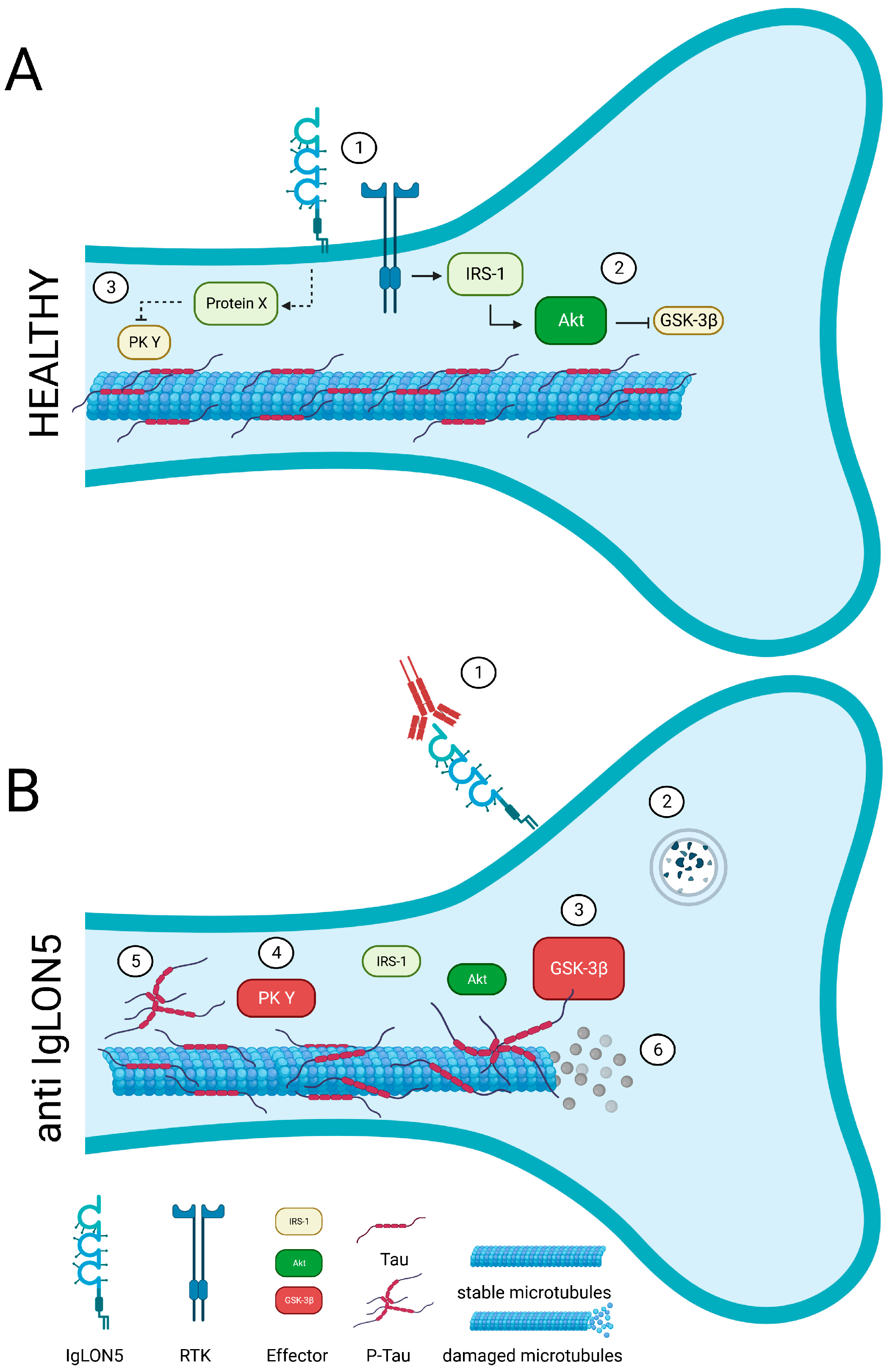
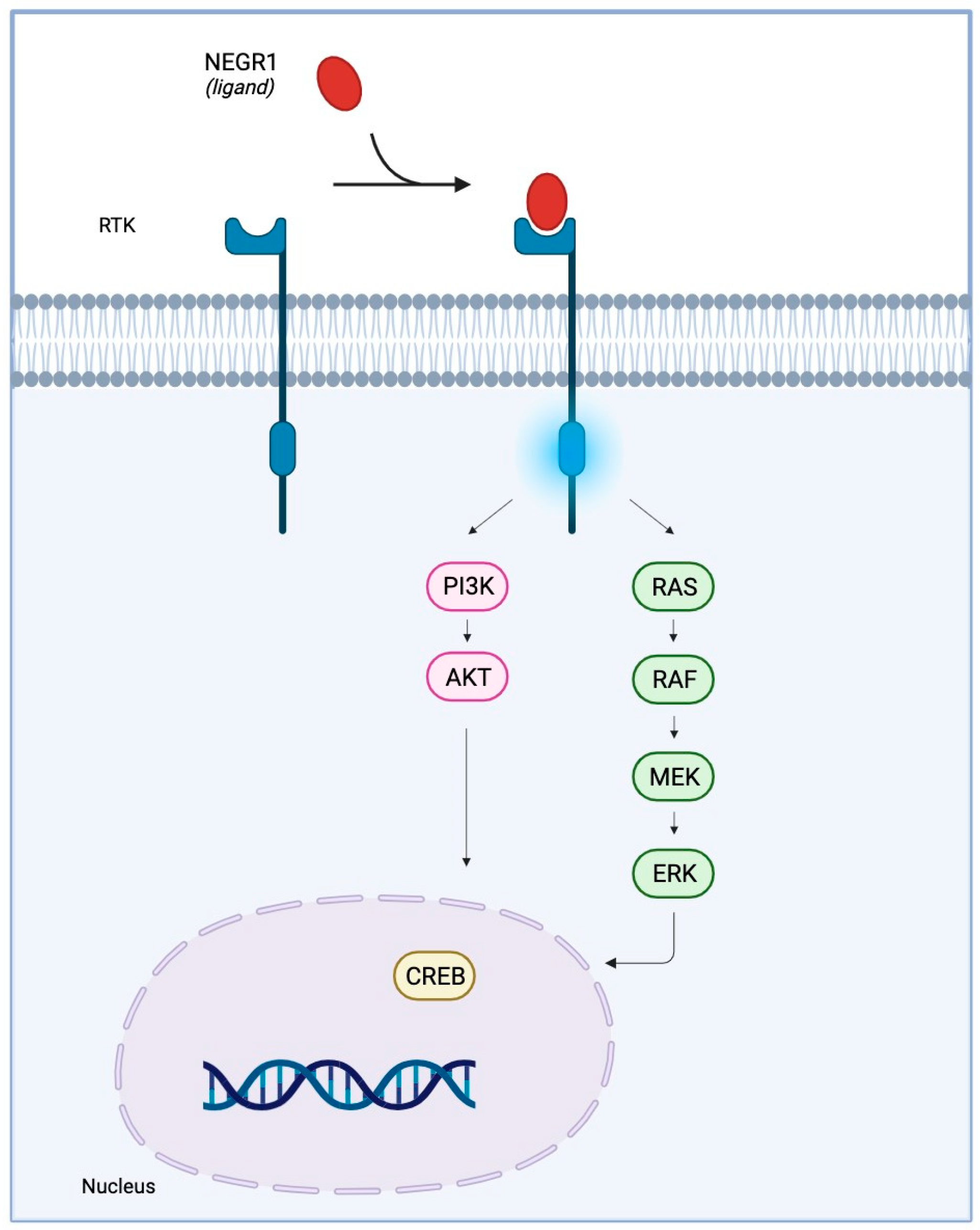
IgLONs interact and regulate receptor tyrosine kinase (RTK) trafficking and, eventually, their signalling impinging on ERK1/2 and AKT phosphorylation [28,29]. Anti-IgLON5 disease is characterised by the aggregation of hyperphosphorylated-Tau, which is phosphorylated by a variety of serine/threonine protein kinases [47]; GSK-3𝛽 is the main kinase responsible for Tau phosphorylation and precipitation [48]. An intricate signalling cascade, encompassing RTKs, insulin receptor substrate 1 (IRS-1), and AKT, has been identified as a key mechanism in curbing Tau phosphorylation by GSK-3𝛽 [49]. Hence, the hypothesis that IgLON5 may interact with and regulate a yet to be identified RTK is exceptionally intriguing (Figure 3). Recent in vitro findings put forth the proposition that antibodies against anti-IgLON5 could potentially interfere with the IgLON5 interactome [50]. It is plausible that these anti-IgLON5 antibodies might impede the IgLON5-RTK interaction, thereby exerting an influence on RTK signalling. This sequence of events could consequently trigger aberrant Tau phosphorylation and accumulation, ultimately culminating in neuronal dysfunction (Figure 3).
In order to pave the way for effective therapeutic interventions geared towards individuals afflicted with IgLON5 deficiency disease, a comprehensive grasp of both the normal and pathological roles of IgLON5, alongside the associated signalling cascade, becomes essential.
3. NEGR1 (Neuronal Growth Regulator 1)
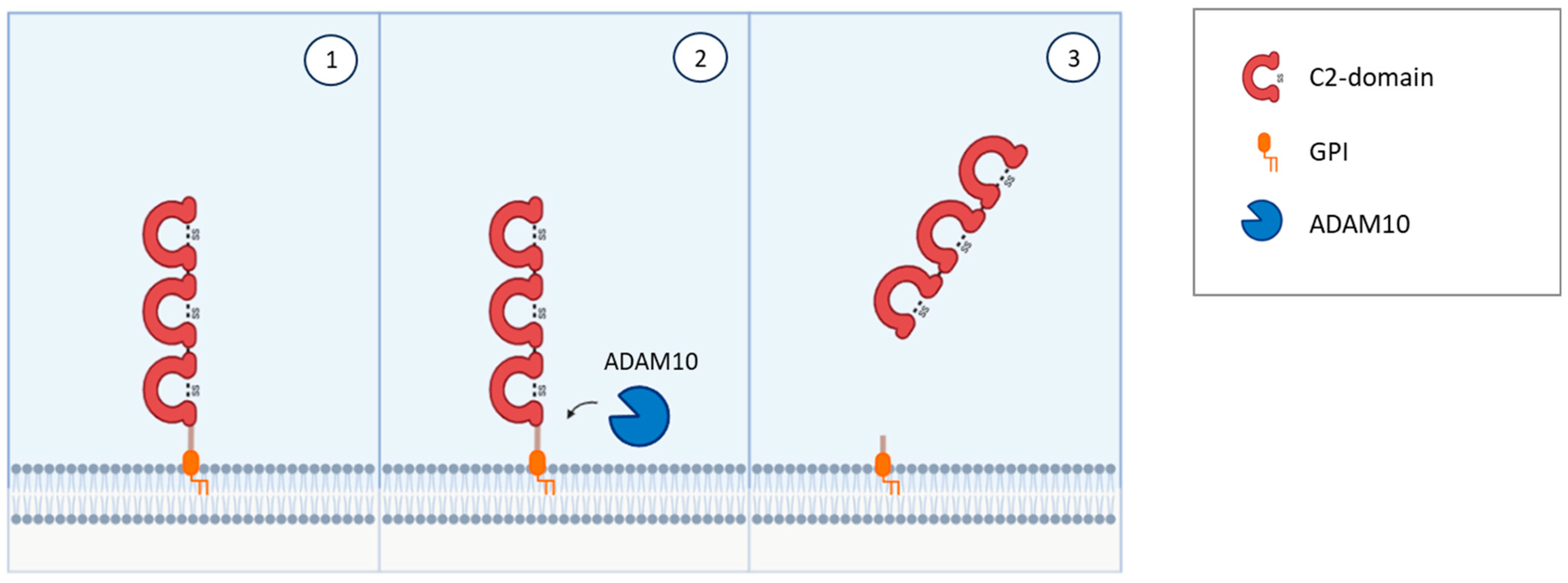
NEGR1 was first isolated and identified as a member of the IgLON family in 1999 in rats [24] and in chicken [25] as a 46–50 kDa protein; it presents the common class features of three amino-terminal C2 domains, six glycosylation sites, and GPI anchoring [24,25,51,52] (Figure 1). Homo- and heterodimerization with other IgLONs in cis and in trans across the synaptic cleft were recently thoroughly investigated [41,53] (Figure 2). Its expression in the brain is widespread, with high levels in the olfactory bulb, cerebral cortex, diencephalon, hippocampus, hypothalamus, and cerebellum [54,55,56,57], in a developmentally-regulated fashion [29,30,58,59]. NEGR1 is able to modulate neurite extension, either upon up- or down-regulation, suggesting that precise regulation is critical for correct functioning and promoting outgrowth in response to CNS injury [28,58,60]. Likewise, NEGR1 was identified as a crucial factor in determining the number of synapses in hippocampal neurons in opposite directions depending on environmental conditions [61], whereas knock-out mice showed impaired axon growth [59]. In cortical cultures, NEGR1 silencing decreased neurites’ total length and number. The reduced basal dendrite arborization induced by NEGR1 downregulation was also demonstrated in vivo in the somatosensory cortex of mice exposed to miRNA-transfection in utero [62]. Ectodomain shedding through the activity of metalloproteinases is a critical mechanism for IgLONs function (Figure 4).
In particular, the Disintegrin and Metalloproteinase Domain-Containing Proteins (ADAM) were demonstrated to be responsible for shedding NEGR1 and other IgLONs from the plasma membrane with a direct impact on neurite outgrowth in cultured cortical neurons [60]. Further research demonstrated that the metalloprotease ADAM10 is specifically involved in NEGR1 shedding and that the generation of soluble NEGR1 was able to regulate neuronal morphology by binding to the FGFR2 receptor and activating its intracellular pathway [28]. Among the latter, a prominent role is exerted by ERK and AKT pathways, which decrease FGFR2 degradation from the plasma membrane [28,29] (Figure 5). NEGR1’s impact on dendritic spine densities of pyramidal neurons has also been reproduced in vivo in the mouse somatosensory cortex [29]. Moreover, a role in hippocampal neurogenesis and long term potentiation are supported by the disruption of these functions in NEGR1 knock-out mice [63]. NEGR1 knock-out mice showed altered serotonergic and dopaminergic neurotransmission [64] and subtle alterations in social behaviour and in learning, as well as increased susceptibility to pentylenetetrazol-induced seizures [59]. In contrast with previous findings reporting no changes in anxiety [59], Noh et al. [63] showed that NEGR1 knock-out mice showed increased anxiety- and depressive-like behaviours, proposing the involvement of lipocalin and Leukaemia Inhibitory Factor (LIF) Receptor in the mechanism of action.
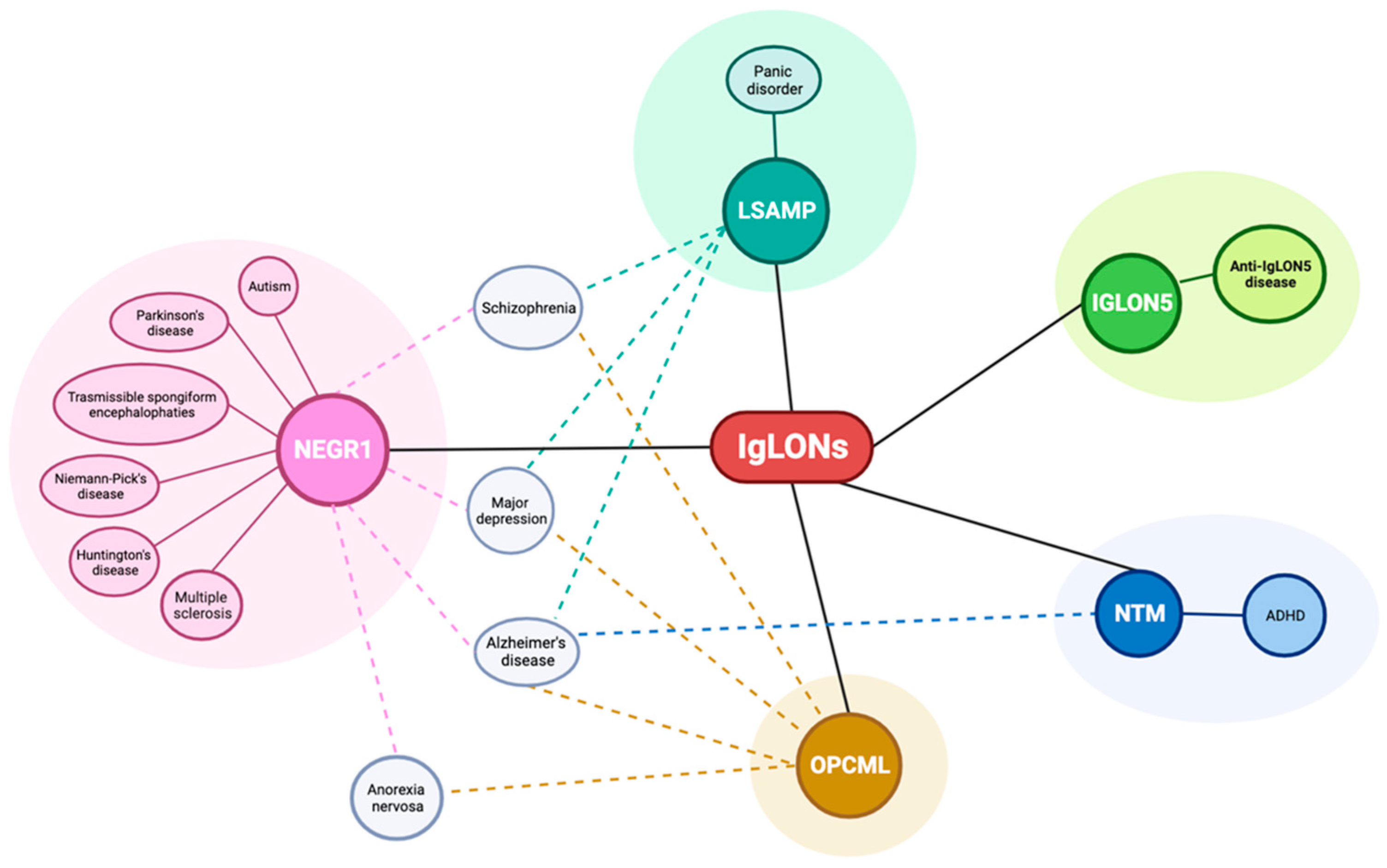
In 2009, a GWAS meta-analysis identified NEGR1 among genetic determinants of the variation in body mass index in adults and children, with a stronger influence on adiposity, which is likely to be mediated by its expression in the hypothalamic nuclei [65,66,67,68,69]. In line with these findings, modulation of NEGR1 in rodents affected body mass index and energy balance in complex, developmentally regulated ways [70,71]. Recently, NEGR1 has been indicated among the principal players in the genetic correlations between obesity and a number of psychiatric disorders, including major depression, schizophrenia, and anorexia nervosa [72].
Within psychiatric disorders, a statistically significant association with major depressive disorder has been discovered in recent meta-analyses of GWAS, with NEGR1 resulting among the strongest signals [73,74,75,76]. By integrating GWAS, brain expression quantitative trait loci data, and transcriptome-wide association studies, the association was confirmed and assigned the highest probability of causality; increased abundance of NEGR1 was significantly associated with an increased depression risk [77,78,79]. Differential regulation of transcription factor binding and the consequential altered expression has been proposed as a potential mechanism of action of the single-nucleotide polymorphisms (SNPs) associated with the increased risk of major depression [80]. Adding further evidence to the significance for depression, NEGR1 levels in the CSF of patients affected by major depression or bipolar disorder were significantly higher than in controls; this biomarker provided a major contribution to the identification of a biosignature, allowing for the correct patient group stratification [81]. In rodent models, treatment with antidepressant medications altered NEGR1 expression in brain regions [82,83], whereas genetic variants have been associated with antidepressant treatment response in obese depressed patients [84]. Supporting a possible involvement in the pathophysiology of schizophrenia, elevated levels of NEGR1 mRNA were detected in the dorsolateral prefrontal cortex of patients, [85], in agreement with previous findings in anterior pre-frontal cortex samples [86]. The association was also demonstrated at the genetic level in human patients [87]. Still within the realm of psychiatric disorders, altered NEGR1 DNA methylation has been discovered in patients affected by anorexia nervosa [88].
Evidence to imply potential NEGR1 connections with the aetiology of neurological and neurodegenerative disorders has been accumulating recently. The existence of an association between NEGR1 and Alzheimer’s disease has been investigated due to the frequent comorbidity with major depression. Ni et al. [89] reported a significant genetic association with Alzheimer’s disease in Han Chinese patients, as well as altered expression in rodent models of the disease. These findings were expanded by a study identifying statistically significant associations with the protective efficacy of SNPs located in the NEGR1 gene to mediate the protective role of educational attainment in Alzheimer’s disease [90]. In healthy subjects’ brains, NEGR1 has been associated with white matter integrity measured as fractional anisotropy. This finding is highly suggestive, since altered white matter integrity characterises several neurodegenerative diseases, comprising Alzheimer’s disease [91,92,93]. In synucleinopathies, including Parkinson’s disease, NEGR1 has been indicated as a potential CSF biomarker in mass spectrometry studies, thus implicating relevance for diagnostic and therapeutic applications [94]. In line with these findings, NEGR1 levels in plasma were downregulated and in correlation with cognitive clinical scores and motor abilities in Parkinson’s disease patients [95]. An association between NEGR1 and transmissible spongiform encephalopathies, fatal neurodegenerative diseases characterised by infectious proteins, has been discovered in rodent models in which substantially decreased levels of NEGR1 have been demonstrated. Interestingly, NEGR1 was the only glycosylated biomarker among those identified [96]. A plausible role for NEGR1 in the neurodegenerative disorder Huntington’s disease has been reported by Kaltenbach et al. [97], who showed that the mutated form of huntingtin interacts with NEGR1 and that the latter is able to modify a neurodegeneration phenotype induced by treatment with a huntingtin fragment in Drosophila. NEGR1 is able to interact with Niemann–Pick disease type C2 protein, increase its stability, and thus modulate cholesterol accumulation and influence its homeostasis. This effect has been implicated in Niemann–Pick disease type C, an autosomal recessive disorder characterised by cholesterol accumulation leading to dementia [98].
Outside the realm of neuropsychiatric disorders, NEGR1 has been implicated in other diseases whose mechanisms may have a bearing on cognitive and intellectual disabilities that often denote neurodegenerative disorders. A possible relevance of NEGR1 in autism is supported by the observation that NEGR1 downregulation in mice reproduces brain development abnormalities resembling the cortical disorganization of neurons and abnormalities in dendritic spines characterising autism spectrum disorder features. In addition, NEGR1 and FGFR2 downregulation alters social skill-related behaviours similarly to autism spectrum disorder, in reducing ultrasound vocalizations in pups, causing sensory deficits in a hot plate test in newborns, and lowering social interactions in adults [29]. Furthermore, NEGR1 has been identified as a candidate susceptibility gene in human genetic studies [99]. An association of NEGR1 with dyslexia emerged in a copy number variation analysis in Indian families. These results suggest a potential role for NEGR1 in this neurogenetic disorder, possibly by influencing the production of dendritic postsynaptic spines in mature neurons [100]. Partial deletion of chromosome 1p31.1, only involving the NEGR1 gene, has been observed in two siblings affected by attention deficit hyperactivity disorder and language impairments, but not affected by dyslexia [101]. Another patient affected by microdeletion 1p31, including NEGR1, showed moderate intellectual disability [102]. Language impairments have been reported in a patient with an interstitial 1p31.1p31.3 deletion affecting several genes including NEGR1 [103]. A meta-analysis approach on gene expression data in multiple sclerosis patients’ tissues showed a significant increase in NEGR1 levels, which was interpreted as a leading cause of impaired synaptogenesis [104]. Additional support to NEGR1 contribution to this disorder derived from a study uncovering a potential impact in the comorbidity with major depression in multiple sclerosis patients [105].
4. OPCML (Opioid Binding Protein/Cell Adhesion Molecule Like)
The IgLON member OPCML (referred also as IgLON1 or OBCAM) binds opioid receptors after shedding from the plasmatic membrane, where it functions as a CAM [53,106] (Figure 2). The OPCML gene, comprising seven exons and spanning approximately 600 Kb, is mapped to human chromosome 11, which also hosts NCAM and other brain-expressed CAMs [107]. As a result of alternative splicing, four different transcripts have been identified [108]. Originally, OPCML was purified from rat brains and presumed to be an opioid binding protein [109], and then later recognised as a member of the IgLON subgroup on the basis of its cDNA sequence [22]. In general, the primary sequence of the protein is very highly conserved between species [22]. OPCML consists of 345 amino acids, with a molecular mass of 38 kDa, harbouring six glycosylation sites at Asn44, Asn70, Asn140, Asn285, Asn293, and Asn306 [22]. Depending on the grade of glycosylation or the presence of other post-translational modifications, the molecular weight of the protein can vary [51,110]. The structure of the OPCML ectodomain is characterised by the presence of three C2 domains [22,53] and the protein is anchored to the plasma membrane through a GPI site [17,53](Figure 1). Therefore, intracellular signalling can be induced by interacting in cis with other transmembrane proteins and through the formation of homo- or heterodimers with other IgLONs [111]. The ability of OPCML to act independently as an opioid receptor has been debated, reaching the conclusion that its playing an accessory role seems more likely, based on primary structure similarity to IgLONs instead of G-protein coupled receptors [22,107]. OPCML transcripts have been widely detected in brain and non-brain tissues in adult and foetal animal models and humans, with brain-specific enrichment of the v2 isoform [108,112]. In line with its high expression in neurons, OPCML takes part in cell adhesion and cell–cell recognition by interacting with and modulating molecules that can promote or inhibit growth, regulating synapse number, synaptogenesis, and plasticity [111,113].
Polymorphisms in the OPCML gene have been linked to schizophrenia in GWAS studies in European patients and the association was later confirmed in a Thai population [114,115,116]. In a genetic study on OPCML SNPs conducted in a Han Chinese population, the association was confirmed again and reduced hippocampal expression was found in risk-allele carriers [117]. However, expression levels were not altered in the prefrontal cortex of adult patients with schizophrenia, suggesting that the control of transcription in the adult brain may not represent a prominent mechanism of the association [118]. More recently, a large meta-analysis combining data from several repositories highlighted another OPCML-associated SNP (rs2917569) with a high grade of association [119]. Although the role of OPCML in the pathophysiology of schizophrenia requires clarification, its crucial function in neurite outgrowth and spine maturation has been implicated through Eph-Cofilin signalling and F-actin polymerization [117]. Still within psychiatric disorders, a major depression study in two independent Dutch populations highlighted a significant linkage with two SNPs on chromosome 11q25 located in the intronic region 1 of the OPCML gene. The finding has been interpreted as due to the relevance of endogenous opioid neurotransmission in depressed patients [120]. Furthermore, a GWAS on anorexia nervosa detected a signal for an intronic SNP in the OPCML gene which was associated with disease risk across all discovery and replication cohorts, although it did not reach genome-wide significance [121]. A case report of two brothers with disruption in the chromosomal region encompassing OPCML and NTM suggested possible links with autistic symptoms [122].
Within neurodegenerative diseases, OPCML has been implicated in Alzheimer’s disease in a GWAS carried out in patients affected by the late-onset form [123]. In line with these findings, altered OPCML mRNA levels were identified in the hippocampus of two mouse models of disease [124]. In addition, reduced OPCML glycosylation in Alzheimer’s disease brains, together with other CAMs, was interpreted as a driver of altered cell adhesion and synaptic function in the disorder [125].
5. LSAMP (Limbic System Associated Membrane Protein)
The limbic system associated membrane protein (LAMP or LSAMP, as indicated in more recent works to distinguish it from the lysosomal associated membrane protein) is an IgLON family member first identified as specifically characterising neurons belonging to the rat limbic system or to areas receiving direct projections from there [21]. Subsequent purification and cloning revealed that LSAMP is an integral membrane protein with a molecular mass of 64–68 KDa containing three internal repeats typical of the Ig-like domain, with conserved pairs of cysteine residues, together with eight putative glycosylation sites [126,127,128] (Figure 1). Like the other IgLONs, it is a membrane protein containing a GPI anchor, which can be released from intact membranes by the action of phosphatidyl inositol-specific phospholipase C [129]. LSAMP nucleotidic and aminoacidic sequences present high sequence identity with those of OPCML and NTM, suggesting similar roles for these proteins [127]. In neurons, most protein expression is post-synaptic, located on neuronal somata and dendrites, while glial cells are not immunoreactive [126]. In the adult rat, LSAMP is expressed in regions related to the limbic system, like the hippocampus, amygdala, perirhinal cortex, anterior and lateral thalamic nuclei, preoptic area of the hypothalamus, septum, nucleus of the solitary tract, and lamina II of the dorsal horn [21,126,128,130]. The specificity of this protein for neurons belonging to the same functional system suggest its possible involvement in brain regionalization during development. Indeed, during development LSAMP mRNA is expressed in the same limbic regions as in the adult, but in a temporal fashion that relates with the time of formation of limbic axonal pathways [128,130,131]. In addition, ultramicroscopic analysis revealed that in embryonic tissue the localization of LSAMP is mainly located in the growth cone of developing axons [126,131]. The axonal staining disappeared during the second post-natal week of development, indicating a role for LSAMP in axonal growth and limbic pathway formation [130,131].
The in vivo administration of anti-LSAMP antibodies altered the formation of mossy fibres projection to pyramidal neurons during hippocampal development, suggesting that LSAMP may serve as a recognition molecule for the proper genesis of limbic connections [127]. LSAMP’s crucial role in establishing limbic connections as well as limbic neural fate was further highlighted by experiments where the connections between neurons belonging to different parts of the brain were manipulated or disrupted [130,132,133,134]. These data suggest that the commitment to limbic fate happens early in development, and that LSAMP represents an early marker of limbic neurons determination. Thus, LSAMP acts as an attractive molecule, guiding the sprouting of limbic axons, and behaving as a repulsive signal for the growing of non-limbic axons [135]. The heterophilic binding both in cis and in trans with other IgLON members mediated by the different IgG domains has been identified as a critical mechanism of action [25,111,136,137,138,139,140] (Figure 2). Another important mechanism of regulation in the development of axonal growth cones involves the molecular shedding of the IgLON ectodomain from the membrane by metalloproteases, with a prominent role played by ADAM10 [60,141] (Figure 4).
In pre-clinical models of disease, evidence of alterations in the molecular expression of LSAMP is available, suggestive of implication in the neurobiological underpinnings of psychiatric disorders. Increased LSAMP expression was found in rats with low exploratory activity in the elevated plus maze test, suggesting a correlation with anxiety [142,143]. Further evidence derives from LSAMP knock-out mice, which demonstrated a heightened responsiveness to novelty in behavioural tests as well as a maladaptive response to environmental stressors and altered interaction with the serotoninergic system, associated with increased sensitivity to psychostimulants [142,144,145,146,147,148]. Moreover, LSAMP deletion impacts synaptogenesis and synaptic plasticity, hippocampus formation, and spatial memory [57,59,149]. In human genetic studies, SNPs in the LSAMP gene have been associated with male suicide [150] when it is well established that the most important risk factor for suicide is depression [151]. Indeed, a significant association with major depression and panic disorder has been detected [152]. In addition, altered LSAMP levels were observed in the dorsolateral prefrontal cortex of schizophrenic patients, albeit with contrasting findings in different patient cohorts [85,153]. In line with these results, significant allelic and haplotypic associations have been reported between SNPs in the LSAMP gene and schizophrenia [154].
Little is known about LSAMP involvement in neurodegenerative diseases. A study aiming at identifying biomarkers for Lewy bodies dementia, Alzheimer’s disease, and Parkinson’s disease in patients’ CSF recognized LSAMP as a novel non-specific biomarker for neurodegeneration due to its significantly increased expression in all three diseases with respect to controls [155]. A similar study, where the relative expression of N-glycosylated proteins in Alzheimer’s disease was analysed through mass spectrometry, showed that LSAMP negatively correlated with the disease, suggesting that decreased post-translational modification can occur [125]. However, neither report provided clues about the molecular mechanisms of LSAMP involvement in neurodegeneration, thus further studies are required.
6. NTM (Neurotrimin)
NTM was characterised in 1995 as a 65 kDa integral protein endowed with three C2 amino-terminal domains, seven glycosylation sites, GPI anchoring, and high sequence homology with other IgLONs [23,156] (Figure 1). Like them, NTM can bind other family members in cis and in trans, as well as form homodimers [41,53,111,136,137,157] (Figure 2). The dimers are essential for NTM function in the regulation of neurite outgrowth and cell adhesion to control neuronal connectivity and synaptogenesis [139,157,158,159,160]. Both membrane-bound and soluble forms are involved in outgrowth regulation, with sensitivity to metalloproteases, particularly ADAM10 [60,141,157,161] (Figure 4). NTM is expressed in the developing and adult brain, with high expression in the sensorimotor cortex, olfactory bulb, thalamus, hypothalamus, hippocampus, basal ganglia, and cerebellum [23,137,156]. Differently from LSAMP knock out mice, NTM deficient mice had no differences in anxiety, social interaction, or locomotor activity, only sharing a lower sensitivity to the locomotor stimulating effect of amphetamine, although to a lower extent, and displaying subtle deficits in cognitively challenging emotional learning tasks [162].
SNPs in the NTM gene have been associated with intelligence in a family-based low-density genome-wide study; these findings are noteworthy since lower cognitive function increases the risk of neuropsychiatric disorders [163]. Autism spectrum disorder patients harboured copy number variants encompassing the NTM gene and two patients with a chromosomal translocation disrupting the NTM gene displayed autistic symptoms [122,164]. Furthermore, a signal for association with NTM has been discovered for childhood aggressiveness in attention deficit hyperactivity disorder, although the signal did not reach statistical significance [165]. A GWAS in a genetically isolated population from the Netherland linked four SNPs placed within intron 1 of the NTM gene to late-onset Alzheimer’s disease [123].
7. Conclusions
A wealth of data is available linking IgLON with neurodegenerative, neurological, and psychiatric disorders (Figure 6); however, since most findings derive from genetic-association studies, the underlying mechanisms of action are poorly understood.
For this family, the available evidence supports functions in nervous system development, neurite extension, synaptic formation and maintenance, blood-brain barrier structure, extracellular matrix protein recognition, and activation of signal transduction within cells able to elicit long-term responses. It is likely that the physiological role of IgLONs exerts a critical function in the pathophysiology of these disorders. However, the neurobiological underpinnings are yet to be investigated. We can speculate that the established function in synaptic plasticity and strength exerts a potential role in learning and memory formation, which can represent a common dysregulation impinging on the molecular underpinning of several disorders, with a prominent impact on neurodegenerative diseases. Dendritic spine pathology is an established hallmark of neurodegenerative diseases [166]. The accumulation of IgLONs in CSF samples gathered from patients affected by neurodegenerative disorders may reflect the spine loss. In addition, the discovery of the autoimmune response associated with anti-IgLON5 disease hints to a possible involvement of autoimmune responses as a contributing mechanism of action in other neurodegenerative and neurodevelopmental disorders associated with this family [167]. Accumulating evidence shows that IgLONs interact and regulate RTK trafficking and, eventually, their signalling impinging on ERK1/2 and Akt phosphorylation [28,29,168]. RTKs are tightly involved in the dynamics of the actin and tubulin cytoskeleton. An intriguing hypothesis is that IgLONs influence the signalling of RTKs and, consequently, cytoskeletal stability. Alteration in the cytoskeleton, such as the tau phosphorylation/accumulation observed in anti-IgLON5 disease, may eventually cause neuronal dysfunction and death.
As a caveat, it should be underlined that the existence of genetic associations does not necessarily imply a prominent role in the neurobiological underpinning of the disorder. In the same line, phenotypes associated with genetic deletions do not automatically imply a general role for the deleted gene in the pathology. Moreover, the partial overlap of disease association with IgLON family members supports the possible cross-disease alterations to be imputed to these CAMs. Further studies will elucidate the potential relevance of IgLON family members as future targets for therapeutic intervention or as biomarkers to aid diagnosis.
Author Contributions
M.S., C.V., S.A., R.R., G.P. and L.C. contributed to literature analysis, L.C. wrote the first draft of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by Fondazione Cariplo, grant number 2019-3415 to G.P.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shapiro, L.; Love, J.; Colman, D.R. Adhesion Molecules in the Nervous System: Structural Insights into Function and Diversity. Annu. Rev. Neurosci. 2007, 30, 451–474. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz, K. Physicochemical Principles of Adhesion Mechanisms in the Brain. Int. J. Mol. Sci. 2023, 24, 5070. [Google Scholar] [CrossRef] [PubMed]
- Kadry, Y.A.; Calderwood, D.A. Chapter 22: Structural and signaling functions of integrins. Biochim. Biophys. Acta—Biomembr. 2020, 1862, 183206. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [PubMed]
- McEver, R.P.; Zhu, C. Rolling cell adhesion. Annu. Rev. Cell Dev. Biol. 2010, 26, 363–396. [Google Scholar] [CrossRef]
- Ley, K.; Kansas, G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004, 4, 325–335. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. [Google Scholar] [CrossRef]
- Angiari, S. Selectin-mediated leukocyte trafficking during the development of autoimmune disease. Autoimmun. Rev. 2015, 14, 984–995. [Google Scholar] [CrossRef]
- Redies, C. Cadherins in the central nervous system. Prog. Neurobiol. 2000, 61, 611–648. [Google Scholar] [CrossRef]
- Takeichi, M. The cadherin superfamily in neuronal connections and interactions. Nat. Rev. Neurosci. 2007, 8, 11–20. [Google Scholar] [CrossRef]
- Cameron, S.; McAllister, A.K. Immunoglobulin-Like Receptors and Their Impact on Wiring of Brain Synapses. Annu. Rev. Genet. 2018, 52, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Barclay, A.N. Membrane proteins with immunoglobulin-like domains—A master superfamily of interaction molecules. Semin. Immunol. 2003, 15, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Gelfand, I.; Kister, A. Structural determinants in the sequences of immunoglobulin variable domain. J. Mol. Biol. 1998, 278, 457–479. [Google Scholar] [CrossRef]
- Walsh, F.S.; Doherty, P. Neural cell adhesion molecules of the immunoglobulin superfamily: Role in axon growth and guidance. Annu. Rev. Cell Dev. Biol. 1997, 13, 425–456. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, T. The role of cell adhesion molecules in brain wiring and neuropsychiatric disorders. Mol. Cell. Neurosci. 2017, 81, 4–11. [Google Scholar] [CrossRef]
- Zinn, K.; Özkan, E. Neural immunoglobulin superfamily interaction networks. Curr. Opin. Neurobiol. 2017, 45, 99–105. [Google Scholar] [CrossRef]
- Tan, R.P.A.; Leshchyns’ka, I.; Sytnyk, V. Glycosylphosphatidylinositol-Anchored Immunoglobulin Superfamily Cell Adhesion Molecules and Their Role in Neuronal Development and Synapse Regulation. Front. Mol. Neurosci. 2017, 10, 378. [Google Scholar] [CrossRef]
- Leshchyns’ka, I.; Sytnyk, V. Reciprocal Interactions between Cell Adhesion Molecules of the Immunoglobulin Superfamily and the Cytoskeleton in Neurons. Front. Cell Dev. Biol. 2016, 4, 9. [Google Scholar] [CrossRef]
- Maness, P.F.; Schachner, M. Neural recognition molecules of the immunoglobulin superfamily: Signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007, 10, 19–26. [Google Scholar] [CrossRef]
- Karagogeos, D. Neural GPI-anchored cell adhesion molecules. Front. Biosci. 2003, 8, s1304–s1320. [Google Scholar] [CrossRef]
- Levitt, P. A monoclonal antibody to limbic system neurons. Science 1984, 223, 299–301. [Google Scholar] [CrossRef]
- Schofield, P.R.; McFarland, K.C.; Hayflick, J.S.; Wilcox, J.N.; Cho, T.M.; Roy, S.; Lee, N.M.; Loh, H.H.; Seeburg, P.H. Molecular characterization of a new immunoglobulin superfamily protein with potential roles in opioid binding and cell contact. EMBO J. 1989, 8, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Struyk, A.F.; Canoll, P.D.; Wolfgang, M.J.; Rosen, C.L.; D’Eustachio, P.; Salzer, J.L. Cloning of neurotrimin defines a new subfamily of differentially expressed neural cell adhesion molecules. J. Neurosci. 1995, 15, 2141–2156. [Google Scholar] [CrossRef]
- Funatsu, N.; Miyata, S.; Kumanogoh, H.; Shigeta, M.; Hamada, K.; Endo, Y.; Sokawa, Y.; Maekawa, S. Characterization of a novel rat brain glycosylphosphatidylinositol- anchored protein (Kilon), a member of the IgLON cell adhesion molecule family. J. Biol. Chem. 1999, 274, 8224–8230. [Google Scholar] [CrossRef]
- Marg, A.; Sirim, P.; Spaltmann, F.; Plagge, A.; Kauselmann, G.; Buck, F.; Rathjen, F.G.; Brümmendorf, T. Neurotractin, a novel neurite outgrowth-promoting Ig-like protein that interacts with CEPU-1 and LAMP. J. Cell Biol. 1999, 145, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Sabater, L.; Gaig, C.; Gelpi, E.; Bataller, L.; Lewerenz, J.; Torres-Vega, E.; Contreras, A.; Giometto, B.; Compta, Y.; Embid, C.; et al. A novel non-rapid-eye movement and rapid-eye-movement parasomnia with sleep breathing disorder associated with antibodies to IgLON5: A case series, characterisation of the antigen, and post-mortem study. Lancet Neurol. 2014, 13, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Takamori, S.; Holt, M.; Stenius, K.; Lemke, E.A.; Grønborg, M.; Riedel, D.; Urlaub, H.; Schenck, S.; Brügger, B.; Ringler, P.; et al. Molecular anatomy of a trafficking organelle. Cell 2006, 127, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Piccoli, G. The IgLON Family Member Negr1 Promotes Neuronal Arborization Acting as Soluble Factor via FGFR2. Front. Mol. Neurosci. 2016, 8, 89. [Google Scholar] [CrossRef]
- Szczurkowska, J.; Pischedda, F.; Pinto, B.; Managò, F.; Haas, C.A.; Summa, M.; Bertorelli, R.; Papaleo, F.; Schäfer, M.K.; Piccoli, G.; et al. NEGR1 and FGFR2 cooperatively regulate cortical development and core behaviours related to autism disorders in mice. Brain 2018, 141, 2772–2794. [Google Scholar] [CrossRef]
- Fearnley, S.; Raja, R.; Cloutier, J.F. Spatiotemporal expression of IgLON family members in the developing mouse nervous system. Sci. Rep. 2021, 11, 19536. [Google Scholar] [CrossRef]
- Kubick, N.; Brösamle, D.; Mickael, M.E. Molecular Evolution and Functional Divergence of the IgLON Family. Evol. Bioinforma. 2018, 14, 1176934318775081. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Sytnyk, V. Synaptic Cell Adhesion Molecules in Alzheimer’s Disease. Neural Plast. 2016, 2016, 6427537. [Google Scholar] [CrossRef] [PubMed]
- Wennström, M.; Nielsen, H.M. Cell adhesion molecules in Alzheimer’s disease. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leshchyns’ka, I.; Liew, H.T.; Shepherd, C.; Halliday, G.M.; Stevens, C.H.; Ke, Y.D.; Ittner, L.M.; Sytnyk, V. Aβ-dependent reduction of NCAM2-mediated synaptic adhesion contributes to synapse loss in Alzheimer’s disease. Nat. Commun. 2015, 6, 8836. [Google Scholar] [CrossRef]
- Tang, X.; Tena, J.; Di Lucente, J.; Maezawa, I.; Harvey, D.J.; Jin, L.-W.; Lebrilla, C.B.; Zivkovic, A.M. Transcriptomic and glycomic analyses highlight pathway-specific glycosylation alterations unique to Alzheimer’s disease. Sci. Rep. 2023, 13, 7816. [Google Scholar] [CrossRef]
- Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Neural Cell Adhesion Molecules of the Immunoglobulin Superfamily Regulate Synapse Formation, Maintenance, and Function. Trends Neurosci. 2017, 40, 295–308. [Google Scholar] [CrossRef]
- Gaig, C.; Graus, F.; Compta, Y.; Högl, B.; Bataller, L.; Brüggemann, N.; Giordana, C.; Heidbreder, A.; Kotschet, K.; Lewerenz, J.; et al. Clinical manifestations of the anti-IgLON5 disease. Neurology 2017, 88, 1736–1743. [Google Scholar] [CrossRef]
- Nissen, M.S.; Blaabjerg, M. Anti-IgLON5 Disease: A Case With 11-Year Clinical Course and Review of the Literature. Front. Neurol. 2019, 10, 1056. [Google Scholar] [CrossRef]
- Werner, J.; Jelcic, I.; Schwarz, E.I.; Probst-Müller, E.; Nilsson, J.; Schwizer, B.; Bloch, K.E.; Lutterotti, A.; Jung, H.-H.; Schreiner, B. Anti-IgLON5 Disease: A New Bulbar-Onset Motor Neuron Mimic Syndrome. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e962. [Google Scholar] [CrossRef]
- Gelpi, E.; Höftberger, R.; Graus, F.; Ling, H.; Holton, J.L.; Dawson, T.; Popovic, M.; Pretnar-Oblak, J.; Högl, B.; Schmutzhard, E.; et al. Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol. 2016, 132, 531–543. [Google Scholar] [CrossRef]
- Ranaivoson, F.M.; Turk, L.S.; Ozgul, S.; Kakehi, S.; von Daake, S.; Lopez, N.; Trobiani, L.; De Jaco, A.; Denissova, N.; Demeler, B.; et al. A Proteomic Screen of Neuronal Cell-Surface Molecules Reveals IgLONs as Structurally Conserved Interaction Modules at the Synapse. Structure 2019, 27, 893–906.e9. [Google Scholar] [CrossRef] [PubMed]
- Sabater, L.; Planagumà, J.; Dalmau, J.; Graus, F. Cellular investigations with human antibodies associated with the anti-IgLON5 syndrome. J. Neuroinflamm. 2016, 13, 226. [Google Scholar] [CrossRef] [PubMed]
- Landa, J.; Gaig, C.; Plagumà, J.; Saiz, A.; Antonell, A.; Sanchez-Valle, R.; Dalmau, J.; Graus, F.; Sabater, L. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann. Neurol. 2020, 88, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Ryding, M.; Gamre, M.; Nissen, M.S.; Nilsson, A.C.; Okarmus, J.; Poulsen, A.A.E.; Meyer, M.; Blaabjerg, M. Neurodegeneration Induced by Anti-IgLON5 Antibodies Studied in Induced Pluripotent Stem Cell-Derived Human Neurons. Cells 2021, 10, 837. [Google Scholar] [CrossRef] [PubMed]
- Honorat, J.A.; Komorowski, L.; Josephs, K.A.; Fechner, K.; St Louis, E.K.; Hinson, S.R.; Lederer, S.; Kumar, N.; Gadoth, A.; Lennon, V.A.; et al. IgLON5 antibody: Neurological accompaniments and outcomes in 20 patients. Neurol.-Neuroimmunol. Neuroinflamm. 2017, 4, e385. [Google Scholar] [CrossRef] [PubMed]
- Erro, M.E.; Sabater, L.; Martínez, L.; Herrera, M.; Ostolaza, A.; García de Gurtubay, I.; Tuñón, T.; Graus, F.; Gelpi, E. Anti-IGLON5 disease: A new case without neuropathologic evidence of brainstem tauopathy. Neurol.-Neuroimmunol. Neuroinflamm. 2020, 7, e651. [Google Scholar] [CrossRef]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar] [CrossRef]
- Hernandez, F.; Lucas, J.J.; Avila, J. GSK3 and tau: Two convergence points in Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33 (Suppl. 1), S141–S144. [Google Scholar] [CrossRef]
- Kanno, T.; Tsuchiya, A.; Tanaka, A.; Nishizaki, T. Combination of PKCε Activation and PTP1B Inhibition Effectively Suppresses Aβ-Induced GSK-3β Activation and Tau Phosphorylation. Mol. Neurobiol. 2016, 53, 4787–4797. [Google Scholar] [CrossRef]
- Landa, J.; Serafim, A.B.; Gaig, C.; Saiz, A.; Koneczny, I.; Hoftberger, R.; Santamaria, J.; Dalmau, J.; Graus, F.; Sabater, L. Patients’ IgLON5 autoantibodies interfere with IgLON5-protein interactions. Front. Immunol. 2023, 14, 1151574. [Google Scholar] [CrossRef]
- Itoh, S.; Hachisuka, A.; Kawasaki, N.; Hashii, N.; Teshima, R.; Hayakawa, T.; Kawanishi, T.; Yamaguchi, T. Glycosylation analysis of IgLON family proteins in rat brain by liquid chromatography and multiple-stage mass spectrometry. Biochemistry 2008, 47, 10132–10154. [Google Scholar] [CrossRef] [PubMed]
- Sim, G.; Jeong, M.; Seo, H.; Kim, J.; Lee, S. The Role of N-Glycosylation in the Intracellular Trafficking and Functionality of Neuronal Growth Regulator 1. Cells 2022, 11, 1242. [Google Scholar] [CrossRef] [PubMed]
- Venkannagari, H.; Kasper, J.M.; Misra, A.; Rush, S.A.; Fan, S.; Lee, H.; Sun, H.; Seshadrinathan, S.; Machius, M.; Hommel, J.D.; et al. Highly Conserved Molecular Features in IgLONs Contrast Their Distinct Structural and Biological Outcomes. J. Mol. Biol. 2020, 432, 5287–5303. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Funatsu, N.; Matsunaga, W.; Kiyohara, T.; Sokawa, Y.; Maekawa, S. Expression of the IgLON cell adhesion molecules Kilon and OBCAM in hypothalamic magnocellular neurons. J. Comp. Neurol. 2000, 424, 74–85. [Google Scholar] [CrossRef]
- Miyata, S.; Matsumoto, N.; Taguchi, K.; Akagi, A.; Iino, T.; Funatsu, N.; Maekawa, S. Biochemical and ultrastructural analyses of IgLON cell adhesion molecules, Kilon and OBCAM in the rat brain. Neuroscience 2003, 117, 645–658. [Google Scholar] [CrossRef]
- Bräuer, A.U.; Savaskan, N.E.; Plaschke, M.; Prehn, S.; Ninnemann, O.; Nitsch, R. IG-molecule kilon shows differential expression pattern from LAMP in the developing and adult rat hippocampus. Hippocampus 2000, 10, 632–644. [Google Scholar] [CrossRef]
- Vanaveski, T.; Singh, K.; Narvik, J.; Eskla, K.-L.L.; Visnapuu, T.; Heinla, I.; Jayaram, M.; Innos, J.; Lilleväli, K.; Philips, M.-A.A.; et al. Promoter-Specific Expression and Genomic Structure of IgLON Family Genes in Mouse. Front. Neurosci. 2017, 11, 38. [Google Scholar] [CrossRef]
- Schäfer, M.; Bräuer, A.U.; Savaskan, N.E.; Rathjen, F.G.; Brümmendorf, T. Neurotractin/kilon promotes neurite outgrowth and is expressed on reactive astrocytes after entorhinal cortex lesion. Mol. Cell. Neurosci. 2005, 29, 580–590. [Google Scholar] [CrossRef]
- Singh, K.; Loreth, D.; Pöttker, B.; Hefti, K.; Innos, J.; Schwald, K.; Hengstler, H.; Menzel, L.; Sommer, C.J.; Radyushkin, K.; et al. Neuronal growth and behavioral alterations in mice deficient for the psychiatric disease-associated negr1 gene. Front. Mol. Neurosci. 2018, 11, 30. [Google Scholar] [CrossRef]
- Sanz, R.; Ferraro, G.B.; Fournier, A.E. IgLON Cell Adhesion Molecules Are Shed from the Cell Surface of Cortical Neurons to Promote Neuronal Growth. J. Biol. Chem. 2015, 290, 4330–4342. [Google Scholar] [CrossRef]
- Hashimoto, T.; Yamada, M.; Maekawa, S.; Nakashima, T.; Miyata, S. IgLON cell adhesion molecule Kilon is a crucial modulator for synapse number in hippocampal neurons. Brain Res. 2008, 1224, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Pischedda, F.; Szczurkowska, J.; Cirnaru, M.D.; Giesert, F.; Vezzoli, E.; Ueffing, M.; Sala, C.; Francolini, M.; Hauck, S.M.; Cancedda, L.; et al. A cell surface biotinylation assay to reveal membrane-associated neuronal cues: Negr1 regulates dendritic arborization. Mol. Cell. Proteom. 2014, 13, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Lee, H.; Choi, T.-Y.; Joo, Y.; Kim, S.-J.; Kim, H.; Kim, J.Y.; Jahng, J.W.; Lee, S.; Choi, S.-Y.; et al. Negr1 controls adult hippocampal neurogenesis and affective behaviors. Mol. Psychiatry 2019, 24, 1189–1205. [Google Scholar] [CrossRef] [PubMed]
- Kaare, M.; Jayaram, M.; Jagomäe, T.; Singh, K.; Kilk, K.; Mikheim, K.; Leevik, M.; Leidmaa, E.; Varul, J.; Nõmm, H.; et al. Depression-Associated Negr1 Gene-Deficiency Induces Alterations in the Monoaminergic Neurotransmission Enhancing Time-Dependent Sensitization to Amphetamine in Male Mice. Brain Sci. 2022, 12, 1696. [Google Scholar] [CrossRef]
- Willer, C.; Speliotes, E.; Loos, R.; Li, S.; Lindgren, C.; Heid, I.; Berndt, S.; Elliott, A.; Jackson, A.; Lamina, C.; et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009, 41, 25–34. [Google Scholar] [CrossRef]
- Renström, F.; Payne, F.; Nordström, A.; Brito, E.C.; Rolandsson, O.; Hallmans, G.; Barroso, I.; Nordström, P.; Franks, P.W. Replication and extension of genome-wide association study results for obesity in 4923 adults from northern Sweden. Hum. Mol. Genet. 2009, 18, 1489–1496. [Google Scholar] [CrossRef]
- Zhao, J.; Bradfield, J.P.; Li, M.; Wang, K.; Zhang, H.; Kim, C.E.; Annaiah, K.; Glessner, J.T.; Thomas, K.; Garris, M.; et al. The role of obesity-associated loci identified in genome-wide association studies in the determination of pediatric BMI. Obesity 2009, 17, 2254–2257. [Google Scholar] [CrossRef]
- Schmid, P.M.; Heid, I.; Buechler, C.; Steege, A.; Resch, M.; Birner, C.; Endemann, D.H.; Riegger, G.A.; Luchner, A. Expression of fourteen novel obesity-related genes in zucker diabetic fatty rats. Cardiovasc. Diabetol. 2012, 11, 48. [Google Scholar] [CrossRef]
- Boender, A.J.; Van Rozen, A.J.; Adan, R.A.H. Nutritional state affects the expression of the obesity-associated genes Etv5, faim2, Fto, and negr1. Obesity 2012, 20, 2420–2425. [Google Scholar] [CrossRef]
- Boender, A.J.; van Gestel, M.A.; Garnerv, K.M.; Luijendijk, M.C.M.; Adan, R.A.H. The Obesity-Associated gene NEGR1 regulates aspects of energy balance in rat hypothalamic areas. Physiol. Rep. 2014, 2, e12083. [Google Scholar] [CrossRef]
- Lee, A.W.S.; Hengstler, H.; Schwald, K.; Berriel-Diaz, M.; Loreth, D.; Kirsch, M.; Kretz, O.; Haas, C.A.; de Angelis, M.H.; Herzig, S.; et al. Functional inactivation of the genome-wide association study obesity gene neuronal growth regulator 1 in mice causes a body mass phenotype. PLoS ONE 2012, 7, e41537. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Ouyang, M.; Wang, J.; Xie, M.; Huang, Y.; Yuan, F.; Jia, Y.; Zhang, X.; Liu, N.; Zhang, N. Shared genetics between classes of obesity and psychiatric disorders: A large-scale genome-wide cross-trait analysis. J. Psychosom. Res. 2022, 162, 111032. [Google Scholar] [CrossRef] [PubMed]
- Hyde, C.L.; Nagle, M.W.; Tian, C.; Chen, X.; Paciga, S.A.; Wendland, J.R.; Tung, J.Y.; Hinds, D.A.; Perlis, R.H.; Winslow, A.R. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat. Genet. 2016, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef]
- Howard, D.M.; Adams, M.J.; Clarke, T.-K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef]
- Levey, D.F.; Stein, M.B.; Wendt, F.R.; Pathak, G.A.; Zhou, H.; Aslan, M.; Quaden, R.; Harrington, K.M.; Nuñez, Y.Z.; Overstreet, C.; et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in >1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 2021, 24, 954–963. [Google Scholar] [CrossRef]
- Dall’Aglio, L.; Lewis, C.M.; Pain, O. Delineating the Genetic Component of Gene Expression in Major Depression. Biol. Psychiatry 2021, 89, 627–636. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, W.; Zhu, J.; Yin, H.; Chang, S.; Yue, W.; Yu, H. Integrating genome-wide association study and expression quantitative trait loci data identifies NEGR1 as a causal risk gene of major depression disorder. J. Affect. Disord. 2020, 265, 679–686. [Google Scholar] [CrossRef]
- Deng, Y.T.; Ou, Y.N.; Wu, B.S.; Yang, Y.X.; Jiang, Y.; Huang, Y.Y.; Liu, Y.; Tan, L.; Dong, Q.; Suckling, J.; et al. Identifying causal genes for depression via integration of the proteome and transcriptome from brain and blood. Mol. Psychiatry 2022, 27, 2849–2857. [Google Scholar] [CrossRef]
- Li, S.; Li, Y.; Li, X.; Liu, J.; Huo, Y.; Wang, J.; Liu, Z.; Li, M.; Luo, X.-J. Regulatory mechanisms of major depressive disorder risk variants. Mol. Psychiatry 2020, 25, 1926–1945. [Google Scholar] [CrossRef]
- Maccarrone, G.; Ditzen, C.; Yassouridis, A.; Rewerts, C.; Uhr, M.; Uhlen, M.; Holsboer, F.; Turck, C.W. Psychiatric patient stratification using biosignatures based on cerebrospinal fluid protein expression clusters. J. Psychiatr. Res. 2013, 47, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Tamási, V.; Petschner, P.; Adori, C.; Kirilly, E.; Ando, R.D.; Tothfalusi, L.; Juhasz, G.; Bagdy, G. Transcriptional evidence for the role of chronic venlafaxine treatment in neurotrophic signaling and neuroplasticity including also glutatmatergic- and insulin-mediated neuronal processes. PLoS ONE 2014, 9, e113662. [Google Scholar] [CrossRef] [PubMed]
- Carboni, L.; Pischedda, F.; Piccoli, G.; Lauria, M.; Musazzi, L.; Popoli, M.; Mathé, A.A.; Domenici, E. Depression-Associated Gene Negr1-Fgfr2 Pathway Is Altered by Antidepressant Treatment. Cells 2020, 9, 1818. [Google Scholar] [CrossRef] [PubMed]
- Amare, A.T.; Schubert, K.O.; Tekola-Ayele, F.; Hsu, Y.H.; Sangkuhl, K.; Jenkins, G.; Whaley, R.M.; Barman, P.; Batzler, A.; Altman, R.B.; et al. The association of obesity and coronary artery disease genes with response to SSRIs treatment in major depression. J. Neural Transm. 2019, 126, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Karis, K.; Eskla, K.L.; Kaare, M.; Täht, K.; Tuusov, J.; Visnapuu, T.; Innos, J.; Jayaram, M.; Timmusk, T.; Weickert, C.S.; et al. Altered expression profile of igLON family of neural cell adhesion molecules in the dorsolateral prefrontal cortex of schizophrenic patients. Front. Mol. Neurosci. 2018, 11, 8. [Google Scholar] [CrossRef]
- Cox, D.A.; Gottschalk, M.G.; Wesseling, H.; Ernst, A.; Cooper, J.D.; Bahn, S. Proteomic systems evaluation of the molecular validity of preclinical psychosis models compared to schizophrenia brain pathology. Schizophr. Res. 2016, 177, 98–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, G. A logical relationship for schizophrenia, bipolar, and major depressive disorder. Part 1: Evidence from chromosome 1 high density association screen. J. Comp. Neurol. 2020, 528, 2620–2635. [Google Scholar] [CrossRef]
- Steiger, H.; Booij, L.; Thaler, L.; St-Hilaire, A.; Israël, M.; Casey, K.F.; Oliverio, S.; Crescenzi, O.; Lee, V.; Turecki, G.; et al. DNA methylation in people with anorexia nervosa: Epigenome-wide patterns in actively ill, long-term remitted, and healthy-eater women. World J. Biol. Psychiatry 2023, 24, 254–259. [Google Scholar] [CrossRef]
- Ni, H.; Xu, M.; Zhan, G.-L.; Fan, Y.; Zhou, H.; Jiang, H.-Y.; Lu, W.-H.; Tan, L.; Zhang, D.-F.; Yao, Y.-G.; et al. The GWAS Risk Genes for Depression May Be Actively Involved in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 1149–1161. [Google Scholar] [CrossRef]
- Raghavan, N.S.; Vardarajan, B.; Mayeux, R. Genomic variation in educational attainment modifies Alzheimer disease risk. Neurol. Genet. 2019, 5, e310. [Google Scholar] [CrossRef]
- Harrison, J.R.; Bhatia, S.; Tan, Z.X.; Mirza-Davies, A.; Benkert, H.; Tax, C.M.W.; Jones, D.K. Imaging Alzheimer’s genetic risk using diffusion MRI: A systematic review. NeuroImage Clin. 2020, 27, 102359. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Pan, P.L.; Song, W.; Huang, R.; Chen, K.; Shang, H.F. A meta-analysis of diffusion tensor imaging studies in amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 1833–1838. [Google Scholar] [CrossRef]
- Ciccarelli, O.; Catani, M.; Johansen-Berg, H.; Clark, C.; Thompson, A. Diffusion-based tractography in neurological disorders: Concepts, applications, and future developments. Lancet Neurol. 2008, 7, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Lachén-Montes, M.; González-Morales, A.; Fernández-Irigoyen, J.; Santamaría, E. Deployment of Label-Free Quantitative Olfactory Proteomics to Detect Cerebrospinal Fluid Biomarker Candidates in Synucleinopathies. Methods Mol. Biol. 2019, 2044, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Abdi, I.Y.; Bartl, M.; Dakna, M.; Abdesselem, H.; Majbour, N.; Trenkwalder, C.; El-Agnaf, O.; Mollenhauer, B. Cross-sectional proteomic expression in Parkinson’s disease-related proteins in drug-naïve patients vs healthy controls with longitudinal clinical follow-up. Neurobiol. Dis. 2023, 177, 105997. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, L.; Simon, S.L.R.; Waitt, B.; Knox, J.D. Proteomic Screen of Brain Glycoproteome Reveals Prion Specific Marker of Pathogenesis. Proteomics 2018, 18, 1700296. [Google Scholar] [CrossRef]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007, 3, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chun, Y.; Che, L.; Kim, J.; Lee, S.; Lee, S. The new obesity-associated protein, neuronal growth regulator 1 (NEGR1), is implicated in Niemann-Pick disease Type C (NPC2)-mediated cholesterol trafficking. Biochem. Biophys. Res. Commun. 2017, 482, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.R.; Noor, A.; Vincent, J.B.; Lionel, A.C.; Feuk, L.; Skaug, J.; Shago, M.; Moessner, R.; Pinto, D.; Ren, Y.; et al. Structural Variation of Chromosomes in Autism Spectrum Disorder. Am. J. Hum. Genet. 2008, 82, 477–488. [Google Scholar] [CrossRef]
- Veerappa, A.M.; Saldanha, M.; Padakannaya, P.; Ramachandra, N.B. Family-based genome-wide copy number scan identifies five new genes of dyslexia involved in dendritic spinal plasticity. J. Hum. Genet. 2013, 58, 539–547. [Google Scholar] [CrossRef]
- Genovese, A.; Cox, D.; Butler, M. Partial Deletion of Chromosome 1p31.1 Including only the Neuronal Growth Regulator 1 Gene in Two Siblings. J. Pediatr. Genet. 2015, 4, 023–028. [Google Scholar] [CrossRef]
- Biswal, S.; Parida, P.; Dubbudu, A.; Sharawat, I.K.; Panda, P.K. Chromosome 1p31.1 Deletion Syndrome: Limited Expression. Ann. Indian Acad. Neurol. 2021, 24, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Tassano, E.; Gamucci, A.; Celle, M.E.; Ronchetto, P.; Cuoco, C.; Gimelli, G. Clinical and Molecular Cytogenetic Characterization of a de novo Interstitial 1p31.1p31.3 Deletion in a Boy with Moderate Intellectual Disability and Severe Language Impairment. Cytogenet. Genome Res. 2015, 146, 39–43. [Google Scholar] [CrossRef]
- Katiyar, A.; Sharma, S.; Singh, T.P.; Kaur, P. Identification of shared molecular signatures indicate the susceptibility of endometriosis to multiple sclerosis. Front. Genet. 2018, 9, 42. [Google Scholar] [CrossRef]
- Brugger, S.W.; Gardner, M.C.; Beales, J.T.; Briggs, F.; Davis, M.F. Depression in multiple sclerosis patients associated with risk variant near NEGR1. Mult. Scler. Relat. Disord. 2020, 46, 102537. [Google Scholar] [CrossRef] [PubMed]
- Lippman, D.A.; Lee, N.M.; Loh, H.H. Opioid-binding cell adhesion molecule (OBCAM)-related clones from a rat brain cDNA library. Gene 1992, 117, 249–254. [Google Scholar] [CrossRef]
- Shark, K.B.; Lee, N.M. Cloning, sequencing and localization to chromosome 11 of a cDNA encoding a human opioid-binding cell adhesion molecule (OBCAM). Gene 1995, 155, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ying, Y.; van Hasselt, A.; Ng, K.M.; Yu, J.; Zhang, Q.; Jin, J.; Liu, D.; Rhim, J.S.; Rha, S.Y.; et al. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS ONE 2008, 3, e2990. [Google Scholar] [CrossRef]
- Cho, T.M.; Hasegawa, J.; Ge, B.L.; Loh, H.H. Purification to apparent homogeneity of a mu-type opioid receptor from rat brain. Proc. Natl. Acad. Sci. USA 1986, 83, 4138–4142. [Google Scholar] [CrossRef]
- Hachisuka, A.; Yamazaki, T.; Sawada, J.; Terao, T. Characterization and tissue distribution of opioid-binding cell adhesion molecule (OBCAM) using monoclonal antibodies. Neurochem. Int. 1996, 28, 373–379. [Google Scholar] [CrossRef]
- Reed, J.; McNamee, C.; Rackstraw, S.; Jenkins, J.; Moss, D. Diglons are heterodimeric proteins composed of IgLON subunits, and Diglon-CO inhibits neurite outgrowth from cerebellar granule cells. J. Cell Sci. 2004, 117, 3961–3973. [Google Scholar] [CrossRef]
- Hachisuka, A.; Nakajima, O.; Yamazaki, T.; Sawada, J. Developmental expression of opioid-binding cell adhesion molecule (OBCAM) in rat brain. Dev. Brain Res. 2000, 122, 183–191. [Google Scholar] [CrossRef]
- Yamada, M.; Hashimoto, T.; Hayashi, N.; Higuchi, M.; Murakami, A.; Nakashima, T.; Maekawa, S.; Miyata, S. Synaptic adhesion molecule OBCAM; synaptogenesis and dynamic internalization. Brain Res. 2007, 1165, 5–14. [Google Scholar] [CrossRef]
- Athanasiu, L.; Mattingsdal, M.; Kähler, A.K.; Brown, A.; Gustafsson, O.; Agartz, I.; Giegling, I.; Muglia, P.; Cichon, S.; Rietschel, M.; et al. Gene variants associated with schizophrenia in a Norwegian genome-wide study are replicated in a large European cohort. J. Psychiatr. Res. 2010, 44, 748–753. [Google Scholar] [CrossRef]
- Panichareon, B.; Nakayama, K.; Thurakitwannakarn, W.; Iwamoto, S.; Sukhumsirichart, W. OPCML gene as a schizophrenia susceptibility locus in Thai population. J. Mol. Neurosci. 2012, 46, 373–377. [Google Scholar] [CrossRef]
- O’Donovan, M.C.; Craddock, N.; Norton, N.; Williams, H.; Peirce, T.; Moskvina, V.; Nikolov, I.; Hamshere, M.; Carroll, L.; Georgieva, L.; et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat. Genet. 2008, 40, 1053–1055. [Google Scholar] [CrossRef]
- Zhang, Z.; Ye, M.; Li, Q.; You, Y.; Yu, H.; Ma, Y.; Mei, L.; Sun, X.; Wang, L.; Yue, W.; et al. The Schizophrenia Susceptibility Gene OPCML Regulates Spine Maturation and Cognitive Behaviors through Eph-Cofilin Signaling. Cell Rep. 2019, 29, 49–61.e7. [Google Scholar] [CrossRef]
- Umeda-Yano, S.; Hashimoto, R.; Yamamori, H.; Weickert, C.S.; Yasuda, Y.; Ohi, K.; Fujimoto, M.; Ito, A.; Takeda, M. Expression analysis of the genes identified in GWAS of the postmortem brain tissues from patients with schizophrenia. Neurosci. Lett. 2014, 568, 12–16. [Google Scholar] [CrossRef]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef]
- Schol-Gelok, S.; Janssens, A.C.J.W.; Tiemeier, H.; Liu, F.; Lopez-Leon, S.; Zorkoltseva, I.V.; Axenovich, T.I.; Van Swieten, J.C.; Uitterlinden, A.G.; Hofman, A.; et al. A genome-wide screen for depression in two independent dutch populations. Biol. Psychiatry 2010, 68, 187–196. [Google Scholar] [CrossRef]
- Huckins, L.M.; Hatzikotoulas, K.; Southam, L.; Thornton, L.M.; Steinberg, J.; Aguilera-McKay, F.; Treasure, J.; Schmidt, U.; Gunasinghe, C.; Romero, A.; et al. Investigation of common, low-frequency and rare genome-wide variation in anorexia nervosa. Mol. Psychiatry 2018, 23, 1169–1180. [Google Scholar] [CrossRef] [PubMed]
- Minhas, H.M.; Pescosolido, M.F.; Schwede, M.; Piasecka, J.; Gaitanis, J.; Tantravahi, U.; Morrow, E.M. An unbalanced translocation involving loss of 10q26.2 and gain of 11q25 in a pedigree with autism spectrum disorder and cerebellar juvenile pilocytic astrocytoma. Am. J. Med. Genet. A 2013, 161A, 787–791. [Google Scholar] [CrossRef]
- Liu, F.; Arias-Vásquez, A.; Sleegers, K.; Aulchenko, Y.S.; Kayser, M.; Sanchez-Juan, P.; Feng, B.-J.J.; Bertoli-Avella, A.M.; van Swieten, J.; Axenovich, T.I.; et al. A genomewide screen for late-onset Alzheimer disease in a genetically isolated Dutch population. Am. J. Hum. Genet. 2007, 81, 17–31. [Google Scholar] [CrossRef]
- Weller, A.E.; Ferraro, T.N.; Doyle, G.A.; Reiner, B.C.; Crist, R.C.; Berrettini, W.H. Single Nucleus Transcriptome Data from Alzheimer’s Disease Mouse Models Yield New Insight into Pathophysiology. J. Alzheimer’s Dis. 2022, 90, 1233–1247. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Chin, L.-S.; Li, L. Integrative glycoproteomics reveals protein N-glycosylation aberrations and glycoproteomic network alterations in Alzheimer’s disease. Sci. Adv. 2020, 6, eabc5802. [Google Scholar] [CrossRef]
- Zacco, A.; Cooper, V.; Chantler, P.; Fisher-Hyland, S.; Horton, H.; Levitt, P. Isolation, biochemical characterization and ultrastructural analysis of the limbic system-associated membrane protein (LAMP), a protein expressed by neurons comprising functional neural circuits. J. Neurosci. 1990, 10, 73–90. [Google Scholar] [CrossRef]
- Pimenta, A.F.; Zhukareva, V.; Barbe, M.F.; Reinoso, B.S.; Grimley, C.; Henzel, W.; Fischer, I.; Levitt, P. The limbic system-associated membrane protein is an Ig superfamily member that mediates selective neuronal growth and axon targeting. Neuron 1995, 15, 287–297. [Google Scholar] [CrossRef]
- Pimenta, A.F.; Reinoso, B.S.; Levitt, P.; Pimenta, A.F.; Levitt, P. Expression of the mRNAs encoding the limbic system-associated membrane protein (LAMP): II. Fetal rat brain. J. Comp. Neurol. 1996, 375, 274–288. [Google Scholar] [CrossRef]
- Zhukareva, V.; Levitt, P. The limbic system-associated membrane protein (LAMP) selectively mediates interactions with specific central neuron populations. Development 1995, 121, 1161–1172. [Google Scholar] [CrossRef]
- Keller, F.; Levitt, P. Developmental and regeneration-associated regulation of the limbic system associated membrane protein in explant cultures of the rat brain. Neuroscience 1989, 28, 455–474. [Google Scholar] [CrossRef]
- Horton, H.; Levitt, P. A unique membrane protein is expressed on early developing limbic system axons and cortical targets. J. Neurosci. 1988, 8, 4653–4661. [Google Scholar] [CrossRef] [PubMed]
- Barbe, M.F.; Levitt, P. The early commitment of fetal neurons to the limbic cortex. J. Neurosci. 1991, 11, 519–533. [Google Scholar] [CrossRef]
- Barbe, M.F.; Levitt, P. Attraction of specific thalamic input by cerebral grafts depends on the molecular identity of the implant. Proc. Natl. Acad. Sci. USA 1992, 89, 3706–3710. [Google Scholar] [CrossRef]
- Ferri, R.T.; Levitt, P. Cerebral Cortical Progenitors Are Fated to Produce Region-specific Neuronal Populations. Cereb. Cortex 1993, 3, 187–198. [Google Scholar] [CrossRef]
- Mann, F.; Zhukareva, V.; Pimenta, A.; Levitt, P.; Bolz, J. Membrane-Associated Molecules Guide Limbic and Nonlimbic Thalamocortical Projections. J. Neurosci. 1998, 18, 9409–9419. [Google Scholar] [CrossRef] [PubMed]
- Lodge, A.P.; Howard, M.R.; McNamee, C.J.; Moss, D.J. Co-localisation, heterophilic interactions and regulated expression of IgLON family proteins in the chick nervous system. Mol. Brain Res. 2000, 82, 84–94. [Google Scholar] [CrossRef]
- Gil, O.D.; Zhang, L.; Chen, S.; Ren, Y.Q.Q.; Pimenta, A.; Zanazzi, G.; Hillman, D.; Levitt, P.; Salzer, J.L. Complementary expression and heterophilic interactions between igLON family members neurotrimin and LAMP. J. Neurobiol. 2002, 51, 190–204. [Google Scholar] [CrossRef]
- McNamee, C.J.; Youssef, S.; Moss, D. IgLONs form heterodimeric complexes on forebrain neurons. Cell Biochem. Funct. 2011, 29, 114–119. [Google Scholar] [CrossRef]
- Hashimoto, T.; Maekawa, S.; Miyata, S. IgLON cell adhesion molecules regulate synaptogenesis in hippocampal neurons. Cell Biochem. Funct. 2009, 27, 496–498. [Google Scholar] [CrossRef]
- Eagleson, K.L.; Pimenta, A.F.; Burns, M.M.; Fairfull, L.D.; Cornuet, P.K.; Zhang, L.; Levitt, P. Distinct domains of the limbic system-associated membrane protein (LAMP) mediate discrete effects on neurite outgrowth. Mol. Cell. Neurosci. 2003, 24, 725–740. [Google Scholar] [CrossRef]
- Sanz, R.L.; Ferraro, G.B.; Girouard, M.-P.P.; Fournier, A.E. Ectodomain shedding of Limbic System-Associated Membrane Protein (LSAMP) by ADAM Metallopeptidases promotes neurite outgrowth in DRG neurons. Sci. Rep. 2017, 7, 7961. [Google Scholar] [CrossRef] [PubMed]
- Nelovkov, A.; Philips, M.A.; Kõks, S.; Vasar, E. Rats with low exploratory activity in the elevated plus-maze have the increased expression of limbic system-associated membrane protein gene in the periaqueductal grey. Neurosci. Lett. 2003, 352, 179–182. [Google Scholar] [CrossRef]
- Nelovkov, A.; Areda, T.; Innos, J.; Kõks, S.; Vasar, E. Rats displaying distinct exploratory activity also have different expression patterns of γ-aminobutyric acid- and cholecystokinin-related genes in brain regions. Brain Res. 2006, 1100, 21–31. [Google Scholar] [CrossRef]
- Catania, E.H.; Pimenta, A.; Levitt, P. Genetic deletion of Lsamp causes exaggerated behavioral activation in novel environments. Behav. Brain Res. 2008, 188, 380–390. [Google Scholar] [CrossRef]
- Innos, J.; Philips, M.-A.A.; Leidmaa, E.; Heinla, I.; Raud, S.; Reemann, P.; Plaas, M.; Nurk, K.; Kurrikoff, K.; Matto, V.; et al. Lower anxiety and a decrease in agonistic behaviour in Lsamp-deficient mice. Behav. Brain Res. 2011, 217, 21–31. [Google Scholar] [CrossRef]
- Bregin, A.; Mazitov, T.; Aug, I.; Philips, M.-A.; Innos, J.; Vasar, E. Increased sensitivity to psychostimulants and GABAergic drugs in Lsamp-deficient mice. Pharmacol. Biochem. Behav. 2019, 183, 87–97. [Google Scholar] [CrossRef]
- Innos, J.; Leidmaa, E.; Philips, M.-A.A.; Sütt, S.; Alttoa, A.; Harro, J.; Kõks, S.; Vasar, E. Lsamp−/− mice display lower sensitivity to amphetamine and have elevated 5-HT turnover. Biochem. Biophys. Res. Commun. 2013, 430, 413–418. [Google Scholar] [CrossRef]
- Innos, J.; Philips, M.-A.; Raud, S.; Lilleväli, K.; Kõks, S.; Vasar, E. Deletion of the Lsamp gene lowers sensitivity to stressful environmental manipulations in mice. Behav. Brain Res. 2012, 228, 74–81. [Google Scholar] [CrossRef]
- Qiu, S.; Champagne, D.L.; Peters, M.; Catania, E.H.; Weeber, E.J.; Levitt, P.; Pimenta, A.F. Loss of limbic system-associated membrane protein leads to reduced hippocampal mineralocorticoid receptor expression, impaired synaptic plasticity, and spatial memory deficit. Biol. Psychiatry 2010, 68, 197–204. [Google Scholar] [CrossRef]
- Must, A.; Tasa, G.; Lang, A.; Vasar, E.; Kõks, S.; Maron, E.; Väli, M. Association of limbic system-associated membrane protein (LSAMP) to male completed suicide. BMC Med. Genet. 2008, 9, 34. [Google Scholar] [CrossRef]
- Miret, M.; Ayuso-Mateos, J.L.; Sanchez-Moreno, J.; Vieta, E. Depressive disorders and suicide: Epidemiology, risk factors, and burden. Neurosci. Biobehav. Rev. 2013, 37, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Traks, T.; Balõtšev, R.; Eller, T.; Must, A.; Koks, S.; Maron, E.; Tõru, I.; Shlik, J.; Vasar, V.; et al. Associations between LSAMP gene polymorphisms and major depressive disorder and panic disorder. Transl. Psychiatry 2012, 2, e152. [Google Scholar] [CrossRef] [PubMed]
- Behan, A.T.; Byrne, C.; Dunn, M.J.; Cagney, G.; Cotter, D.R.; Behan, Á.; Byrne, C.; Dunn, M.J.; Cagney, G.; Cotter, D.R. Proteomic analysis of membrane microdomain-associated proteins in the dorsolateral prefrontal cortex in schizophrenia and bipolar disorder reveals alterations in LAMP, STXBP1 and BASP1 protein expression. Mol. Psychiatry 2009, 14, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Koido, K.; Janno, S.; Traks, T.; Parksepp, M.; Ljubajev, Ü.; Veiksaar, P.; Must, A.; Shlik, J.; Vasar, V.; Vasar, E. Associations between polymorphisms of LSAMP gene and schizophrenia. Psychiatry Res. 2014, 215, 797–798. [Google Scholar] [CrossRef]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef]
- Spaltmann, F.; Brümmendorf, T. CEPU-1, a novel immunoglobulin superfamily molecule, is expressed by developing cerebellar Purkinje cells. J. Neurosci. 1996, 16, 1770–1779. [Google Scholar] [CrossRef]
- Gil, O.D.; Zanazzi, G.; Struyk, A.F.; Salzer, J.L. Neurotrimin mediates bifunctional effects on neurite outgrowth via homophilic and heterophilic interactions. J. Neurosci. 1998, 18, 9312–9325. [Google Scholar] [CrossRef]
- McNamee, C.J.; Reed, J.E.; Howard, M.R.; Lodge, A.P.; Moss, D.J. Promotion of neuronal cell adhesion by members of the IgLON family occurs in the absence of either support or modification of neurite outgrowth. J. Neurochem. 2002, 80, 941–948. [Google Scholar] [CrossRef]
- Chen, S.; Gil, O.; Ren, Y.Q.; Zanazzi, G.; Salzer, J.L.; Hillman, D.E. Neurotrimin expression during cerebellar development suggests roles in axon fasciculation and synaptogenesis. J. Neurocytol. 2001, 30, 927–937. [Google Scholar] [CrossRef]
- Singh, K.; Lilleväli, K.; Gilbert, S.F.; Bregin, A.; Narvik, J.; Jayaram, M.; Rahi, M.; Innos, J.; Kaasik, A.; Vasar, E.; et al. The combined impact of IgLON family proteins Lsamp and Neurotrimin on developing neurons and behavioral profiles in mouse. Brain Res. Bull. 2018, 140, 5–18. [Google Scholar] [CrossRef]
- Lodge, A.P.; McNamee, C.J.; Howard, M.R.; Reed, J.E.; Moss, D.J. Identification and characterization of CEPU-Se-A secreted isoform of the IgLON family protein, CEPU-1. Mol. Cell. Neurosci. 2001, 17, 746–760. [Google Scholar] [CrossRef]
- Mazitov, T.; Bregin, A.; Philips, M.A.; Innos, J.; Vasar, E. Deficit in emotional learning in neurotrimin knockout mice. Behav. Brain Res. 2017, 317, 311–318. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, K.S.; Aragam, N. NTM and NR3C2 polymorphisms influencing intelligence: Family-based association studies. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Maruani, A.; Huguet, G.; Beggiato, A.; ElMaleh, M.; Toro, R.; Leblond, C.S.; Mathieu, A.; Amsellem, F.; Lemière, N.; Verloes, A.; et al. 11q24.2-25 micro-rearrangements in autism spectrum disorders: Relation to brain structures. Am. J. Med. Genet. A 2015, 167A, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Brevik, E.J.; van Donkelaar, M.M.J.; Weber, H.; Sánchez-Mora, C.; Jacob, C.; Rivero, O.; Kittel-Schneider, S.; Garcia-Martínez, I.; Aebi, M.; van Hulzen, K.; et al. Genome-wide analyses of aggressiveness in attention-deficit hyperactivity disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Dorostkar, M.M. Dendritic Spine Pathology in Neurodegenerative Diseases. Annu. Rev. Pathol. 2016, 11, 221–250. [Google Scholar] [CrossRef]
- Kayser, M.S.; Dalmau, J. The emerging link between autoimmune disorders and neuropsychiatric disease. J. Neuropsychiatry Clin. Neurosci. 2011, 23, 90–97. [Google Scholar] [CrossRef] [PubMed]
- McKie, A.B.; Vaughan, S.; Zanini, E.; Okon, I.S.; Louis, L.; de Sousa, C.; Greene, M.I.; Wang, Q.; Agarwal, R.; Shaposhnikov, D.; et al. The OPCML tumor suppressor functions as a cell surface repressor-adaptor, negatively regulating receptor tyrosine kinases in epithelial ovarian cancer. Cancer Discov. 2012, 2, 156–171. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Structure of IgLON family members.

Figure 2.
IgLONs can form homo- and hetero-dimers to interact in trans (in different cells) or in cis (in the same cell). They can be cleaved from the membrane by metalloproteases to form soluble interacting proteins.
Figure 2.
IgLONs can form homo- and hetero-dimers to interact in trans (in different cells) or in cis (in the same cell). They can be cleaved from the membrane by metalloproteases to form soluble interacting proteins.

Figure 3.
A potential link between IgLON5 and the aggregation of hyperphosphorylated-Tau. (A) In healthy conditions, IgLON5 may interact with and regulate a yet to be identified receptor tyrosine kinase (RTK, 1). A signalling cascade involving such RTK, insulin receptor substrate 1 (IRS-1), and AKT (2) prevents Tau phosphorylation by GSK-3β (3). A different RTK-triggered pathway may control Tau phosphorylation (4) and assure microtubule stability. (B) In patients, the anti-IgLON5 antibodies sequester IgLON5 (1) and trigger the degradation of IgLON5 together with the RTK, potentially (2). As such, RTK signalling pathways are altered (3–4). This may result in pathological tau phosphorylation and accumulation (5) and, eventually, neurotoxic microtubule disassembly (6).
Figure 3.
A potential link between IgLON5 and the aggregation of hyperphosphorylated-Tau. (A) In healthy conditions, IgLON5 may interact with and regulate a yet to be identified receptor tyrosine kinase (RTK, 1). A signalling cascade involving such RTK, insulin receptor substrate 1 (IRS-1), and AKT (2) prevents Tau phosphorylation by GSK-3β (3). A different RTK-triggered pathway may control Tau phosphorylation (4) and assure microtubule stability. (B) In patients, the anti-IgLON5 antibodies sequester IgLON5 (1) and trigger the degradation of IgLON5 together with the RTK, potentially (2). As such, RTK signalling pathways are altered (3–4). This may result in pathological tau phosphorylation and accumulation (5) and, eventually, neurotoxic microtubule disassembly (6).

Figure 4.
The metalloprotease ADAM10 can cleave IgLONs to generate soluble proteins able to bind RTKs and start intracellular signal transduction.
Figure 4.
The metalloprotease ADAM10 can cleave IgLONs to generate soluble proteins able to bind RTKs and start intracellular signal transduction.

Figure 5.
Soluble NEGR1 generated by shedding binds FGFR2 and activates intracellular signal transduction pathways.
Figure 5.
Soluble NEGR1 generated by shedding binds FGFR2 and activates intracellular signal transduction pathways.

Figure 6.
Associations between IgLONs and neuropsychiatric disorders.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Salluzzo, M.; Vianello, C.; Abdullatef, S.; Rimondini, R.; Piccoli, G.; Carboni, L. The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes 2023, 14, 1886. https://doi.org/10.3390/genes14101886
AMA Style
Salluzzo M, Vianello C, Abdullatef S, Rimondini R, Piccoli G, Carboni L. The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases. Genes. 2023; 14(10):1886. https://doi.org/10.3390/genes14101886
Chicago/Turabian StyleSalluzzo, Marco, Clara Vianello, Sandra Abdullatef, Roberto Rimondini, Giovanni Piccoli, and Lucia Carboni. 2023. "The Role of IgLON Cell Adhesion Molecules in Neurodegenerative Diseases" Genes 14, no. 10: 1886. https://doi.org/10.3390/genes14101886
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.