

Genetic Distinctness and Diversity of American Aberdeen Cattle Compared to Common Beef Breeds in the United States

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
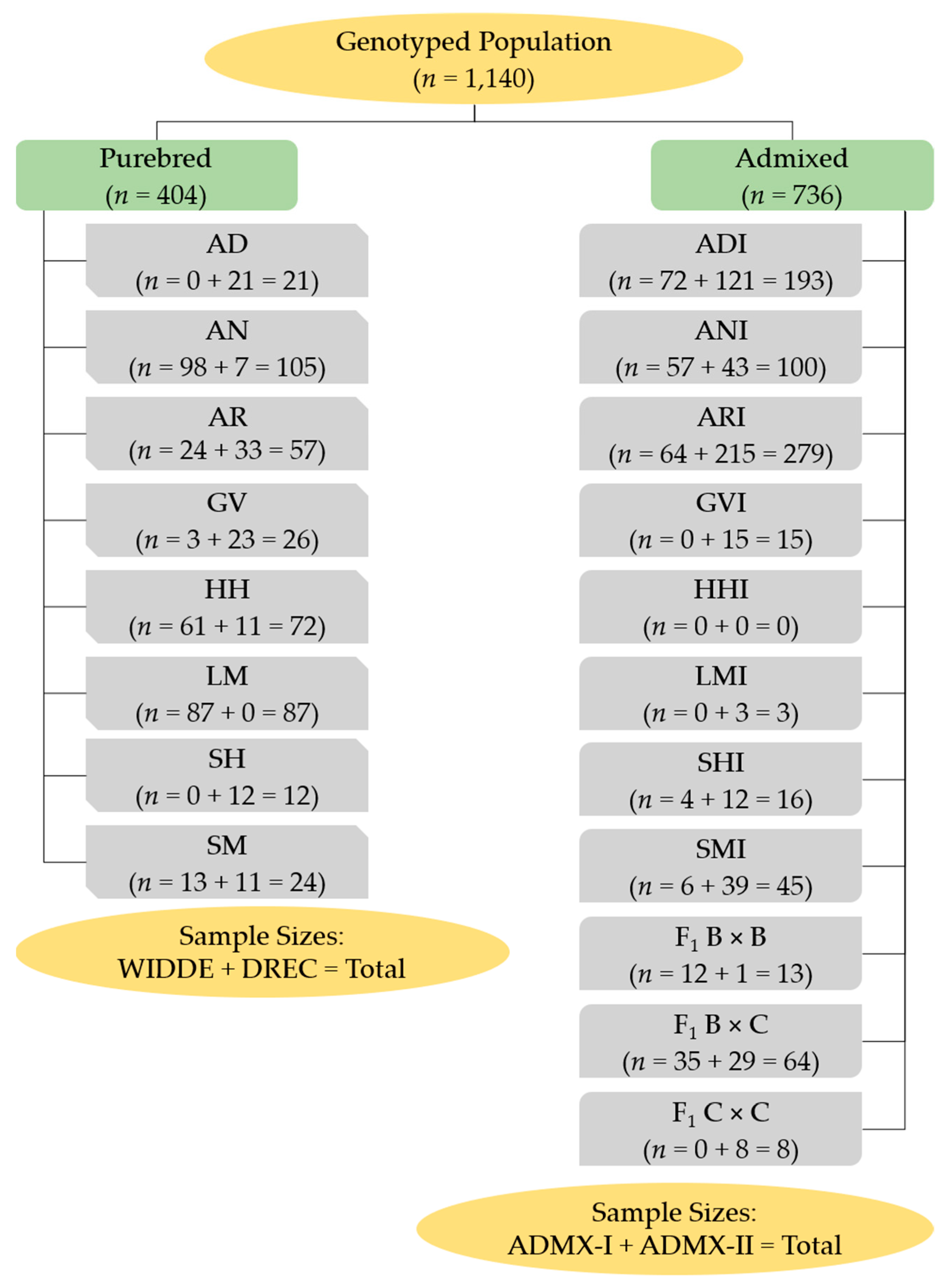
2.1. Animals
2.2. NDSU DREC Population DNA, Genotyping, and Parentage Testing
2.3. Population Strata
- 8 known purebred types (AD, AN, AR, GV, HH, LM, SH, SM),
- 7 known influenced types (ADI, ANI, ARI, GVI, LMI, SHI, SMI), and
- 3 F1 cross types.
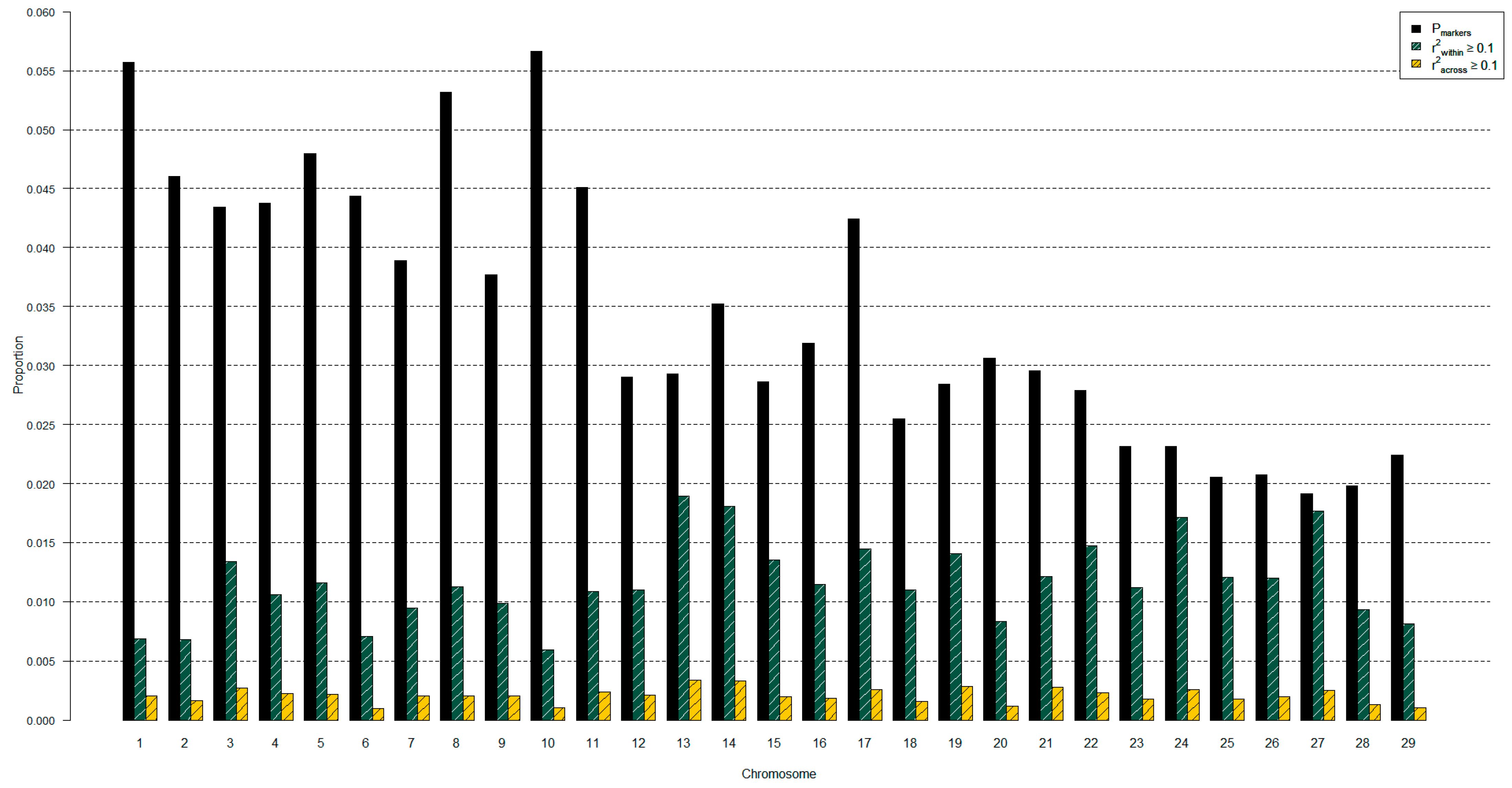
2.4. Genotype Preparation for Diversity Analyses
2.5. Genetic Diversity Analyses
2.5.1. Intrapopulation Measures
2.5.2. Interpopulation Measures
3. Results
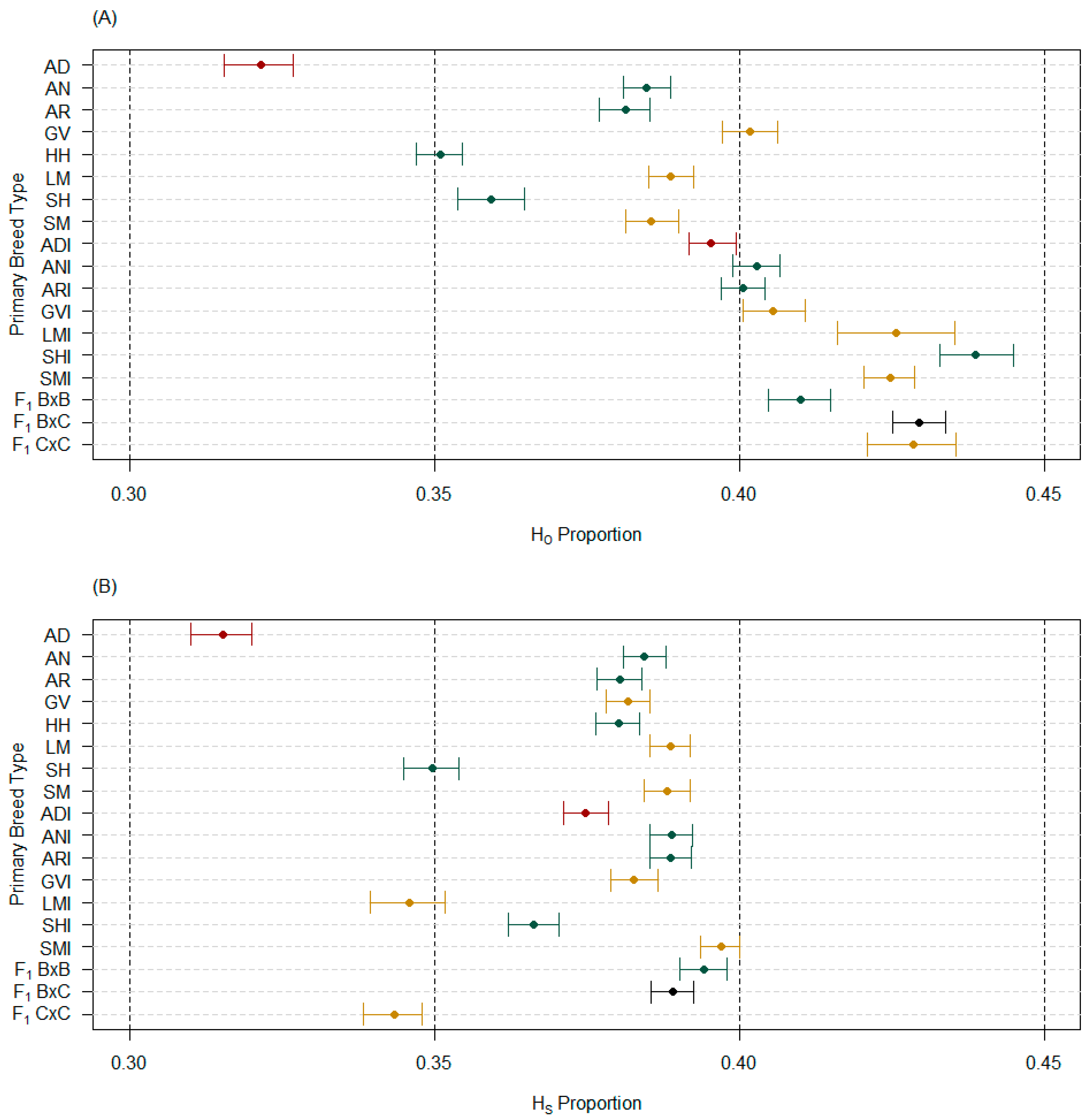
3.1. Average Population Measures
3.2. Intrapopulation Measures
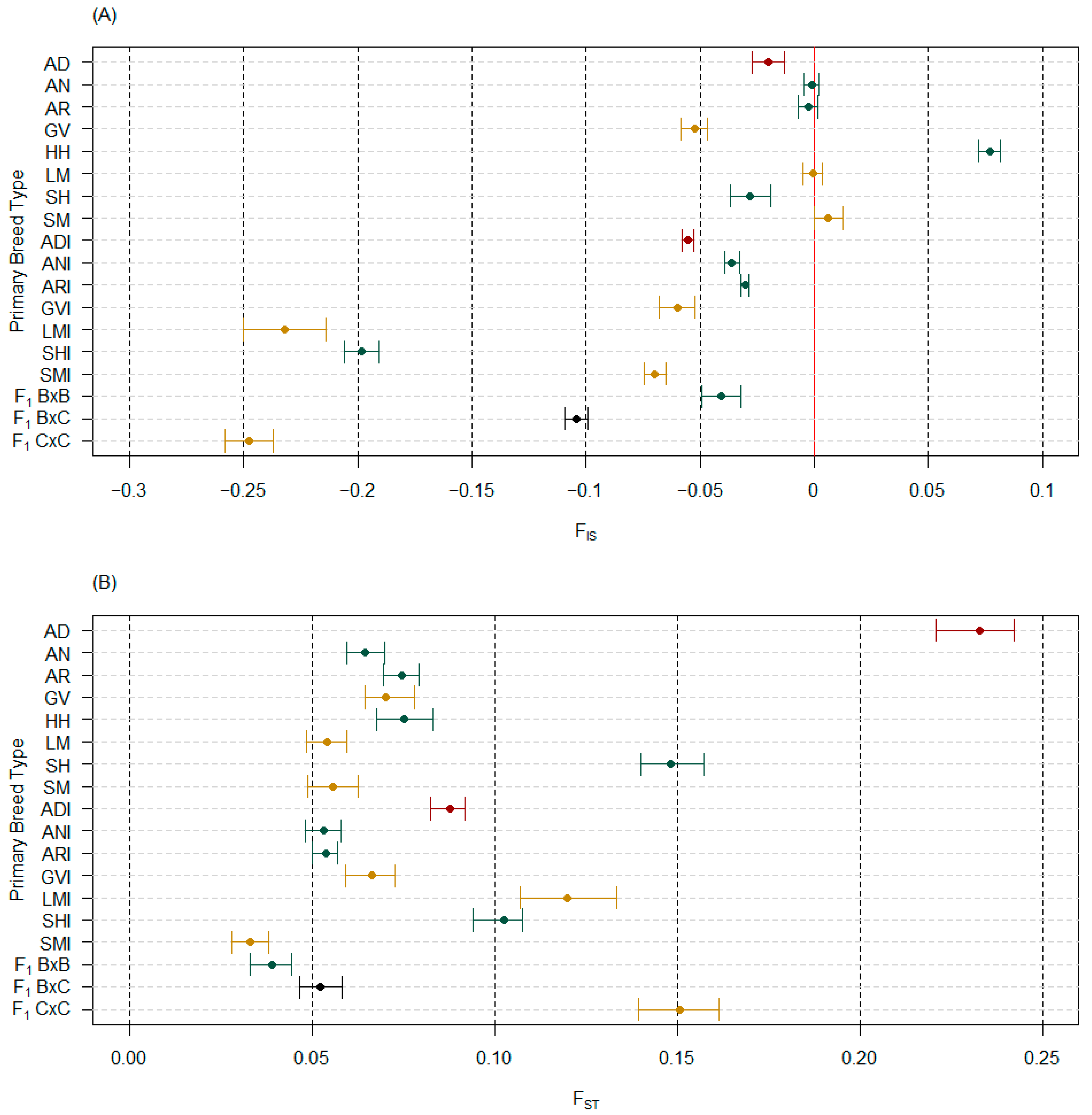
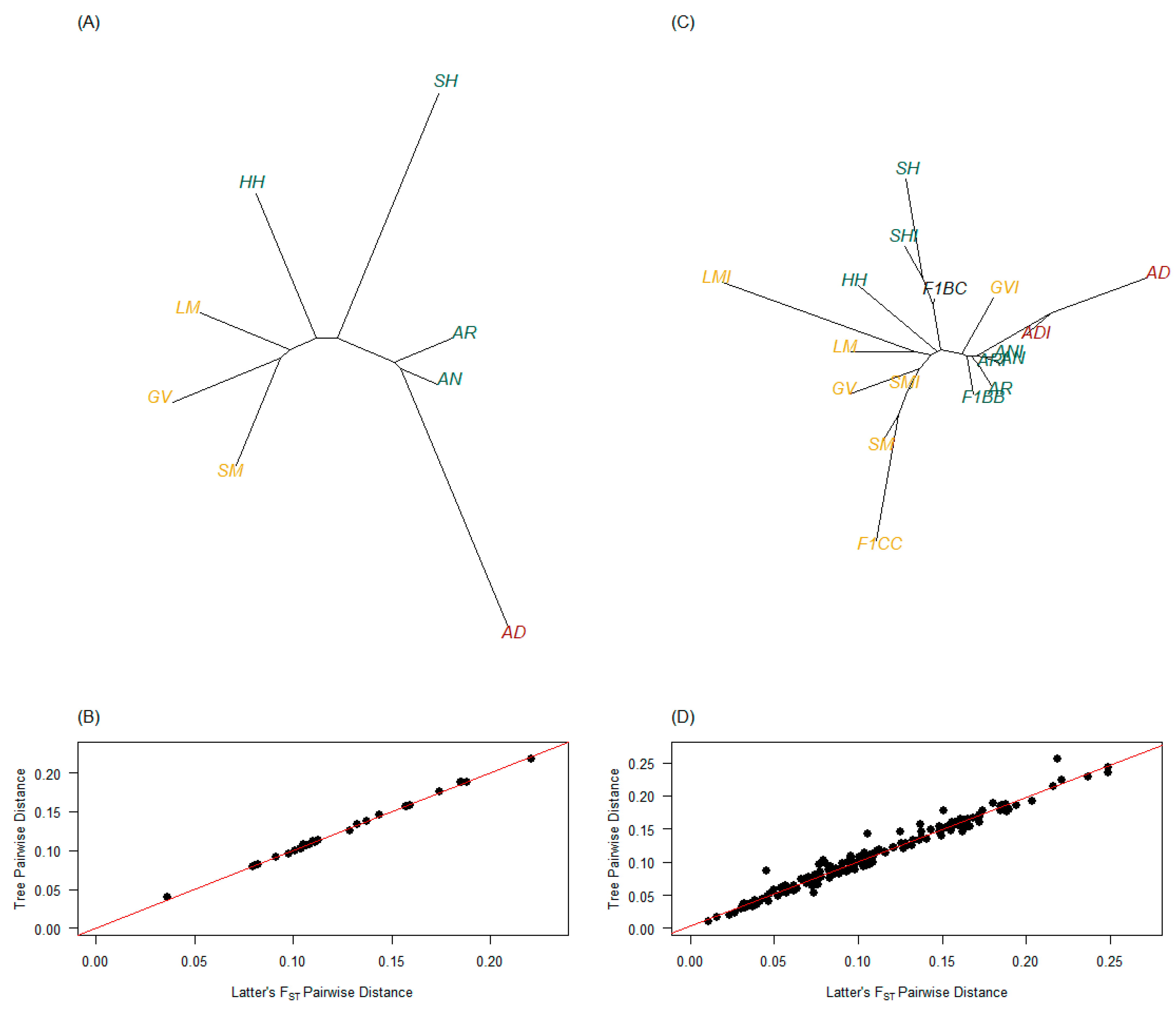
3.3. Interpopulation Measures
3.3.1. Genetic Distances and FST
3.3.2. Analysis of Molecular Variance
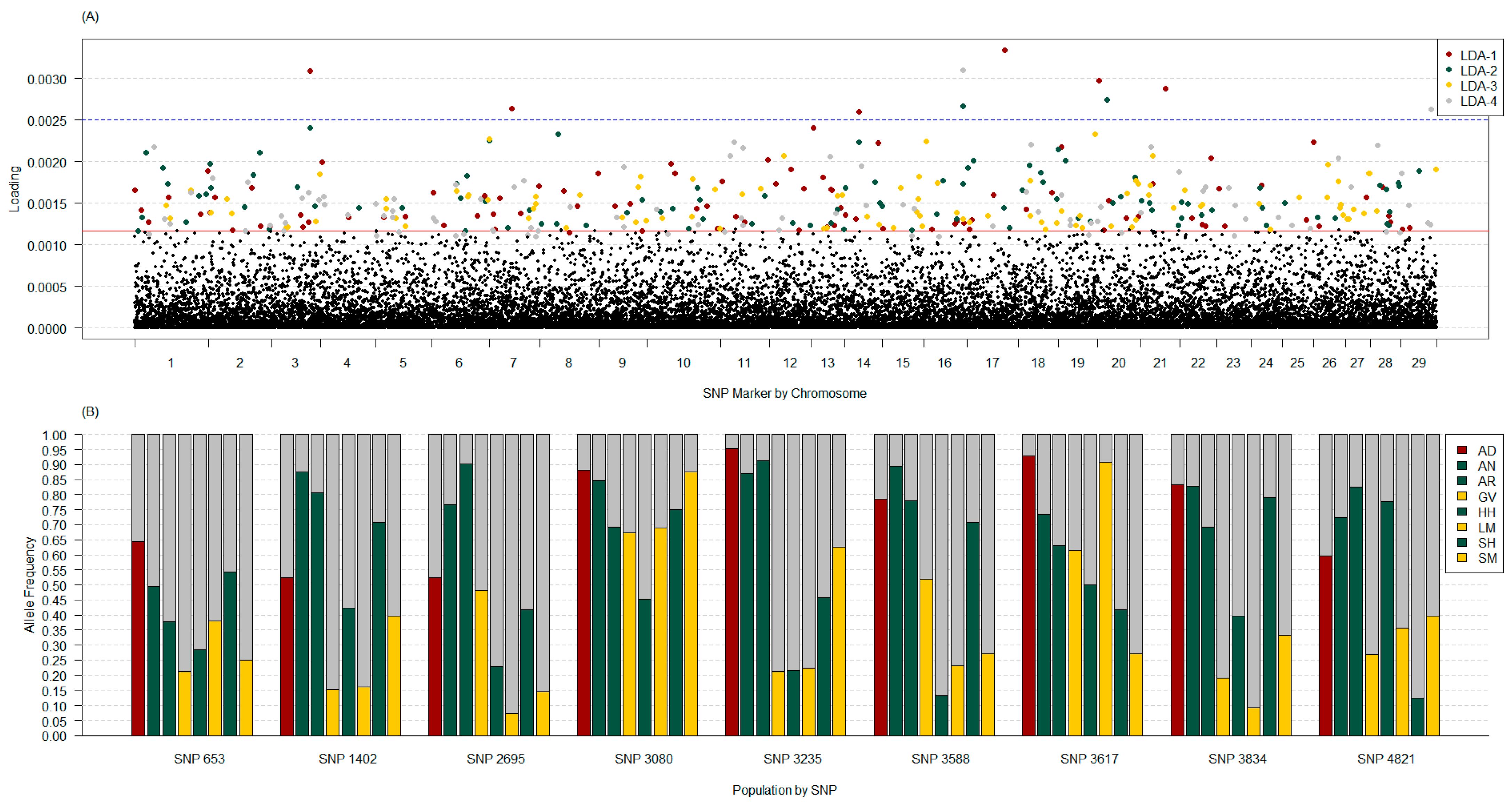
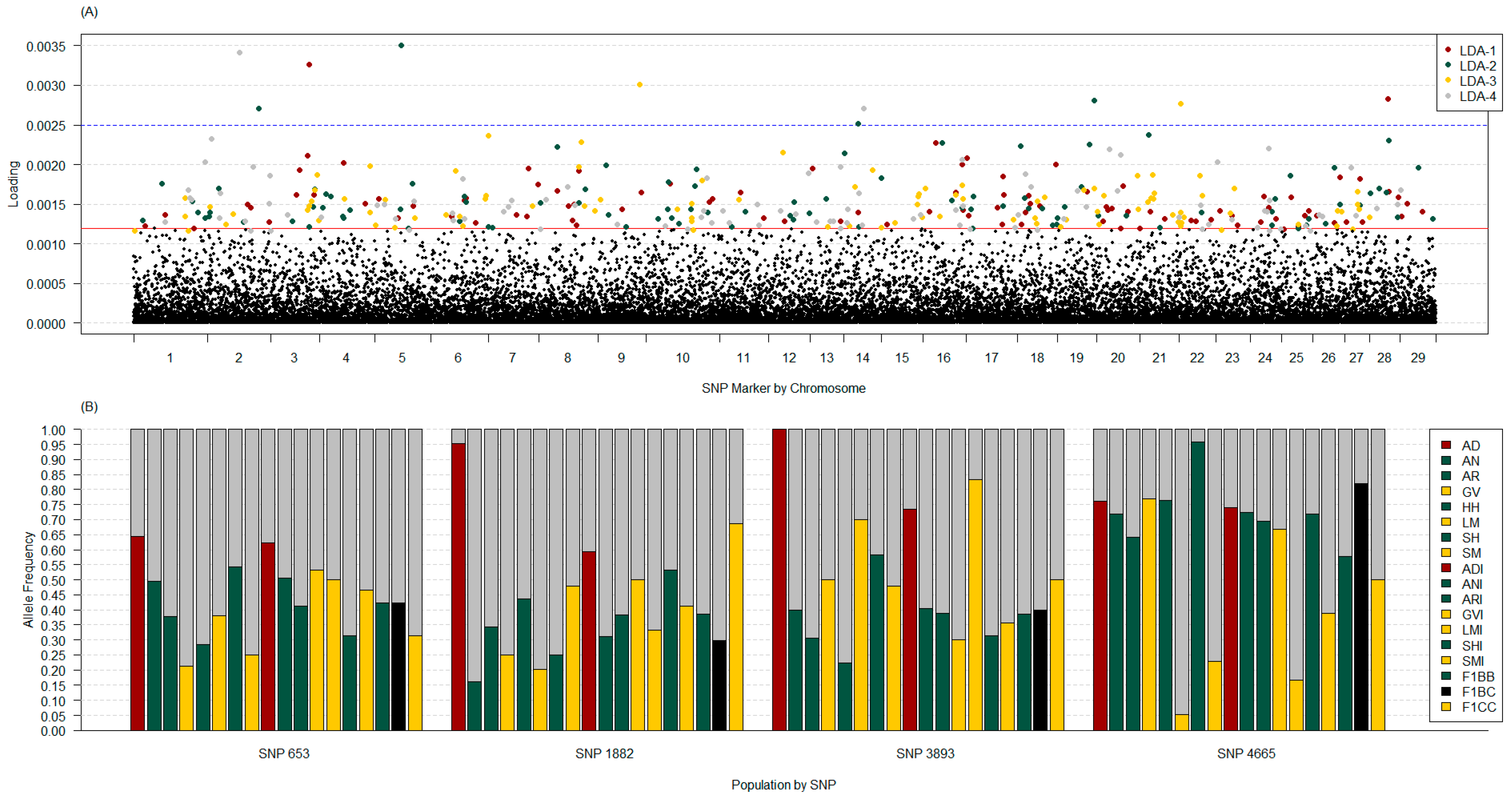
3.3.3. Discriminant Analysis of Principal Components
- Group 1: LM;
- Group 2: GV, HH, SH, and SM;
- Group 3: AD; and
- Group 4: AN and AR.
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zimin, A.V.; Delcher, A.L.; Florea, L.; Kelley, D.R.; Schatz, M.C.; Puiu, D.; Hanrahan, F.; Pertea, G.; Van Tassell, C.P.; Sonstegard, T.S.; et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009, 10, R42. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M.; Lien, S.; et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.Z.; Sasazaki, S.; Mannen, H. Genetic diversity and structure in Bos taurus and Bos indicus populations analyzed by SNP markers. Anim. Sci. J. 2010, 81, 281–289. [Google Scholar] [CrossRef] [PubMed]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.D.; Gill, C.A.; Gao, C.; et al. An assessment of population structure in eight breeds of cattle using a whole genome SNP panel. BMC Genet. 2008, 9, 37. [Google Scholar] [CrossRef]
- Campos, B.M.; do Carmo, A.S.; do Egito, A.A.; da Mariante, A.S.; do Albuquerque, M.S.M.; de Gouveia, J.J.S.; Malhado, C.H.M.; Verardo, L.L.; da Silva, M.V.G.B.; Carneiro, P.L.S. Genetic diversity, population structure, and correlations between locally adapted zebu and taurine breeds in Brazil using SNP markers. Trop. Anim. Health Prod. 2017, 49, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Edea, Z.; Bhuiyan, M.S.A.; Dessie, T.; Rothschild, M.F.; Dadi, H.; Kim, K.S. Genome-wide genetic diversity, population structure and admixture analysis in African and Asian cattle breeds. Animal 2015, 9, 218–226. [Google Scholar] [CrossRef]
- Makina, S.O.; Muchadeyi, F.C.; van Marle-Köster, E.; MacNeil, M.D.; Maiwashe, A. Genetic diversity and population structure among six cattle breeds in South Africa using a whole genome SNP panel. Front. Genet. 2014, 5, 333. [Google Scholar] [CrossRef]
- Notter, D.R. The importance of genetic diversity in livestock populations of the future. J. Anim. Sci. 1999, 77, 61–69. [Google Scholar] [CrossRef]
- Sermyagin, A.A.; Dotsev, A.V.; Gladyr, E.A.; Traspov, A.A.; Deniskova, T.E.; Kostyunina, O.V.; Reyer, H.; Wimmers, K.; Barbato, M.; Paronyan, I.A.; et al. Whole-genome SNP analysis elucidates the genetic structure of Russian cattle and its relationship with Eurasian taurine breeds. Genet. Sel. Evol. 2018, 50, 37. [Google Scholar] [CrossRef]
- Alam, M.Z.; Lee, Y.-M.; Son, H.-J.; Hanna, L.H.; Riley, D.G.; Mannen, H.; Sasazaki, S.; Park, S.P.; Kim, J.-J. Genetic characteristics of Korean Jeju Black cattle with high density single nucleotide polymorphisms. Anim. Biosci. 2020, 34, 789–800. [Google Scholar] [CrossRef]
- Cañas-Álvarez, J.J.; González-Rodríguez, A.; Munilla, S.; Varona, L.; Díaz, C.; Baro, J.A.; Altarriba, J.; Molina, A.; Piedrafita, J. Genetic diversity and divergence among Spanish beef cattle breeds assessed by a bovine high-density SNP chip. J. Anim. Sci. 2015, 93, 5164–5174. [Google Scholar] [CrossRef]
- Edea, Z.; Dadi, H.; Kim, S.W.; Dessie, T.; Lee, T.; Kim, H.; Kim, J.J.; Kim, K.S. Genetic diversity, population structure and relationships in indigenous cattle populations of Ethiopia and Korean Hanwoo breeds using SNP markers. Front. Genet. 2013, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Karimi, K.; Strucken, E.M.; Moghaddar, N.; Ferdosi, M.H.; Esmailizadeh, A.; Gondro, C. Local and global patterns of admixture and population structure in Iranian native cattle. BMC Genet. 2016, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, X.; Zhang, Y.; Zhao, Y.; Zhang, J.; Jia, Y.; Zhu, B.; Xu, L.; Zhang, L.; Gao, H.; et al. Genome-wide assessment of genetic diversity and population structure insights into admixture and introgression in Chinese indigenous cattle. BMC Genet. 2018, 19, 114. [Google Scholar] [CrossRef] [PubMed]
- Parnell, P.F.; Arthur, P.F.; Barlow, R. Direct response to divergent selection for yearling growth rate in Angus cattle. Livest. Prod. Sci. 1997, 49, 297–304. [Google Scholar] [CrossRef]
- Arthur, P.F.; Parnell, P.F.; Richardson, E.C. Correlated responses in calf body weight and size to divergent selection for yearling growth rate in Angus cattle. Livest. Prod. Sci. 1997, 49, 305–312. [Google Scholar] [CrossRef]
- Archer, J.A.; Herd, R.M.; Arthur, P.F.; Parnell, P.F. Correlated responses in rate of maturation and mature ssssize of cows and steers to divergent selection for yearling growth rate in Angus cattle. Livest. Prod. Sci. 1998, 54, 183–192. [Google Scholar] [CrossRef]
- Perry, D.; Arthur, P.F. Correlated responses in body composition and fat partitioning to divergent selection for yearling growth rate in Angus cattle. Livest. Prod. Sci. 2000, 62, 143–153. [Google Scholar] [CrossRef]
- Barnett, D. History of Lowlines. In Australian Lowline Cattle Association Foundation Herd Book; Australian Lowline Cattle Association: Armidale, NSW, Australia, 1998; pp. 1–4. [Google Scholar]
- Effertz EZ Ranch Who Are, We. Available online: https://effertzezranch.com/aboutus.html (accessed on 16 April 2023).
- American Aberdeen Association. The Ledger; Blueprint Media LLC: Parker, CO, USA, 2021; pp. 11–31. [Google Scholar]
- Hanotte, O.; Dessie, T.; Kemp, S. Time to tap Africa’s livestock genomes. Science 2010, 328, 1640–1641. [Google Scholar] [CrossRef]
- Sempéré, G.; Moazami-Goudarzi, K.; Eggen, A.; Laloë, D.; Gautier, M.; Flori, L. WIDDE: A Web-Interfaced next Generation Database for Genetic Diversity Exploration, with a first application in cattle. BMC Genomics 2015, 16, 940. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Matukumalli, L.K.; Lawley, C.T.; Schnabel, R.D.; Taylor, J.F.; Allan, M.F.; Heaton, M.P.; O’Connell, J.; Moore, S.S.; Smith, T.P.L.; Sonstegard, T.S.; et al. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 2009, 4, e5350. [Google Scholar] [CrossRef] [PubMed]
- Illumina Inc. Agrigenomics. Available online: https://www.illumina.com/areas-of-interest/agrigenomics.html (accessed on 26 April 2023).
- Rosen, B.D.; Bickhart, D.M.; Schnabel, R.D.; Koren, S.; Elsik, C.G.; Tseng, E.; Rowan, T.N.; Low, W.Y.; Zimin, A.; Couldrey, C.; et al. De novo assembly of the cattle reference genome with single-molecule sequencing. Gigascience 2020, 9, giaa021. [Google Scholar] [CrossRef]
- Wigginton, J.E.; Cutler, D.J.; Abecasis, R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005, 76, 887–893. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing. Available online: https://www.R-project.org/ (accessed on 26 April 2023).
- Posit Team RStudio: Integrated Development Environment for R. Available online: http://www.posit.co/ (accessed on 26 April 2023).
- Hayes, B.J. Technical Note: Efficient parentage assignment and pedigree reconstruction with dense single nucleotide polymorphism data. J. Dairy Sci. 2011, 94, 2114–2117. [Google Scholar] [CrossRef]
- Dowle, M.; Srinivasan, A. data.table: Extension of “data.frame”. Available online: https://CRAN.R-project.org/package=data.table (accessed on 26 April 2023).
- Clayton, D. SnpStats: SnpMatrix and XSnpMatrix Classes and Methods. Available online: https://www.bioconductor.org/packages/release/bioc/html/snpStats.html (accessed on 26 April 2023).
- Price, A.L.; Weale, M.E.; Patterson, N.; Myers, S.R.; Need, A.C.; Shianna, K.V.; Ge, D.; Rotter, J.I.; Torres, E.; Taylor, K.D.; et al. Long-range LD can confound genome scans in admixed populations. Am. J. Hum. Genet. 2008, 83, 132–135. [Google Scholar] [CrossRef]
- Abdellaoui, A.; Hottenga, J.-J.; de Knijff, P.; Nivard, M.G.; Xiao, X.; Scheet, P.; Brooks, A.; Ehli, E.A.; Hu, Y.; Davies, G.E.; et al. Population structure, migration, and diversifying selection in the Netherlands. Eur. J. Hum. Genet. 2013, 21, 1277–1285. [Google Scholar] [CrossRef]
- Rogers, A.R. How population growth affects linkage disequilibrium. Genetics 2014, 197, 1329–1341. [Google Scholar] [CrossRef]
- Tong, X. PlinkFile: “PLINK” (and ’GCTA’) File Helpers. Available online: https://CRAN.R-project.org/package=plinkFile (accessed on 26 April 2023).
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef]
- Goudet, J.; Jombart, T. Hierfstat: Estimation and Tests of Hierarchical F-Statistics. Available online: https://CRAN.R-project.org/package=hierfstat (accessed on 26 April 2023).
- Li, Z.; Löytynoja, A.; Fraimout, A.; Merilä, J. Effects of marker type and filtering criteria on QST-FST comparisons. R. Soc. Open Sci. 2019, 6, 190666. [Google Scholar] [CrossRef] [PubMed]
- Dementieva, N.V.; Mitrofanova, O.V.; Dysin, A.P.; Kudinov, A.A.; Stanishevskaya, O.I.; Larkina, T.A.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N.; Smaragdov, M.G. Assessing the effects of rare alleles and linkage disequilibrium on estimates of genetic diversity in the chicken populations. Animal 2021, 15, 100171. [Google Scholar] [CrossRef] [PubMed]
- Takezaki, N.; Nei, M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics 1996, 144, 389–399. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. Ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2019, 35, 526–528. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef]
- Dray, S.; Dufour, A.-B. The Ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef]
- Bougeard, S.; Dray, S. Supervised multiblock analysis in R with the Ade4 package. J. Stat. Softw. 2018, 86, 1–17. [Google Scholar] [CrossRef]
- Thioulouse, J.; Dray, S.; Dufour, A.-B.; Siberchicot, A.; Jombart, T.; Pavoine, S. Multivariate Analysis of Ecological Data with Ade4; Springer: New York, NY, USA, 2018; ISBN 978-1-4939-8848-8. [Google Scholar]
- Ojala, M.; Garriga, G.C. Permutations tests for studying classifier performance. J. Mach. Learn. Res. 2010, 11, 1833–1863. [Google Scholar]
- Thia, J.A. Guidelines for standardizing the application of discriminant analysis of principal components to genotype data. Mol. Ecol. Resour. 2023, 23, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, D.; Adler, D. Rgl: 3D Visualization Using OpenGL. Available online: https://CRAN.R-project.org/package=rgl (accessed on 6 June 2023).
- American Aberdeen Association Bylaws and Policies. Available online: https://americanaberdeen.com/about/bylaws-policies/ (accessed on 19 June 2023).
- Red Angus Association of America Rules and Regulations. Available online: https://redangus.org/herd-management/breeders-guide/ (accessed on 19 June 2023).
- American Gelbvieh Association Rules and Bylaws. Available online: https://gelbvieh.org/membership/aga-rules (accessed on 19 June 2023).
- North American Limousin Foundation Member Center. Available online: https://nalf.org/management/member-center/ (accessed on 19 June 2023).
- American Shorthorn Association Member Services. Available online: https://shorthorn.org/ (accessed on 19 June 2023).
- American Simmental Association Rules and Bylaws. Available online: https://simmental.org/site/index.php/about/board-of-trustees#rules-bylaws-annual-report-financial-statements (accessed on 19 June 2023).
- World Angus Secretariat Members. Available online: https://worldangussecretariat.com/members/ (accessed on 19 June 2023).
- American Angus Association Rules and Forms. Available online: https://www.angus.org/General/NewMem (accessed on 19 June 2023).
- American Hereford Association Rules and Forms. Available online: https://hereford.org/member-services/register-an-animal/rules-forms/ (accessed on 19 June 2023).
- Neogen GeneSeek® Genomic ProfilerTM Bovine 100K. Available online: https://www.neogen.com/globalassets/pim/assets/original/10000/official_40-ggp-100k-beef_technical-specifications-sheet.pdf (accessed on 19 June 2023).
- Illumina, Inc. GeneSeek GGP Array Kit Support Resources. Available online: https://support.illumina.com/array/array_kits/geneseek-ggp-arrays.html (accessed on 19 June 2023).
- Reist-Marti, S.B.; Simianer, H.; Gibson, J.; Hanotte, O.; Rege, J.E.O. Weitzman’s approach and conservation of breed diversity: An application to African cattle breeds. Conserv. Biol. 2003, 17, 1299–1311. [Google Scholar] [CrossRef]
- Márquez, G.C.; Speidel, S.E.; Enns, R.M.; Garrick, D.J. Genetic diversity and population structure of American Red Angus cattle. J. Anim. Sci. 2010, 88, 59–68. [Google Scholar] [CrossRef]
- Lawson, D.J.; van Dorp, L.; Falush, D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat. Commun. 2018, 9, 3258. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Cullingham, C.I.; Peery, R.M. The influence of a priori grouping on inference of genetic clusters: Simulation study and literature review of the DAPC method. Heredity 2020, 125, 269–280. [Google Scholar] [CrossRef]
- Gouveia, J.J.d.S.; da Silva, M.V.G.B.; Paiva, S.R.; de Oliveira, S.M.P. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Iso-Touru, T.; Tapio, M.; Vilkki, J.; Kiseleva, T.; Ammosov, I.; Ivanova, Z.; Popov, R.; Ozerov, M.; Kantanen, J. Genetic diversity and genomic signatures of selection among cattle breeds from Siberia, Eastern and Northern Europe. Anim. Genet. 2016, 47, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Bhati, M.; Kadri, N.K.; Crysnanto, D.; Pausch, H. Assessing genomic diversity and signatures of selection in original Braunvieh cattle using whole-genome sequencing data. BMC Genomics 2020, 21, 27. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, S.; Zhang, H.; Zhang, Z.; Chen, N.; Li, Z.; Sun, H.; Liu, X.; Lyu, S.; Wang, X.; et al. Assessing genomic diversity and signatures of selection in Jiaxian Red cattle using whole-genome sequencing data. BMC Genomics 2021, 22, 43. [Google Scholar] [CrossRef]
- Taye, M.; Yoon, J.; Dessie, T.; Cho, S.; Oh, S.J.; Lee, H.-K.; Kim, H. Deciphering signature of selection affecting beef quality traits in Angus cattle. Genes Genom. 2018, 40, 63–75. [Google Scholar] [CrossRef]
- Bickhart, D.M.; Xu, L.; Hutchison, J.L.; Cole, J.B.; Null, D.J.; Schroeder, S.G.; Song, J.; Garcia, J.F.; Sonstegard, T.S.; Van Tassell, C.P.; et al. Diversity and population-genetic properties of copy number variations and multicopy genes in cattle. DNA Res. 2016, 23, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Dimauro, C.; Cellesi, M.; Steri, R.; Gaspa, G.; Sorbolini, S.; Stella, A.; Macciotta, N.P.P. Use of the canonical discriminant analysis to select SNP markers for bovine breed assignment and traceability purposes. Anim. Genet. 2013, 44, 377–382. [Google Scholar] [CrossRef]
- Bertolini, F.; Galimberti, G.; Calò, D.G.; Schiavo, G.; Matassino, D.; Fontanesi, L. Combined use of principal component analysis and random forests identify population-informative single nucleotide polymorphisms: Application in cattle breeds. J. Anim. Breed. Genet. 2015, 132, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Judge, M.M.; Kelleher, M.M.; Kearney, J.F.; Sleator, R.D.; Berry, D.P. Ultra-low-density genotype panels for breed assignment of Angus and Hereford cattle. Animal 2017, 11, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Hulsegge, I.; Schoon, M.; Windig, J.; Neuteboom, M.; Hiemstra, S.J.; Schurink, A. Development of a genetic tool for determining breed purity of cattle. Livest. Sci. 2019, 223, 60–67. [Google Scholar] [CrossRef]
- Reverter, A.; Hudson, N.J.; McWilliam, S.; Alexandre, P.A.; Li, Y.; Barlow, R.; Welti, N.; Daetwyler, H.; Porto-Neto, L.R.; Dominik, S. A low-density SNP genotyping panel for the accurate prediction of cattle breeds. J. Anim. Sci. 2020, 98, skaa337. [Google Scholar] [CrossRef] [PubMed]
- Gebrehiwot, N.Z.; Strucken, E.M.; Marshall, K.; Aliloo, H.; Gibson, J.P. SNP panels for the estimation of dairy breed proportion and parentage assignment in African crossbred dairy cattle. Genet. Sel. Evol. 2021, 53, 21. [Google Scholar] [CrossRef]
- Kumar, H.; Panigrahi, M.; Chhotaray, S.; Parida, S.; Chauhan, A.; Bhushan, B.; Gaur, G.K.; Mishra, B.P.; Singh, R.K. Comparative analysis of five different methods to design a breed-specific SNP panel for cattle. Anim. Biotechnol. 2021, 32, 130–136. [Google Scholar] [CrossRef]
- Wilmot, H.; Bormann, J.; Soyeurt, H.; Hubin, X.; Glorieux, G.; Mayeres, P.; Bertozzi, C.; Gengler, N. Development of a genomic tool for breed assignment by comparison of different classification models: Application to three local cattle breeds. J. Anim. Breed. Genet. 2022, 139, 40–61. [Google Scholar] [CrossRef]
- Wilmot, H.; Niehoff, T.; Soyeurt, H.; Gengler, N.; Calus, M.P.L. The use of a genomic relationship matrix for breed assignment of cattle breeds: Comparison and combination with a machine learning method. J. Anim. Sci. 2023, 101, skad172. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purebred PBT | All PBT | |||||
|---|---|---|---|---|---|---|
| Estimate 2 | Unpruned | Pruned | Change, % 3 | Unpruned | Pruned | Change, % 3 |
| HO | 0.3565 | 0.3718 | 4.12 | 0.3833 | 0.3965 | 3.33 |
| HS | 0.3560 | 0.3713 | 4.12 | 0.3629 | 0.3756 | 3.38 |
| HT | 0.4006 | 0.4118 | 2.72 | 0.3986 | 0.4090 | 2.54 |
| DST | 0.0449 | 0.0405 | −9.80 | 0.0357 | 0.0334 | −6.44 |
| FST | 0.1119 | 0.0983 | 12.15 | 0.0897 | 0.0816 | −9.03 |
| HT’ | 0.4073 | 0.4175 | 2.44 | 0.4007 | 0.4109 | 2.48 |
| DST’ | 0.0513 | 0.0463 | −9.75 | 0.0378 | 0.0353 | −6.61 |
| FST’ | 0.1259 | 0.1108 | −11.99 | 0.0944 | 0.0859 | −9.00 |
| FIS | −0.0012 | −0.0014 | 16.67 | −0.0564 | −0.0556 | −1.44 |
| Dest | 0.0796 | 0.0736 | −7.54 | 0.0594 | 0.0566 | −4.71 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhowmik, N.; Seaborn, T.; Ringwall, K.A.; Dahlen, C.R.; Swanson, K.C.; Hulsman Hanna, L.L. Genetic Distinctness and Diversity of American Aberdeen Cattle Compared to Common Beef Breeds in the United States. Genes 2023, 14, 1842. https://doi.org/10.3390/genes14101842
Bhowmik N, Seaborn T, Ringwall KA, Dahlen CR, Swanson KC, Hulsman Hanna LL. Genetic Distinctness and Diversity of American Aberdeen Cattle Compared to Common Beef Breeds in the United States. Genes. 2023; 14(10):1842. https://doi.org/10.3390/genes14101842
Chicago/Turabian StyleBhowmik, Nayan, Travis Seaborn, Kris A. Ringwall, Carl R. Dahlen, Kendall C. Swanson, and Lauren L. Hulsman Hanna. 2023. "Genetic Distinctness and Diversity of American Aberdeen Cattle Compared to Common Beef Breeds in the United States" Genes 14, no. 10: 1842. https://doi.org/10.3390/genes14101842