
Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Modifier Genes in Alport Spectrum Diseases
2.1. Slit Diaphragm NPHS2 and NPHS1 Gene Variants
2.2. NEPH3 (Filtrin, Kirrel2)
2.3. LAMA5 (Laminin α-5)
2.4. MYH9 (Non-Muscle Myosin Heavy Chain-9)
2.5. HBEGF (Heparin Binding Epidermal Growth Factor)
2.6. MYO1E (Non-Muscle Membrane-Associated Class I Myosin)
2.7. SYNPO (Synaptopodin)
2.8. Fmn1 (Formin 1)
2.9. DDR1 (Discoidin Domain Receptor-1)
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Gregorio, V.; Caparali, E.B.; Shojaei, A.; Ricardo, S.; Barua, M. Alport Syndrome: Clinical Spectrum and Therapeutic Advances. Kidney Med. 2023, 5, 100631. [Google Scholar] [CrossRef] [PubMed]
- Flinter, F.A.; Cameron, J.S.; Chantler, C.; Houston, I.; Bobrow, M. Genetics of classic Alport’s syndrome. Lancet 1988, 2, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Savige, J. Alport syndrome: Its effects on the glomerular filtration barrier and implications for future treatment. J. Physiol. 2014, 592, 4013–4023. [Google Scholar] [CrossRef]
- Gross, O.; Kashtan, C.E.; Rheault, M.N.; Flinter, F.; Savige, J.; Miner, J.H.; Torra, R.; Ars, E.; Deltas, C.; Savva, I.; et al. Advances and unmet needs in genetic, basic and clinical science in Alport syndrome: Report from the 2015 International Workshop on Alport Syndrome. Nephrol. Dial. Transplant. 2016, 32, 916–924. [Google Scholar] [CrossRef]
- Aypek, H.; Krisp, C.; Lu, S.; Liu, S.; Kylies, D.; Kretz, O.; Wu, G.; Moritz, M.; Amann, K.; Benz, K.; et al. Loss of the collagen IV modifier prolyl 3-hydroxylase 2 causes thin basement membrane nephropathy. J. Clin. Investig. 2022, 132, e147253. [Google Scholar] [CrossRef]
- Deltas, C. Thin basement membrane lesion is not only a collagen IV nephropathy: Do not underestimate “decorative” additions to collagens. Kidney Int. 2022, 102, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Trevillian, P.; May, S.; Diakumis, P.; Wang, Y.; Colville, D.; Bahlo, M.; Greferath, U.; Fletcher, E.; Young, B.; et al. KCTD1 and Scalp-Ear-Nipple (‘Finlay–Marks’) syndrome may be associated with myopia and Thin basement membrane nephropathy through an effect on the collagen IV α3 and α4 chains. Ophthalmic Genet. 2023, 44, 19–27. [Google Scholar] [CrossRef]
- Savige, J.; Lipska-Zietkiewicz, B.S.; Watson, E.; Hertz, J.M.; Deltas, C.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; Cerkauskaite, A.; et al. Guidelines for Genetic Testing and Management of Alport Syndrome. Clin. J. Am. Soc. Nephrol. 2022, 17, 143–154. [Google Scholar] [CrossRef]
- Savige, J.; Storey, H.; Watson, E.; Hertz, J.M.; Deltas, C.; Renieri, A.; Mari, F.; Hilbert, P.; Plevova, P.; Byers, P.; et al. Consensus statement on standards and guidelines for the molecular diagnostics of Alport syndrome: Refining the ACMG criteria. Eur. J. Hum. Genet. 2021, 29, 1186–1197. [Google Scholar] [CrossRef]
- Matthaiou, A.; Poulli, T.; Deltas, C. Prevalence of clinical, pathological and molecular features of glomerular basement membrane nephropathy caused by COL4A3 or COL4A4 mutations: A systematic review. Clin. Kidney J. 2020, 13, 1025–1036. [Google Scholar] [CrossRef]
- Longo, I.; Porcedda, P.; Mari, F.; Giachino, D.; Meloni, I.; Deplano, C.; Brusco, A.; Bosio, M.; Massella, L.; Lavoratti, G.; et al. COL4A3/COL4A4 mutations: From familial hematuria to autosomal-dominant or recessive Alport syndrome. Kidney Int. 2002, 61, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C.; Savva, I.; Voskarides, K.; Papazachariou, L.; Pierides, A. Carriers of Autosomal Recessive Alport Syndrome with Thin Basement Membrane Nephropathy Presenting as Focal Segmental Glomerulosclerosis in Later Life. Nephron 2015, 130, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Voskarides, K.; Damianou, L.; Neocleous, V.; Zouvani, I.; Christodoulidou, S.; Hadjiconstantinou, V.; Ioannou, K.; Athanasiou, Y.; Patsias, C.; Alexopoulos, E.; et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2007, 18, 3004–3016. [Google Scholar] [CrossRef]
- Becherucci, F.; Landini, S.; Palazzo, V.; Cirillo, L.; Raglianti, V.; Lugli, G.; Tiberi, L.; Dirupo, E.; Bellelli, S.; Mazzierli, T.; et al. A Clinical Workflow for Cost-Saving High-Rate Diagnosis of Genetic Kidney Diseases. J. Am. Soc. Nephrol. 2023, 34, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Pierides, A.; Voskarides, K.; Kkolou, M.; Hadjigavriel, M.; Deltas, C. X-linked, COL4A5 hypomorphic Alport mutations such as G624D and P628L may only exhibit thin basement membrane nephropathy with microhematuria and late onset kidney failure. Hippokratia 2013, 17, 207–213. [Google Scholar]
- Hadjipanagi, D.; Papagregoriou, G.; Koutsofti, C.; Polydorou, C.; Alivanis, P.; Andrikos, A.; Christodoulidou, S.; Dardamanis, M.; Diamantopoulos, A.A.; Fountoglou, A.; et al. Novel and Founder Pathogenic Variants in X-Linked Alport Syndrome Families in Greece. Genes 2022, 13, 2203. [Google Scholar] [CrossRef]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.; Tönshoff, B.; Höcker, B.; Wygoda, S.; Ehrich, J.H.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef]
- Gross, O.; Tonshoff, B.; Weber, L.T.; Pape, L.; Latta, K.; Fehrenbach, H.; Lange-Sperandio, B.; Zappel, H.; Hoyer, P.; Staude, H.; et al. A multicenter, randomized, placebo-controlled, double-blind phase 3 trial with open-arm comparison indicates safety and efficacy of nephroprotective therapy with ramipril in children with Alport’s syndrome. Kidney Int. 2020, 97, 1275–1286. [Google Scholar] [CrossRef]
- Yamamura, T.; Horinouchi, T.; Nagano, C.; Omori, T.; Sakakibara, N.; Aoto, Y.; Ishiko, S.; Nakanishi, K.; Shima, Y.; Nagase, H.; et al. Genotype-phenotype correlations influence the response to angiotensin-targeting drugs in Japanese patients with male X-linked Alport syndrome. Kidney Int. 2020, 98, 1605–1614. [Google Scholar] [CrossRef]
- Boeckhaus, J.; Hoefele, J.; Riedhammer, K.M.; Nagel, M.; Beck, B.B.; Choi, M.; Gollasch, M.; Bergmann, C.; Sonntag, J.E.; Troesch, V.; et al. Lifelong effect of therapy in young patients with the COL4A5 Alport missense variant p.(Gly624Asp): A prospective cohort study. Nephrol. Dial. Transplant. 2022, 37, 2496–2504. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C. Digenic inheritance and genetic modifiers. Clin. Genet. 2018, 93, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Tonna, S.; Wang, Y.Y.; Wilson, D.; Rigby, L.; Tabone, T.; Cotton, R.; Savige, J. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr. Nephrol. 2008, 23, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Voskarides, K.; Arsali, M.; Athanasiou, Y.; Elia, A.; Pierides, A.; Deltas, C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr. Nephrol. 2012, 27, 675–679. [Google Scholar] [CrossRef]
- Stefanou, C.; Pieri, M.; Savva, I.; Georgiou, G.; Pierides, A.; Voskarides, K.; Deltas, C. Co-Inheritance of Functional Podocin Variants with Heterozygous Collagen IV Mutations Predisposes to Renal Failure. Nephron 2015, 130, 200–212. [Google Scholar] [CrossRef]
- Frese, J.; Kettwig, M.; Zappel, H.; Hofer, J.; Gröne, H.-J.; Nagel, M.; Sunder-Plassmann, G.; Kain, R.; Neuweiler, J.; Gross, O. Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis. Int. J. Mol. Sci. 2019, 20, 519. [Google Scholar] [CrossRef]
- Daga, S.; Fallerini, C.; Furini, S.; Pecoraro, C.; Scolari, F.; Ariani, F.; Bruttini, M.; Mencarelli, M.A.; Mari, F.; Renieri, A.; et al. Non-collagen genes role in digenic Alport syndrome. BMC Nephrol. 2019, 20, 70. [Google Scholar] [CrossRef]
- Voskarides, K.; Stefanou, C.; Pieri, M.; Demosthenous, P.; Felekkis, K.; Arsali, M.; Athanasiou, Y.; Xydakis, D.; Stylianou, K.; Daphnis, E.; et al. A functional variant in NEPH3 gene confers high risk of renal failure in primary hematuric glomerulopathies. Evidence for predisposition to microalbuminuria in the general population. PLoS ONE 2017, 12, e0174274. [Google Scholar] [CrossRef]
- Voskarides, K.; Papagregoriou, G.; Hadjipanagi, D.; Petrou, I.; Savva, I.; Elia, A.; Athanasiou, Y.; Pastelli, A.; Kkolou, M.; Hadjigavriel, M.; et al. COL4A5 and LAMA5 variants co-inherited in familial hematuria: Digenic inheritance or genetic modifier effect? BMC Nephrol. 2018, 19, 114. [Google Scholar] [CrossRef]
- Strasser, K.; Hoefele, J.; Bergmann, C.; Büscher, A.K.; Büscher, R.; Hoyer, P.F.; Weber, S. COL4A5-associated X-linked Alport syndrome in a female patient with early inner ear deafness due to a mutation in MYH9. Nephrol. Dial. Transplant. 2012, 27, 4236–4240. [Google Scholar] [CrossRef]
- Papagregoriou, G.; Erguler, K.; Dweep, H.; Voskarides, K.; Koupepidou, P.; Athanasiou, Y.; Pierides, A.; Gretz, N.; Felekkis, K.N.; Deltas, C. A miR-1207-5p Binding Site Polymorphism Abolishes Regulation of HBEGF and Is Associated with Disease Severity in CFHR5 Nephropathy. PLoS ONE 2012, 7, e31021. [Google Scholar] [CrossRef] [PubMed]
- Lennon, R.; Stuart, H.M.; Bierzynska, A.; Randles, M.J.; Kerr, B.; Hillman, K.A.; Batra, G.; Campbell, J.; Storey, H.; Flinter, F.A.; et al. Coinheritance of COL4A5 and MYO1E mutations accentuate the severity of kidney disease. Pediatr. Nephrol. 2015, 30, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Voskarides, K.; Demosthenous, P.; Papazachariou, L.; Arsali, M.; Athanasiou, Y.; Zavros, M.; Stylianou, K.; Xydakis, D.; Daphnis, E.; Gale, D.P.; et al. Epistatic role of the MYH9/APOL1 region on familial hematuria genes. PLoS ONE 2013, 8, e57925. [Google Scholar] [CrossRef] [PubMed]
- Ning, L.; Suleiman, H.Y.; Miner, J.H. Synaptopodin deficiency exacerbates kidney disease in a mouse model of Alport syndrome. Am. J. Physiol. Physiol. 2021, 321, F12–F25. [Google Scholar] [CrossRef]
- Takemon, Y.; Wright, V.; Davenport, B.; Gatti, D.M.; Sheehan, S.M.; Letson, K.; Savage, H.S.; Lennon, R.; Korstanje, R. Uncovering Modifier Genes of X-Linked Alport Syndrome Using a Novel Multiparent Mouse Model. J. Am. Soc. Nephrol. 2021, 32, 1961–1973. [Google Scholar] [CrossRef]
- Gross, O.; Girgert, R.; Beirowski, B.; Kretzler, M.; Kang, H.G.; Kruegel, J.; Miosge, N.; Busse, A.C.; Segerer, S.; Vogel, W.F.; et al. Loss of collagen-receptor DDR1 delays renal fibrosis in hereditary type IV collagen disease. Matrix Biol. 2010, 29, 346–356. [Google Scholar] [CrossRef]
- Jarad, G.; Knutsen, R.H.; Mecham, R.P.; Miner, J.H. Albumin contributes to kidney disease progression in Alport syndrome. Am. J. Physiol. Physiol. 2016, 311, F120–F130. [Google Scholar] [CrossRef]
- Cosgrove, D.; Rodgers, K.; Meehan, D.; Miller, C.; Bovard, K.; Gilroy, A.; Gardner, H.; Kotelianski, V.; Gotwals, P.; Amatucci, A.; et al. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am. J. Pathol. 2000, 157, 1649–1659. [Google Scholar] [CrossRef]
- Rubel, D.; Frese, J.; Martin, M.; Leibnitz, A.; Girgert, R.; Miosge, N.; Eckes, B.; Müller, G.-A.; Gross, O. Collagen receptors integrin alpha2beta1 and discoidin domain receptor 1 regulate maturation of the glomerular basement membrane and loss of integrin alpha2beta1 delays kidney fibrosis in COL4A3 knockout mice. Matrix Biol. 2014, 34, 13–21. [Google Scholar] [CrossRef]
- Hahm, K.; Lukashev, M.E.; Luo, Y.; Yang, W.J.; Dolinski, B.M.; Weinreb, P.H.; Simon, K.J.; Wang, L.C.; Leone, D.R.; Lobb, R.R.; et al. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am. J. Pathol. 2007, 170, 110–125. [Google Scholar] [CrossRef]
- Funk, S.D.; Bayer, R.H.; Malone, A.F.; McKee, K.K.; Yurchenco, P.D.; Miner, J.H. Pathogenicity of a Human Laminin beta2 Mutation Revealed in Models of Alport Syndrome. J. Am. Soc. Nephrol. 2018, 29, 949–960. [Google Scholar] [CrossRef]
- Andrews, K.L.; Betsuyaku, T.; Rogers, S.; Shipley, J.M.; Senior, R.M.; Miner, J.H. Gelatinase B (MMP-9) Is Not Essential in the Normal Kidney and Does Not Influence Progression of Renal Disease in a Mouse Model of Alport Syndrome. Am. J. Pathol. 2000, 157, 303–311. [Google Scholar] [CrossRef]
- Ding, W.; Yousefi, K.; Goncalves, S.; Goldstein, B.J.; Sabater, A.L.; Kloosterboer, A.; Ritter, P.; Lambert, G.; Mendez, A.J.; Shehadeh, L.A. Osteopontin deficiency ameliorates Alport pathology by preventing tubular metabolic deficits. JCI Insight 2018, 3, e94818. [Google Scholar] [CrossRef]
- Fukuda, R.; Suico, M.A.; Kai, Y.; Omachi, K.; Motomura, K.; Koga, T.; Komohara, Y.; Koyama, K.; Yokota, T.; Taura, M.; et al. Podocyte p53 Limits the Severity of Experimental Alport Syndrome. J. Am. Soc. Nephrol. 2016, 27, 144–157. [Google Scholar] [CrossRef]
- Tanaka, M.; Asada, M.; Higashi, A.Y.; Nakamura, J.; Oguchi, A.; Tomita, M.; Yamada, S.; Asada, N.; Takase, M.; Okuda, T.; et al. Loss of the BMP antagonist USAG-1 ameliorates disease in a mouse model of the progressive hereditary kidney disease Alport syndrome. J. Clin. Investig. 2010, 120, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Boute, N.; Gribouval, O.; Roselli, S.; Benessy, F.; Lee, H.; Fuchshuber, A.; Dahan, K.; Gubler, M.-C.; Niaudet, P.; Antignac, C. Faculty Opinions recommendation of NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat. Genet. 2000, 24, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Roselli, S.; Gribouval, O.; Boute, N.; Sich, M.; Benessy, F.; Attié, T.; Gubler, M.-C.; Antignac, C. Podocin Localizes in the Kidney to the Slit Diaphragm Area. Am. J. Pathol. 2002, 160, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; de Jorge, E.G.; Cook, H.T.; Martinez-Barricarte, R.; Hadjisavvas, A.; McLean, A.G.; Pusey, C.D.; Pierides, A.; Kyriacou, K.; Athanasiou, Y.; et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010, 376, 794–801. [Google Scholar] [CrossRef]
- Deltas, C.; Gale, D.; Cook, T.; Voskarides, K.; Athanasiou, Y.; Pierides, A. C3 glomerulonephritis/CFHR5 nephropathy is an endemic disease in Cyprus: Clinical and molecular findings in 21 families. Complement Ther. 2013, 735, 189–196. [Google Scholar] [CrossRef]
- Jones, L.K.; Lam, R.; McKee, K.K.; Aleksandrova, M.; Dowling, J.; Alexander, S.I.; Mallawaarachchi, A.; Cottle, D.L.; Short, K.M.; Pais, L.; et al. A mutation affecting laminin alpha 5 polymerisation gives rise to a syndromic developmental disorder. Development 2020, 147, dev189183. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Nagano, C.; Sekiguchi, K.; Tashiro, A.; Sugawara, N.; Sakaguchi, H.; Umeda, C.; Aoto, Y.; Ishiko, S.; Rossanti, R.; et al. Clear Evidence of LAMA5 Gene Biallelic Truncating Variants Causing Infantile Nephrotic Syndrome. Kidney360 2021, 2, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Anesthesia Analg. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Pierides, A.; Voskarides, K.; Athanasiou, Y.; Ioannou, K.; Damianou, L.; Arsali, M.; Zavros, M.; Pierides, M.; Vargemezis, V.; Patsias, C.; et al. Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/ COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2009, 24, 2721–2729. [Google Scholar] [PubMed]
- Sevillano, A.M.; Gutierrez, E.; Morales, E.; Hernandez, E.; Molina, M.; Gonzalez, E.; Praga, M. Multiple kidney cysts in thin basement membrane disease with proteinuria and kidney function impairment. Clin. Kidney J. 2014, 7, 251–256. [Google Scholar] [CrossRef]
- Shannon, M.B.; Patton, B.L.; Harvey, S.J.; Miner, J.H. A Hypomorphic Mutation in the Mouse Laminin α5 Gene Causes Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2006, 17, 1913–1922. [Google Scholar] [CrossRef]
- Pecci, A.; Ma, X.; Savoia, A.; Adelstein, R.S. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene 2018, 664, 152–167. [Google Scholar] [CrossRef]
- Dai, S.; Wang, Z.; Pan, X.; Wang, W.; Chen, X.; Ren, H.; Hao, C.; Bin Han, B.; Chen, N. Functional analysis of promoter mutations in the ACTN4 and SYNPO genes in focal segmental glomerulosclerosis. Nephrol. Dial. Transplant. 2010, 25, 824–835. [Google Scholar] [CrossRef]
- Ning, L.; Suleiman, H.Y.; Miner, J.H. Synaptopodin Is Dispensable for Normal Podocyte Homeostasis but Is Protective in the Context of Acute Podocyte Injury. J. Am. Soc. Nephrol. 2020, 31, 2815–2832. [Google Scholar] [CrossRef]
- Richter, H.; Satz, A.L.; Bedoucha, M.; Buettelmann, B.; Petersen, A.C.; Harmeier, A.; Hermosilla, R.; Hochstrasser, R.; Burger, D.; Gsell, B.; et al. DNA-Encoded Library-Derived DDR1 Inhibitor Prevents Fibrosis and Renal Function Loss in a Genetic Mouse Model of Alport Syndrome. ACS Chem. Biol. 2019, 14, 37–49. [Google Scholar] [CrossRef]
- Sannomiya, Y.; Kaseda, S.; Kamura, M.; Yamamoto, H.; Yamada, H.; Inamoto, M.; Kuwazuru, J.; Niino, S.; Shuto, T.; Suico, M.A.; et al. The role of discoidin domain receptor 2 in the renal dysfunction of alport syndrome mouse model. Ren. Fail. 2021, 43, 510–519. [Google Scholar] [CrossRef]
- Falcone, S.; Wisby, L.; Nicol, T.; Blease, A.; Starbuck, B.; Parker, A.; Sanderson, J.; Brown, S.D.M.; Scudamore, C.L.; Pusey, C.D.; et al. Modification of an aggressive model of Alport Syndrome reveals early differences in disease pathogenesis due to genetic background. Sci. Rep. 2019, 9, 20398. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Wang, X.P.; Miner, J.H.; Morello, R.; Sado, Y.; Abrahamson, D.R.; Borza, D.B. Loss of alpha3/alpha4(IV) collagen from the glomerular basement membrane induces a strain-dependent isoform switch to alpha5alpha6(IV) collagen associated with longer renal survival in Col4a3−/− Alport mice. J. Am. Soc. Nephrol. 2006, 17, 1962–1969. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Mudd, J.L.; Li, C.; Miner, J.H. Quantitative Trait Loci Influence Renal Disease Progression in a Mouse Model of Alport Syndrome. Am. J. Pathol. 2002, 160, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, S.; Deltas, C. A Comparative Presentation of Mouse Models That Recapitulate Most Features of Alport Syndrome. Genes 2022, 13, 1893. [Google Scholar] [CrossRef]
- Deltas, C.; Pierides, A.; Voskarides, K. Molecular genetics of familial hematuric diseases. Nephrol. Dial. Transplant. 2013, 28, 2946–2960. [Google Scholar] [CrossRef]
- Voskarides, K.; Pierides, A.; Deltas, C. COL4A3/COL4A4 mutations link familial hematuria and focal segmental glomerulosclerosis. glomerular epithelium destruction via basement membrane thinning? Connect. Tissue Res. 2008, 49, 283–288. [Google Scholar] [CrossRef]
- Wickman, L.; Hodgin, J.B.; Wang, S.Q.; Afshinnia, F.; Kershaw, D.; Wiggins, R.C. Podocyte Depletion in Thin GBM and Alport Syndrome. PLoS ONE 2016, 11, e0155255. [Google Scholar] [CrossRef]
- Ding, F.; Wickman, L.; Wang, S.Q.; Zhang, Y.; Wang, F.; Afshinnia, F.; Hodgin, J.; Ding, J.; Wiggins, R.C. Accelerated podocyte detachment and progressive podocyte loss from glomeruli with age in Alport Syndrome. Kidney Int. 2017, 92, 1515–1525. [Google Scholar] [CrossRef]
- Nishizono, R.; Kikuchi, M.; Wang, S.Q.; Chowdhury, M.; Nair, V.; Hartman, J.; Fukuda, A.; Wickman, L.; Hodgin, J.B.; Bitzer, M.; et al. FSGS as an Adaptive Response to Growth-Induced Podocyte Stress. J. Am. Soc. Nephrol. 2017, 28, 2931–2945. [Google Scholar] [CrossRef]
- Mencarelli, M.A.; Heidet, L.; Storey, H.; van Geel, M.; Knebelmann, B.; Fallerini, C.; Miglietti, N.; Antonucci, M.F.; Cetta, F.; Sayer, J.A.; et al. Faculty Opinions recommendation of Evidence of digenic inheritance in Alport syndrome. J. Med. Genet. 2015, 52, 163–174. [Google Scholar] [CrossRef]
- Schäffer, A.A. Digenic inheritance in medical genetics. J. Med. Genet. 2013, 50, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Deltas, C. Thin basement membrane nephropathy: Is there genetic predisposition to more severe disease? Pediatr. Nephrol. 2009, 24, 877–879. [Google Scholar] [CrossRef] [PubMed]
- Gallati, S. Disease-modifying genes and monogenic disorders: Experience in cystic fibrosis. Appl. Clin. Genet. 2014, 7, 133–146. [Google Scholar] [CrossRef] [PubMed]


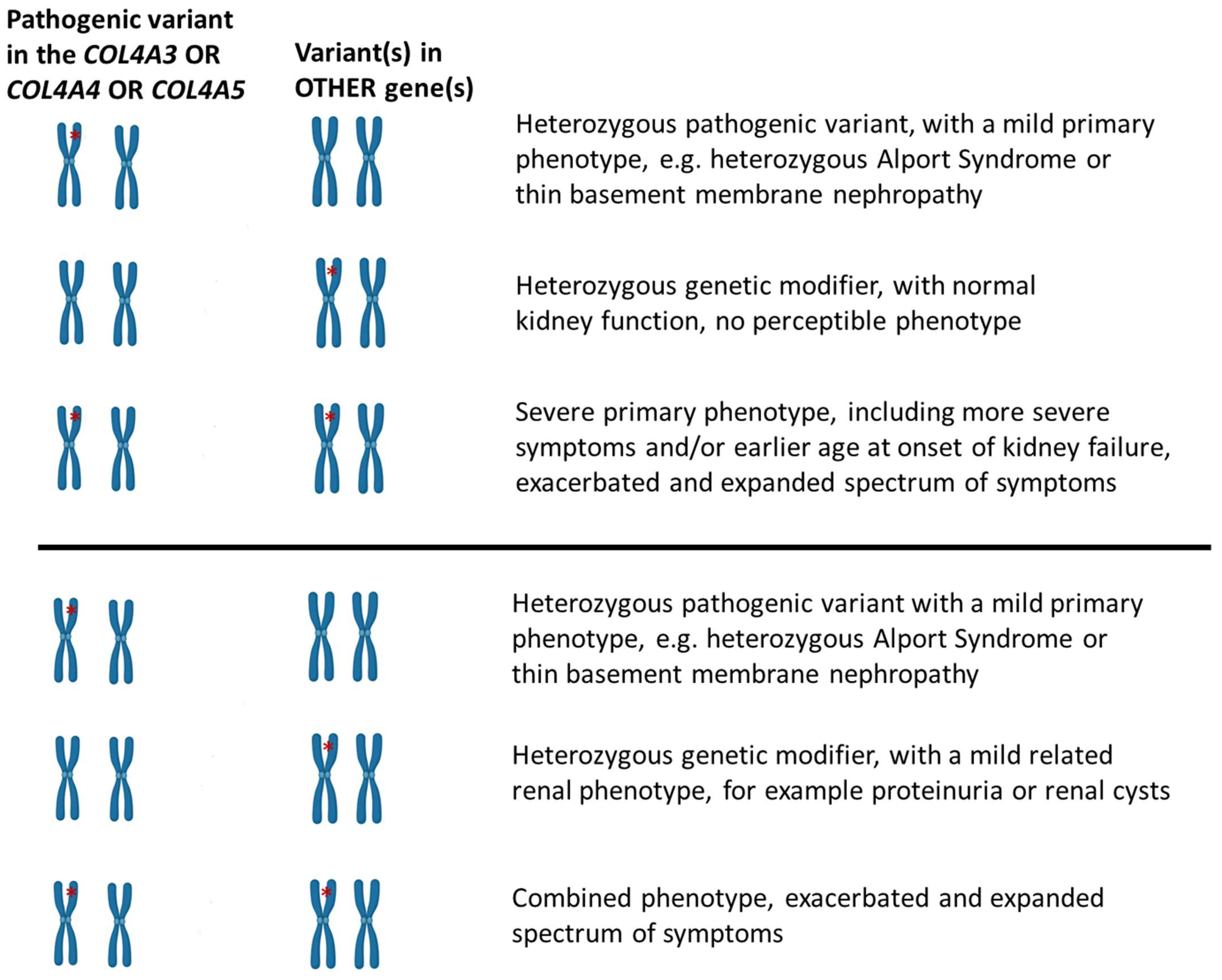{kind=link}
| Primary Gene Defect | Co-Inherited Likely Modifier Gene Variant(s) | MAF (gnomAD) | MAF (CYPROME) a | Comments | Reference |
|---|---|---|---|---|---|
| COL4A3 | NPHS2:p.Arg229Gln (rs61747728) | 0.03025 | 0.0166 | Tested in a cohort of patients carrying several pathogenic variants and in functional studies in cell culture | [23,24,25,26,27] * |
| COL4A3 | NPHS2:p.Glu237Gln (rs146906190) | 0.0007370 | 0.0055 | Tested in families and functional studies in cell culture | [25] * |
| COL4A5 | NPHS1:p.Arg408Gln (rs33950747) | 0.04859 | 0.0151 | Tested in families | [26] |
| COL4A4:c.1109G>A p.Gly370Glu (rs779604374) | LAMA5:c.5149C>T, p.His1717Tyr (rs875379) | 0.07186 | 0.1039 | Tested in a family that the authors diagnosed with ADAS | [27] |
| COL4A5:c.3319G>A p.Gly1107Arg (rs104886225) | LAMA5:c.9388C>T, p.His3130Tyr (rs201154340) | 0.0001200 | 0.0005 | Tested in a family with XLAS | |
| COL4A4:c.4444del p.Leu1482Trpfs*70 (NM_000092.5) | LAMA5:c.2321C>T, p.Thr774Ile (rs145721906), co-inherited with NPHS2:c.686G>A, p.Arg229Gln (rs61747728) | 0.001418 0.03025 | 0.004 0.016 | Tested in a family that the authors diagnosed with ADAS | |
| COL4A3 and COL4A4 | NEPH3:p.Val353Met (rs35423326) | 0.02863 | 0.0306 | Tested in a large cohort of patients carrying several pathogenic variants and in cohorts of the general population. Also, tested with extensive cell culture assays. | [28] * |
| COL4A5 | LAMA5:p.Pro1243Leu (rs756101090) | 0.00004538 | Not detected | Tested in a family segregating XLAS | [29] |
| COL4A5:c.2605G>A, p.Gly869Arg (rs104886189) | MYH9:c.4952T>G; p.Met1651Arg (NM_002473.6) | Not detected? | Not detected | Found in one female patient | [30] |
| CFHR5 | HBEGF (Heparin Binding Epidermal Growth Factor) SNP C1936T in the 3′-UTR of HBEGF, 2nd position of seed region of miRNA hsa-miR-1207-5p c.*1006C>T (rs13385) The asterik denotes that the position is 1006 nucleotides past the translation STOP codon | 0.1988 | Information Not available | Tested in patients with CFHR5 nephropathy/C3 glomerulopathy, due to a founder CFHR5 exon 2-3 duplication. Also tested in cell culture functional studies. | [31] * |
| COL4A5:c.2858G>T; p.(Gly953Val) (rs78972735) AND in cis c.3097G>C; p.(Gly1033Arg) (NM_033380.3) | MYO1E:c.352A>G; p.(Lys118Glu) (NM_004998.4) Homozygous MYO1E:c.2627C>G; p.(Thr876Arg) (rs147596471) Homozygous | Not detected? | Not detected 0.0010 | Found in a brother and sister in a family segregating XLAS | [32] |
| COL4A and CFHR5 | MYH9:c.-19-5801G>T (rs11089788) | 0.4604 | Information Not available | Tested in a cohort of patients carrying several COL4A variants or the CFHR5 exon 2-3 duplication | [33] |
| Potential Modifier Gene (Knocked-Out or Heterozygous Mutant) | Impact on Kidney Disease Progression | Reference |
|---|---|---|
| SYNPO (Synaptopodin) | Accelerated kidney disease progression due to podocyte cytoskeletal defects and easier detachment from the GBM | [34] |
| Fmn1 (Formin 1) | Fmn1 heterozygosity reduced albuminuria, podocyte foot process efffacement and podocyte protrusions into the GBM | [35] |
| Ddr1 (Discoidin Domain Receptor-1) | Slowed kidney disease progression; extended lifespan | [36] |
| Alb (Albumin) | Reduced glomerular and tubulointerstitial pathology and increased lifespan by 64% on the C57BL/6J background | [37] |
| Itga1 (Integrin α1) | Delayed onset of proteinuria and preservation of podocyte foot process architecture. Inhibiting TGF activity or expression synergized with Itga1 knockout to further slow kidney disease progression. | [38] |
| Itga2 (Integrin α2) | Delayed glomerulosclerosis and tubulointerstitial fibrosis, reduced proteinuria, improved GBM architecture, increased lifespan by 20%. | [39] |
| Itgb6 (integrin β6) | Decreased renal fibrosis and smooth muscle actin+ cells, consistent with reduced TGFb expression | [40] |
| Lamb2 (laminin β2) null and p.S83R mutations | Worsened kidney disease progression | [41] |
| Mmp9 (Matrix metalloproteinase 9) | No discernible impact on kidney disease progression or GBM architecture | [42] |
| Spp1 (Osteopontin/OPN) | Reduced proteinuria, high blood pressure and GBM thickening and increaased lifespan | [43] |
| Trp53 (p53); podocyte-specific mutation | Increased foot process effacement and renal dysfunction | [44] |
| Usag1 (Uterine-sensitization-associated gene 1) | Attenuation of disease progression, normalization of GBM ultrastructure, preservation of renal function and extension of life span | [45] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deltas, C.; Papagregoriou, G.; Louka, S.F.; Malatras, A.; Flinter, F.; Gale, D.P.; Gear, S.; Gross, O.; Hoefele, J.; Lennon, R.; et al. Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice. Genes 2023, 14, 1686. https://doi.org/10.3390/genes14091686
Deltas C, Papagregoriou G, Louka SF, Malatras A, Flinter F, Gale DP, Gear S, Gross O, Hoefele J, Lennon R, et al. Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice. Genes. 2023; 14(9):1686. https://doi.org/10.3390/genes14091686
Chicago/Turabian StyleDeltas, Constantinos, Gregory Papagregoriou, Stavroula F. Louka, Apostolos Malatras, Frances Flinter, Daniel P. Gale, Susie Gear, Oliver Gross, Julia Hoefele, Rachel Lennon, and et al. 2023. "Genetic Modifiers of Mendelian Monogenic Collagen IV Nephropathies in Humans and Mice" Genes 14, no. 9: 1686. https://doi.org/10.3390/genes14091686