
The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder
by
 , and
, and
Camilla Ferrari
1,*,
Elena Capacci
1,
Silvia Bagnoli
1,
Assunta Ingannato
1,
Sandro Sorbi
1,2 and
Benedetta Nacmias
1,2 1
Department of Neuroscience, Psychology, Drug Research and Child Health (NEUROFARBA), University of Florence, 50139 Florence, Italy
2
IRCCS Fondazione Don Carlo Gnocchi, 50143 Florence, Italy
*
Author to whom correspondence should be addressed.
Genes 2023, 14(9), 1681; https://doi.org/10.3390/genes14091681
Submission received: 4 August 2023
/
Revised: 21 August 2023
/
Accepted: 23 August 2023
/
Published: 25 August 2023
(This article belongs to the Section Human Genomics and Genetic Diseases)
Abstract
:Background and objectives: Huntington’s disease (HD) is characterized by motor, cognitive and psychiatric manifestations and caused by an expansion of CAG repeats over 35 triplets on the huntingtin (HTT) gene. However, expansions in the range 27–35 repeats (intermediate allele) can be associated with pathological phenotypes. The onset of HD is conventionally defined by the onset of motor symptoms, but psychiatric disturbances can precede the motor phase by up to twenty years. The aims of the present study are to identify HD patients in the pre-motor phase of the disease among patients diagnosed with bipolar disorders and evaluate any differences between bipolar patients carrying the normal HTT allele and patients with the expanded HTT gene. Methods: We assessed the HTT genotype in an Italian cohort of 69 patients who were affected by either type 1 or type 2 bipolar disorder. Results: No patient was found to be a carrier of the pathological HTT allele, but 10% of bipolar subjects carried an intermediate allele. Carriers of the intermediate allele were older at the onset of psychiatric symptoms than non-carriers. Conclusion: The pathological HTT gene was not associated with bipolar disorder, while we found a higher frequency of the intermediate allele among the bipolar population with respect to healthy controls. The identification of this subset of bipolar subjects has implications for the clinical management of patients and their family members and promotes further investigation into possible pathological mechanisms common to both HD and bipolar disorder.
1. Introduction
Expansion of CAG triplets in the huntingtin gene (HTT) on chromosome 4 is responsible for Huntington’s disease (HD). The presence of 40 or more CAG triplets is invariably associated with manifest disease, while expansions in the range 36–39 repeats are considered to confer a reduced disease penetrance [1,2]. HD is characterized by motor, cognitive and psychiatric disorders [1]. Conventionally, HD onset is defined as the age of the onset of motor signs; however, psychiatric disturbances, especially mood disturbances, can often occur up to twenty years before the manifest motor phase [3,4]. Observational studies report psychiatric symptoms to be the first manifestation of HD in about 20% of cases [5].
HD is transmitted in an autosomal dominant manner, but new mutations can be generated in the offspring via the elongation of unstable alleles falling in the range 27–35 repeats, which are defined as intermediate alleles (IA) [6]. The percentage of IA has been variably estimated to be between 0.45 and 8.7% [7,8] in the healthy population. The reported frequency of IA in European-based cohorts is about 6% [7]. Although the HTT gene with less than 36 CAG repeats is considered to be normal, a growing number of studies have reported that subjects carrying IA are at higher risk of behavioral problems and can even develop the clinical phenotype of HD [9,10,11,12,13]. Bipolar disorder is a multifactorial disease with a high heritability rate, which is estimated at 60–90% in twin studies. However, the genetic architecture of the disorder is still far from being elucidated [14,15]. Hypomanic and manic episodes have been reported in up to 10% of HD patients [16,17], and the pathogenesis of HD and bipolar disorder is connected to the same brain structures [18,19,20,21,22,23]. This finding could suggest the presence of common mechanisms underlying the development of HD and bipolar disease. Recent studies have demonstrated that the length of CAG repeats can influence the risk of the development of depressive symptoms [24], and major depression could be a pre-motor phenotype of HD [25]. Subjects with expanded HTT alleles and pre-motor psychiatric symptoms could be misdiagnosed as having primary psychiatric disease, while their early identification would imply different clinical management, such as specific clinical follow-up, tailored treatment [26] and the genetic counseling of family members [6,11,13]. In the present study, we analyzed the HTT gene in a psychiatric cohort of patients affected by bipolar disorder in order to identify pre-motor HD patients and evaluate whether the length of CAG repeats may influence or contribute to the development of bipolar disorder.
2. Materials and Methods
2.1. Population
The blood samples of 69 unrelated patients affected by bipolar disorder type I and type II were genotyped to determine the presence of the HTT gene. The diagnosis of bipolar disorder was made according to the DSM IV criteria [27].
Demographic–clinical data included the following information: age at the time of withdrawal, age at disease onset, gender, first symptom at onset, educational level and family history of psychiatric disorders. The genetic results were compared to a cohort of 104 healthy controls that we recently described, i.e., 45 males (43.3%) and 59 females (56.7%), with the mean age being 64.4 (±8.5) years old [28].
2.2. Genetic Analysis
Genomic DNA was isolated from peripheral blood samples using a QIAamp DNA Blood Mini QIAcube kit (cat. No 51126 QIAGEN) and quality-checked via a QIAxpert spectrophotometer. DNA samples were stored at +4° until use. CAG repeat expansion of the HTT gene was investigated via a polymerase chain reaction (PCR) amplification assay, using the primers 5′-[6-FAM] GACCCTGGAAAAGCTGATGA-3′ and 5′-GGCTGAGGAAGCTGAGGAG-3′. The forward primer was modified using the fluorescent dye 6-carboxyfluorescein (6-FAM) [29].
The PCR was performed using 10 ng of genomic DNA, a 1xTaq MegamùMix (Microzone with Buffer master mix) and 1.5 µL of each primer and water up to a final volume of 15 µL. The PCR was performed via 35 cycles of 30 s denaturation at 94 °C, 30 s of annealing at 60 °C, 30 s of elongation at 72 °C and 2 min of elongation at 74 °C. Every PCR included a negative control without genomic DNA and two positive control samples with predetermined 19/20 and 23/27 HTT CAG repeats. The PCR products were run via a capillary electrophoresis using the SeqStudio Genetic Analyzer (ThermoFisher, Monza, Italy) and analyzed using the GeneMapper software (v.4.0, Applied Biosystems, Waltham, MA, USA). A set of known length HTT CAG alleles was used as the size standard. A CAG trinucleotide expansion under 27 repetitions was considered to be normal allele, IAs with repeats were in the range 27–35 and pathologic alleles had expansion sizes >35 repeats.
2.3. Statistical Analysis
The demographic–clinical features of bipolar patients were described via the mean and standard deviation in case of continuous variables and via percentages in the case of categorical variables. Due to the identification of IA, we performed a comparison between bipolar patients carrying IA (IA carriers) and bipolar patients carrying the normal alleles (non-IA carriers). The comparisons between the demographic–clinical features were performed through non-parametric analysis: Fisher’s exact test was used for categorical variables and Mann–Whitney test was used for continuous variables. The study population was then compared to the healthy control group in terms of the HTT CAG distribution and IA frequency. The Mann–Whitney test and Fisher’s exact test were used if appropriate. Statistical analyses were performed using IBM SPSS Statistics 20.0 (IBM Corp., New York, NY, USA).
3. Results
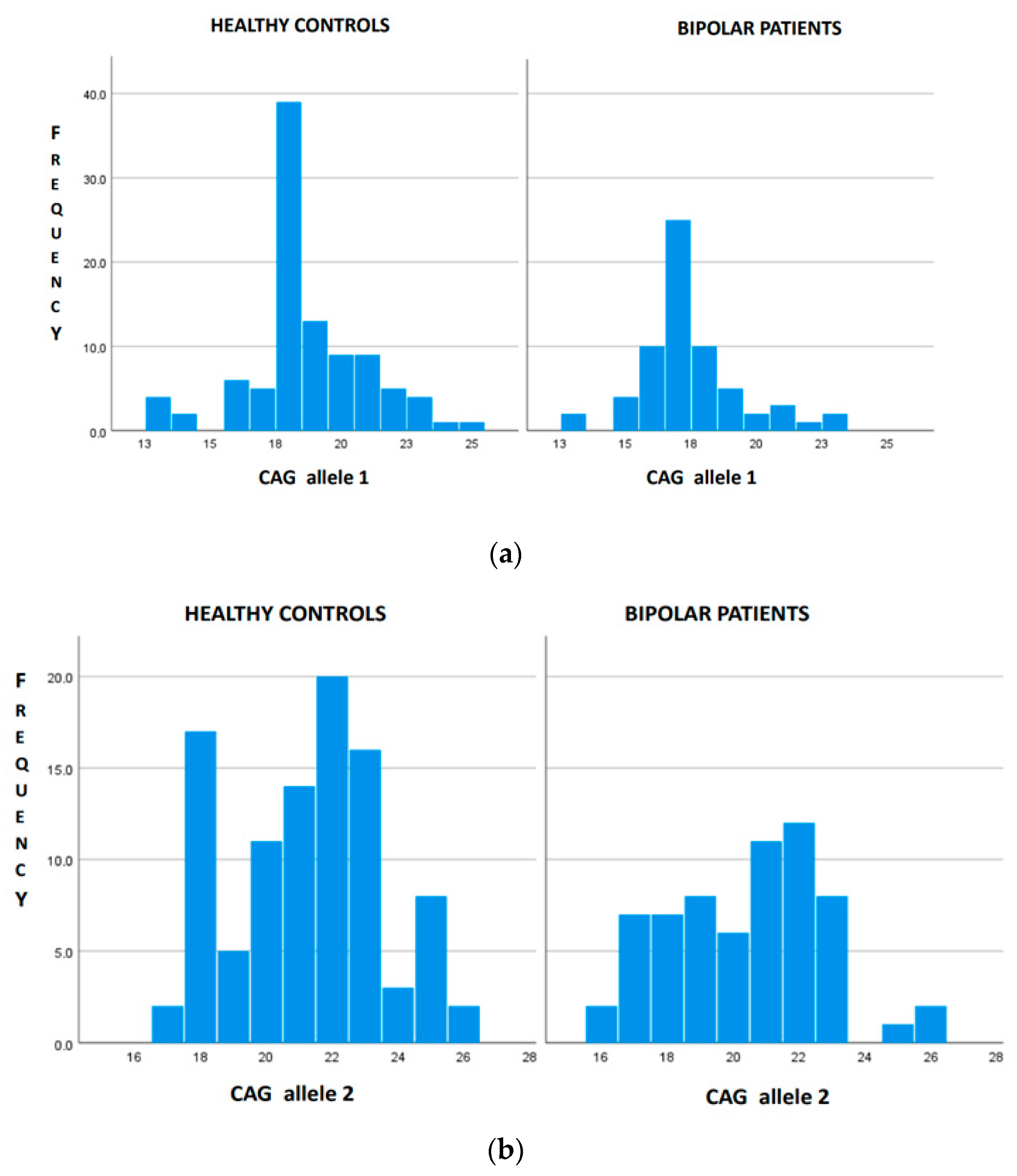
There were 69 bipolar patients with a mean age at disease onset of 34 years old and a tendency to be female (60.9%) (Table 1). The onset of bipolar disorder occurred in 49.2% of cases with depressive symptoms, in 15.9% of cases with maniacal symptoms and in the remaining cases with a mixture of both symptoms. More than 60% of patients had type 1 bipolar disorder. A family history of psychiatric disorders was present in 43.5% of cases. The mean number of CAG repeats in the HTT gene was 17.7 for the shorter allele and 21.03 for the longer allele (Table 1). No patient was found to be a carrier of a pathological allele. We identified seven bipolar patients carrying IA (IA carriers). The mean length of IA was 28.6 (ranging 27–33). The comparison between bipolar patients IA carriers and non-IA carriers is reported in Table 2. IA carriers were older at disease onset than non-IA carriers (Table 2). The frequency of IA was statistically significant higher in bipolar patients, i.e., 10.14% (7 out of 69), than in the healthy control population, i.e., 5.8% (6 out of 104) (p < 0.001), while the range of normal CAG repeats was 13–26 in both groups. The distribution of the HTT CAG repeat length among the two cohorts is shown in Figure 1. The distribution of the normal-range alleles differs in the two cohorts, as healthy subjects have higher numbers of triplets in both alleles (Figure 1).
4. Discussion
The study of the HTT gene in a cohort of 69 patients with bipolar disorder did not identify any subjects in a pre-motor stage of HD; in fact, no patient was a carrier of the pathological allele. The IA was detected in 10.14% of cases. IA carriers were older than non-IA carriers at the disease’s onset. Although psychiatric symptoms may often precede the motor phase of HD, only three previous studies evaluated the frequency of the pathological expansion in the HTT gene in psychiatric cohorts [24,25,30]: two studies in patients affected by major depression [24,25] and one study in a Brazilian cohort affected by bipolar disorder [30]. Major depression was frequent in the pre-motor phase of HD [25], while Ramos et al. [30], in line with our results, did not identify any association between the pathological HTT allele and bipolar disorder. The frequency of IA carriers in our psychiatric cohort was almost the double that detected in the local reference population. Data from the literature have already shown a higher risk of behavioral disturbances in subjects carrying IA [10], such as apathy, obsessive disorder, anxiety, depression and suicidal ideation. However, the mechanism through which the IA of HTT gene is associated with psychiatric manifestations remains unclear [10]: one hypothesis states that the association occurs through mechanisms unrelated to those leading to HD pathology, such as interaction with other genes [10]. In that case, the difference in the genetic structure between ethnic groups could be explained based on the lack of association between IA and bipolar disorder in the Brazilian population [30,31]. The analysis of the distribution of the HTT normal alleles revealed a statistically significant difference between bipolar patients and controls, with shorter normal alleles among patients. These data support the hypothesis of a non-linear association between the CAG repeat length and the risk of bipolar disorder, which means that both the normal alleles in the shorter range and the IA could increase the risk of developing bipolar disorder. This non-linear association between the HTT CAG length and the risk of developing major depression was previously described [24], as it was between the HTT size and intelligence in the general population [32]. Therefore, it seems that the increasing size of the non-pathological HTT gene confers an advantage up to the values that determine pathology. In fact, the increasing number of HTT triplets is part of the evolutionary process [33,34,35], and the variation in the number of repeats across the normal range influences the brain’s structure in healthy subjects [36]: longer non-pathological alleles have been found to be associated with an increasing volume of basal ganglia [36]. Studies of HD patients [37,38,39] demonstrated that the increasing sizes of normal alleles could be protective by mitigating the effects of the pathological allele and delaying disease onset. Interestingly, in the present cohort, we found that bipolar patients carrying IA were older at disease onset than non-IA carriers. Basal ganglia are also involved in the pathophysiology of bipolar disorder [20,40,41]; thus, IA, via its effect on basal ganglia [36], could have a protective role and delay the onset of bipolar symptoms.
Due to the multiplicity of functions of the HTT gene, the neuroprotective effect of IA could also depend on other mechanisms: the regulation of genes transcription, the modulation of synaptic transmission and the anti-apoptotic and anti-oxidant functions [23,34].
The number of HTT triplets may also modulate the individuals’ predisposition to the effects of other genetic and environmental factors.
Another hypothesis could be that when the development of bipolar symptoms is driven by the IA of HTT, the age at onset is later than when it is caused by other genetic factors.
The main limitations of the present study are the small sample sizes used, the mono-centric setting and the lack of information about the genetic status of the patients’ family members. Thus, the generalizability of the results is questionable, and the interpretation of the findings, while intriguing, can only be speculative. The suggestions that the IA of HTT plays a role in the development of bipolar disorder or is associated with a peculiar new non-motor phenotype in the spectrum of HD disorder are both fascinating hypotheses that need to be clarified through a larger study of bipolar patients and their relatives. Regardless, beyond the interpretation of the role played by the HTT gene in the development of bipolar symptoms, the identification of IA carriers among this Italian cohort of bipolar patients has important implications for both patients and their families: patients can be at risk of developing a complete HD phenotype [11,13], and family members should be made aware of their increased risk of developing behavioral disturbances [10] or inheriting a pathological expanded HTT gene [6].
5. Conclusions
To the best of our knowledge, there is only one other study that evaluated the HTT genotype among bipolar patients [26], and similar to our study, bipolar disorder was not associated with the pathological HTT allele. In our cohort, IA was associated with bipolar disorder, albeit with a delayed age at onset. The findings of the present brief report are intended to be proof of concept that justify further investigation into the role played by the IA of the HTT gene in the onset of bipolar disorder.
Author Contributions
Conceptualization, C.F.; Formal analysis, S.B. and A.I.; Investigation, E.C.; Writing—original draft, C.F.; Writing—review and editing, S.S. and B.N. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Fondi Ricerca UNIFI 2023 (C.F., S.S., B.N.).
Institutional Review Board Statement
The study has been conducted in accordance with the ethical standards of the institutional research committee and the Helsinki Declaration, and all patients signed an informed consent form.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data of this study are available from the corresponding author, C.F., upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Kay, C.; Hayden, M.R.; Leavitt, B.R. Epidemiology of Huntington disease. Handb. Clin. Neurol. 2017, 144, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Reilmann, R.; Cardoso, F.; McCusker, E.A.; Testa, C.M.; Stout, J.C.; Leavitt, B.R.; Pei, Z.; Landwehrmeyer, B.; Martinez, A.; et al. Movement Disorder Society Task Force Viewpoint: Huntington’s Disease Diagnostic Categories. Mov. Disord. Clin. Pract. 2019, 6, 541–546. [Google Scholar] [CrossRef]
- Julien, C.L.; Thompson, J.C.; Wild, S.; Yardumian, P.; Snowden, J.S.; Turner, G.; Craufurd, D. Psychiatric disorders in preclinical Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 939–943. [Google Scholar] [CrossRef] [PubMed]
- Orth, M.; Handley, O.J.; Schwenke, C.; Dunnett, S.B.; Craufurd, D.; Ho, A.K.; Wild, E.; Tabrizi, S.J.; Landwehrmeyer, G.B. The investigators of the European Huntington’s Disease Network Observing Huntington’s Disease: The European Huntington’s Disease Network’s REGISTRY. PLoS Curr. 2010, 2, RRN1184. [Google Scholar] [CrossRef]
- Hammer, M.B.; Singleton, A.B. Common Premutations in the General Population. JAMA Neurol. 2019, 76, 639–640. [Google Scholar] [CrossRef] [PubMed]
- Apolinário, T.; Paiva, C.; Agostinho, L. REVIEW-ARTICLE Intermediate alleles of Huntington’s disease HTT gene in different populations worldwide: A systematic review. Genet. Mol. Res. 2017, 16, gmr16029648. [Google Scholar] [CrossRef]
- Sundblom, J.; Niemelä, V.; Ghazarian, M.; Strand, A.S.; Bergdahl, I.A.; Jansson, J.H.; Söderberg, S.; Stattin, E.L. High frequency of intermediary alleles in the HTT gene in Northern Sweden—The Swedish Huntingtin Alleles and Phenotype (SHAPE) study. Sci. Rep. 2020, 10, 9853. [Google Scholar] [CrossRef]
- Oosterloo, M.; Van Belzen, M.J.; Bijlsma, E.K.; Roos, R.A. Is There Convincing Evidence that Intermediate Repeats in the HTT Gene Cause Huntington’s Disease? J. Huntingt. Dis. 2015, 4, 141–148. [Google Scholar] [CrossRef]
- Killoran, A.; Biglan, K.M.; Jankovic, J.; Eberly, S.; Kayson, E.; Oakes, D.; Young, A.B.; Shoulson, I. Characterization of the Huntington intermediate CAG repeat expansion phenotype in PHAROS. Neurology 2013, 80, 2022–2027. [Google Scholar] [CrossRef]
- Kenney, C.; Powell, S.; Jankovic, J. Autopsy-proven Huntington’s disease with 29 trinucleotide repeats. Mov. Disord. 2007, 22, 127–130. [Google Scholar] [CrossRef]
- Savitt, D.; Jankovic, J. Clinical phenotype in carriers of intermediate alleles in the huntingtin gene. J. Neurol. Sci. 2019, 402, 57–61. [Google Scholar] [CrossRef]
- Jevtic, S.D.; Provias, J.P. Case report and literature review of Huntington disease with intermediate CAG expansion. BMJ Neurol. Open 2020, 2, e000027. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, K.S.; Coombes, B.J. Genetic contributions to bipolar disorder: Current status and future directions. Psychol. Med. 2021, 51, 2156–2167. [Google Scholar] [CrossRef] [PubMed]
- Gordovez, F.J.A.; McMahon, F.J. The genetics of bipolar disorder. Mol. Psychiatry 2020, 25, 544–559. [Google Scholar] [CrossRef]
- Peyser, C.E.; Folstein, S.E. Huntington’s disease as a model for mood disorders. Clues from neuropathology and neurochemistry. Mol. Chem. Neuropathol. 1990, 12, 99–119. [Google Scholar] [CrossRef] [PubMed]
- Mendez, M.F. Huntington’s disease: Update and review of neuropsychiatric aspects. Int. J. Psychiatry Med. 1994, 24, 189–208. [Google Scholar] [CrossRef]
- Blumenstock, S.; Dudanova, I. Cortical and Striatal Circuits in Huntington’s Disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Garcia-Gorro, C.; Llera, A.; Martinez-Horta, S.; Perez-Perez, J.; Kulisevsky, J.; Rodriguez-Dechicha, N.; Vaquer, I.; Subira, S.; Calopa, M.; Muñoz, E.; et al. Specific patterns of brain alterations underlie distinct clinical profiles in Huntington’s disease. NeuroImage Clin. 2019, 23, 101900. [Google Scholar] [CrossRef]
- Phillips, M.L.; Swartz, H.A.; Sharma, A.; Wolf, D.H.; Ciric, R.; Kable, J.W.; Moore, T.M.; Vandekar, S.N.; Katchmar, N.; Daldal, A.; et al. A Critical Appraisal of Neuroimaging Studies of Bipolar Disorder: Toward a New Conceptualization of Underlying Neural Circuitry and a Road Map for Future Research. Am. J. Psychiatry 2014, 171, 829–843. [Google Scholar] [CrossRef]
- Diaz, A.P.; Bauer, I.E.; Sanches, M.; Soares, J.C. Neuroanatomic and Functional Neuroimaging Findings. Curr. Top. Behav. Neurosci. 2021, 48, 173–196. [Google Scholar] [CrossRef] [PubMed]
- Young, A.H.; Juruena, M.F. The Neurobiology of Bipolar Disorder. Curr. Top. Behav. Neurosci. 2021, 48, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zuccato, C.; Valenza, M.; Cattaneo, E. Molecular Mechanisms and Potential Therapeutical Targets in Huntington’s Disease. Physiol. Rev. 2010, 90, 905–981. [Google Scholar] [CrossRef]
- Gardiner, S.L.; Van Belzen, M.J.; Boogaard, M.W.; van Roon-Mom, W.M.; Rozing, M.P.; Van Hemert, A.M.; Smit, J.H.; Beekman, A.T.; Van Grootheest, G.; Schoevers, R.A.; et al. Huntingtin gene repeat size variations affect risk of lifetime depression. Transl. Psychiatry 2017, 7, 1277. [Google Scholar] [CrossRef] [PubMed]
- Perlis, R.H.; Smoller, J.W.; Mysore, J.; Sun, M.; Gillis, T.; Purcell, S.; Rietschel, M.; Nöthen, M.M.; Witt, S.; Maier, W.; et al. Prevalence of incompletely penetrant Huntington’s disease alleles among individuals with major depressive disorder. Am. J. Psychiatry 2010, 167, 574–579. [Google Scholar] [CrossRef]
- Pang, T.Y.; Du, X.; Zajac, M.S.; Howard, M.L.; Hannan, A.J. Altered serotonin receptor expression is associated with depression-related behavior in the R6/1 transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2009, 18, 753–766. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 4th ed.; American Psychiatric Association: Washington, DC, USA, 2000. [Google Scholar] [CrossRef]
- Ingannato, A.; Bagnoli, S.; Bessi, V.; Ferrari, C.; Mazzeo, S.; Sorbi, S.; Nacmias, B. Intermediate alleles of HTT: A new pathway in longevity. J. Neurol. Sci. 2022, 438, 120274. [Google Scholar] [CrossRef]
- Jama, M.; Millson, A.; Miller, C.E.; Lyon, E. Triplet Repeat Primed PCR Simplifies Testing for Huntington Disease. J. Mol. Diagn. 2013, 15, 255–262. [Google Scholar] [CrossRef]
- Ramos, E.M.; Gillis, T.; Mysore, J.S.; Lee, J.M.; Alonso, I.; Gusella, J.F.; Smoller, J.W.; Sklar, P.; MacDonald, M.E.; Perlis, R.H. Prevalence of Huntington’s disease gene CAG trinucleotide repeat alleles in patients with bipolar disorder. Bipolar. Disord. 2015, 17, 403–408. [Google Scholar] [CrossRef]
- Raskin, S.; Allan, N.; Teive, H.A.; Cardoso, F.; Haddad, M.S.; Levi, G.; Boy, R.; Lerena, J.U.A.N., Jr.; Sotomaior, V.S.; Janzen-Dück, M.Ô.N.I.C.A.; et al. Huntington disease: DNA analysis in Brazilian population. Arq. Neuropsiquiatr. 2000, 58, 977–985. [Google Scholar] [CrossRef]
- Lee, J.K.; Conrad, A.; Epping, E.; Mathews, K.; Magnotta, V.; Dawson, J.D.; Nopoulos, P. Effect of Trinucleotide Repeats in the Huntington’s Gene on Intelligence. EBioMedicine 2018, 3, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Hannan, A.J. Tandem Repeat Polymorphisms: Genetic Plasticity, Neural Diversity and Disease; Landes Bioscience and Springer Scienc: New York, NY, USA, 2012. [Google Scholar]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to Huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Tartari, M.; Gissi, C.; Sardo, V.L.; Zuccato, C.; Picardi, E.; Pesole, G.; Cattaneo, E. Phylogenetic Comparison of Huntingtin Homologues Reveals the Appearance of a Primitive polyQ in Sea Urchin. Mol. Biol. Evol. 2008, 25, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Mühlau, M.; Winkelmann, J.; Rujescu, D.; Giegling, I.; Koutsouleris, N.; Gaser, C.; Arsic, M.; Weindl, A.; Reiser, M.; Meisenzahl, E.M. Variation within the Huntington’s Disease Gene Influences Normal Brain Structure. PLoS ONE 2012, 7, e29809. [Google Scholar] [CrossRef]
- Aziz, N.A.; Jurgens, C.K.; Landwehrmeyer, G.B.; Van Roon-Mom, W.M.C.; Van Ommen, G.J.B.; Stijnen, T.; Roos, R.A.C.; EHDN Registry Study Group. Normal and mutant HTT interact to affect clinical severity and progression in Huntington disease. Neurology 2009, 73, 1280–1285, Erratum in: Neurology 2009, 73, 1608. Erratum in: Neurology 2011, 76, 202. [Google Scholar] [CrossRef]
- Djousse, L.; Knowlton, B.; Hayden, M.; Almqvist, E.W.; Brinkman, R.; Ross, C.; Margolis, R.; Rosenblatt, A.; Durr, A.; Dode, C.; et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am. J. Med. Genet. A 2003, 119, 279–282. [Google Scholar] [CrossRef]
- Volpi, E.; Terenzi, F.; Bagnoli, S.; Latorraca, S.; Nacmias, B.; Sorbi, S.; Piacentini, S.; Ferrari, C. Late-onset Huntington disease: An Italian cohort. J. Clin. Neurosci. 2021, 86, 58–63. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Jia, Y.; Zhong, S.; Sun, Y.; Zhou, Z.; Zhang, Z.; Huang, L. Microstructural Abnormalities of Basal Ganglia and Thalamus in Bipolar and Unipolar Disorders: A Diffusion Kurtosis and Perfusion Imaging Study. Psychiatry Investig. 2017, 14, 471–482. [Google Scholar] [CrossRef]
- Aylward, E.H.; Roberts-Twillie, J.V.; Barta, P.E.; Kumar, A.J.; Harris, G.J.; Geer, M.; Peyser, C.E.; Pearlson, G.D. Basal ganglia volumes and white matter hyperintensities in patients with bipolar disorder. Am. J. Psychiatry 1994, 151, 687–693. [Google Scholar] [CrossRef]
Figure 1.
Distribution of HTT gene length in bipolar patients (n = 62) and healthy controls (n = 98) with the exclusion of IA carriers. (a) The distribution of CAG in allele 1 differs between healthy controls and bipolar patients (p < 0.001). (b) The distribution of CAG in allele 2 differs between healthy controls and bipolar patients (p = 0.028).
Figure 1.
Distribution of HTT gene length in bipolar patients (n = 62) and healthy controls (n = 98) with the exclusion of IA carriers. (a) The distribution of CAG in allele 1 differs between healthy controls and bipolar patients (p < 0.001). (b) The distribution of CAG in allele 2 differs between healthy controls and bipolar patients (p = 0.028).


{kind=link}
Table 1.
Clinical–demographic and genetic features of the studied patients.
| Patients (n = 69) | |
|---|---|
| Age (SD) | 53.91 (10.2) |
| Age at disease onset (SD) | 34.81 (13.4) |
| Sex | |
| Male (%) | 27 (39.1) |
| Female (%) | 42 (60.9) |
| Education in years (SD) | 10.25 (3.1) |
| Symptoms at onset | |
| Maniacal (%) | 11 (15.9) |
| Depression (%) | 34 (49.2) |
| Mix (%) | 24 (34.7) |
| Type 1 bipolar disorder (%) | 44 (63.7) |
| Family history of psychiatric disorders (%) | 30 (43.5) |
| HTT gene * | |
| 17.7 (2.1) |
| 21.03 (3.3) |
| - |
| 0 |
| 7 (10.2) |
* Pathological allele > 35 CAG repeats; intermediate allele 27–35 CAG repeats; normal allele < 27 CAG repeats; allele CAG 1 = shorter allele; allele CAG 2 = longer allele.
Table 2.
Characteristics of bipolar patients by the genetic status of IA carriers.
| No * IA Carriers (n = 62) | * IA Carriers (n = 7) | p | |
|---|---|---|---|
| Age at disease onset (SD) | 33.7 (13.5) | 43.3 (9.9) | 0.048 |
| Sex | |||
| Male (%) | 23 (37.1%) | 4 (57.3%) | 0.38 |
| Female (%) | 39 (62.9%) | 3 (42.8) | |
| Symptoms at onset | |||
| Maniacal (%) | 9 (14.5) | 2 (28.5) | 0.89 |
| Depression (%) | 30 (48.4) | 4 (57.1) | |
| Mix (%) | 23 (37.1) | 1 (14.2) | |
| Family history of psychiatric disorder (%) | 39 (45.1) | 2 (28.5) | 0.09 |
| CAG 1 (SD) | 17.37 (1.8) | 20.6 (2.3) | 0.09 |
| CAG 2 (SD) | 20.2 (2.08) | 28.6 (1.4) | <0.001 |
* IA = intermediate allele.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferrari, C.; Capacci, E.; Bagnoli, S.; Ingannato, A.; Sorbi, S.; Nacmias, B. The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder. Genes 2023, 14, 1681. https://doi.org/10.3390/genes14091681
AMA Style
Ferrari C, Capacci E, Bagnoli S, Ingannato A, Sorbi S, Nacmias B. The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder. Genes. 2023; 14(9):1681. https://doi.org/10.3390/genes14091681
Chicago/Turabian StyleFerrari, Camilla, Elena Capacci, Silvia Bagnoli, Assunta Ingannato, Sandro Sorbi, and Benedetta Nacmias. 2023. "The Huntington’s Disease Gene in an Italian Cohort of Patients with Bipolar Disorder" Genes 14, no. 9: 1681. https://doi.org/10.3390/genes14091681
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.