
Comparative Genome-Wide Analysis of MicroRNAs and Their Target Genes in Roots of Contrasting Indica Rice Cultivars under Reproductive-Stage Drought

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. Processing and Analysis of Small RNA-Seq Data
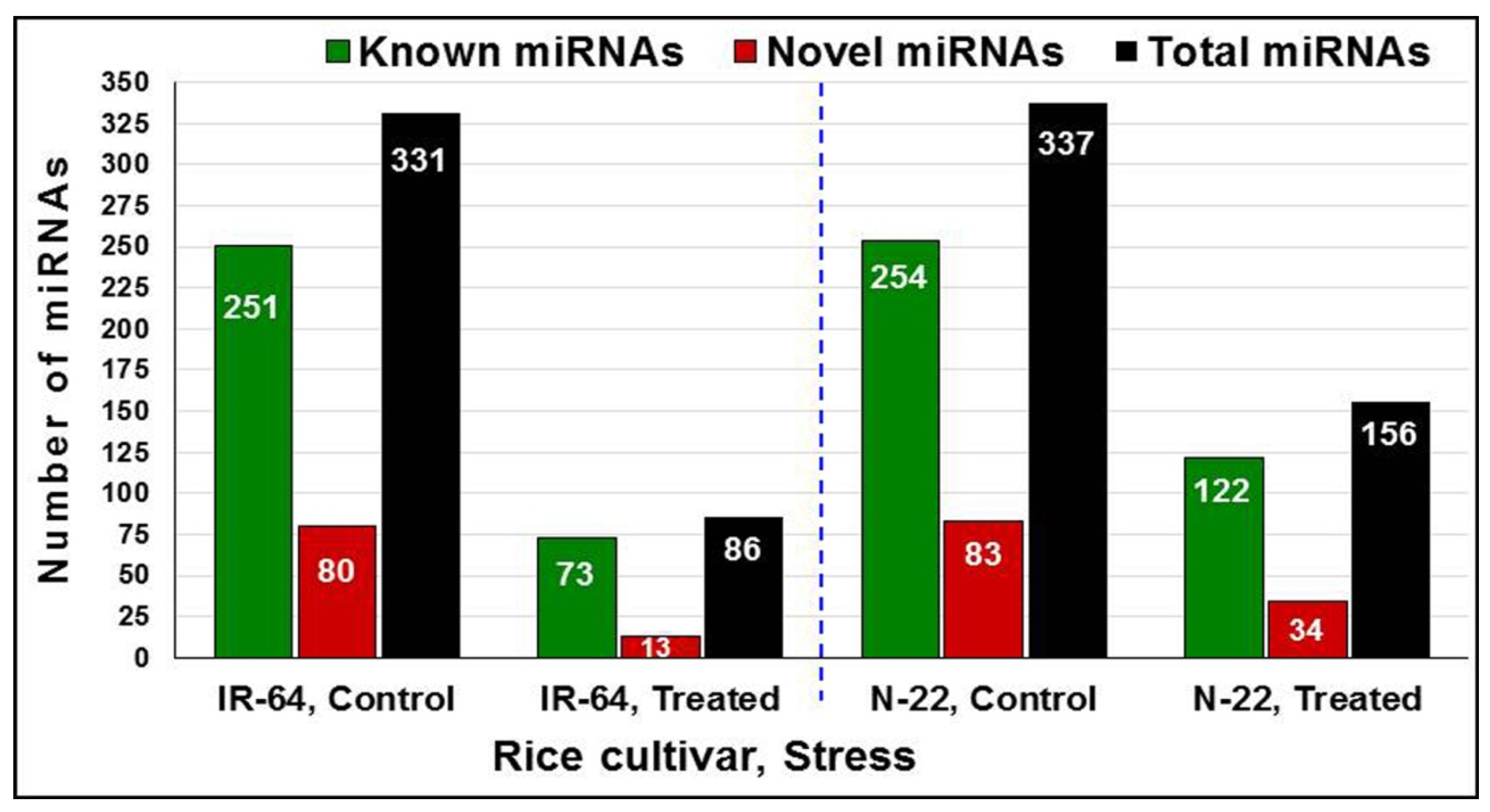
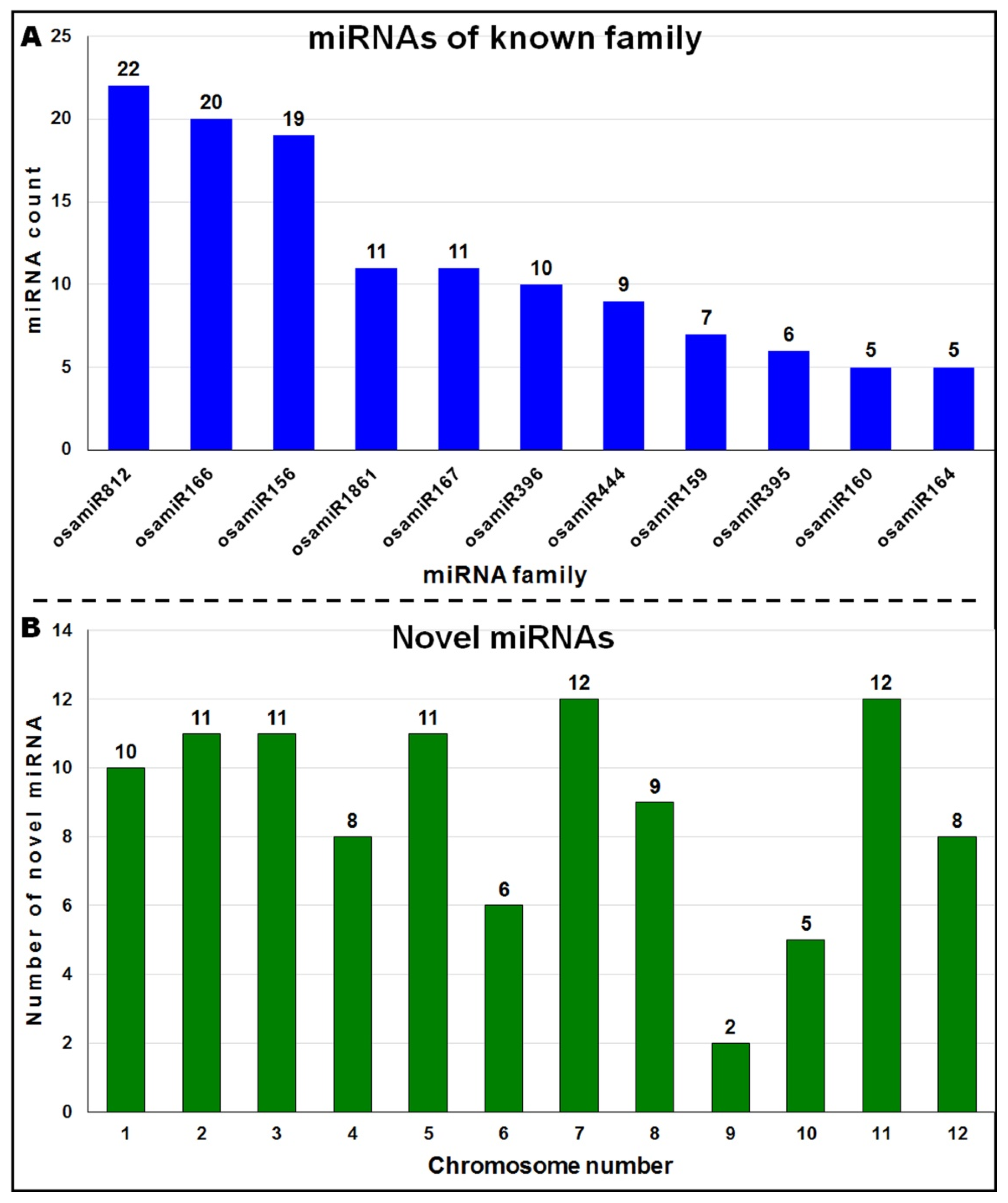
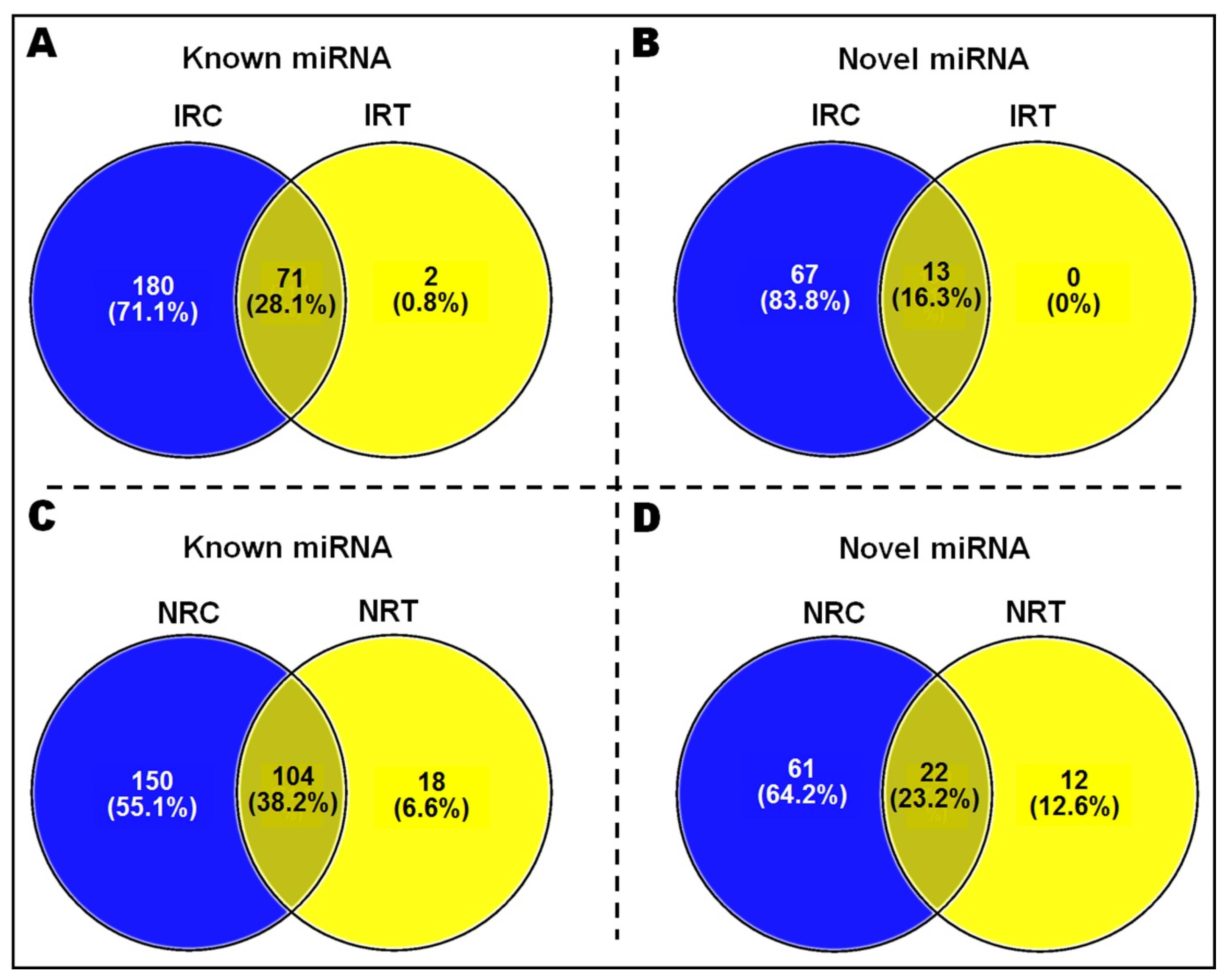
2.2. Identification of miRNAs Expressed in the Rice Cultivars
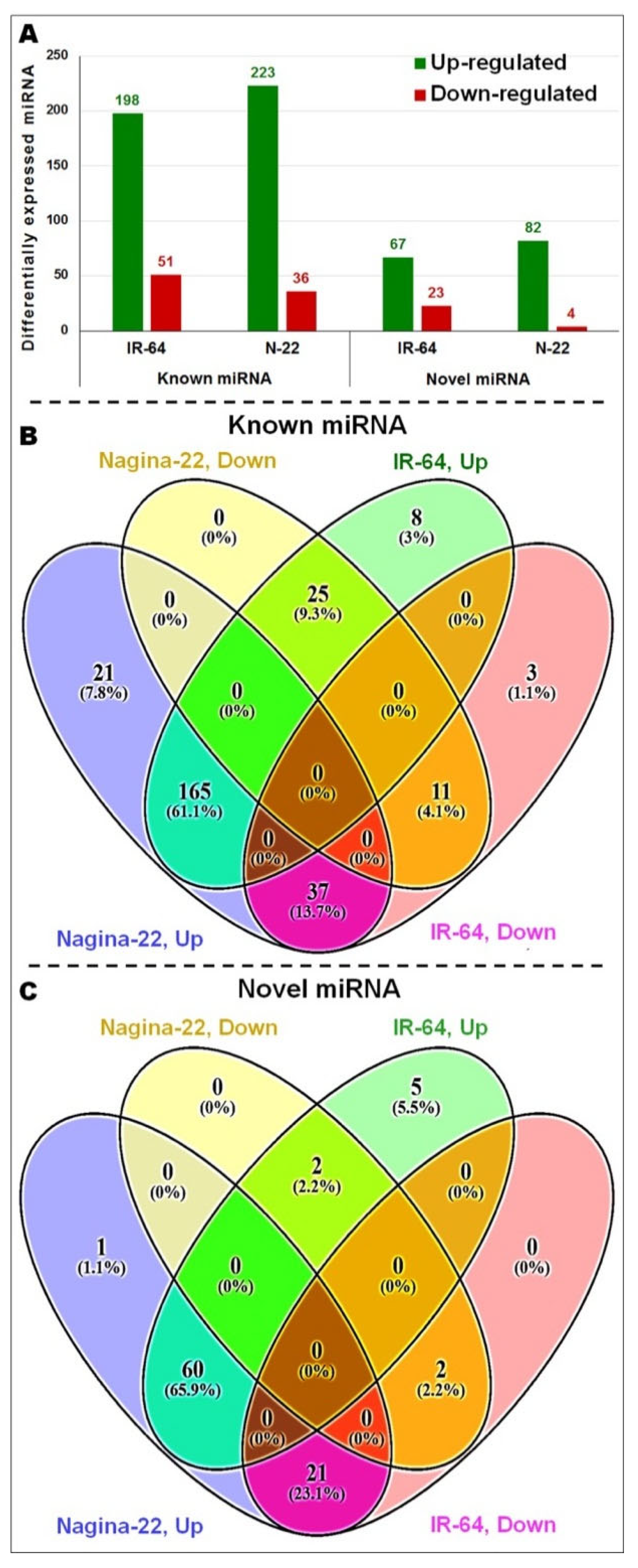
2.3. Differential Expression of miRNA in Root
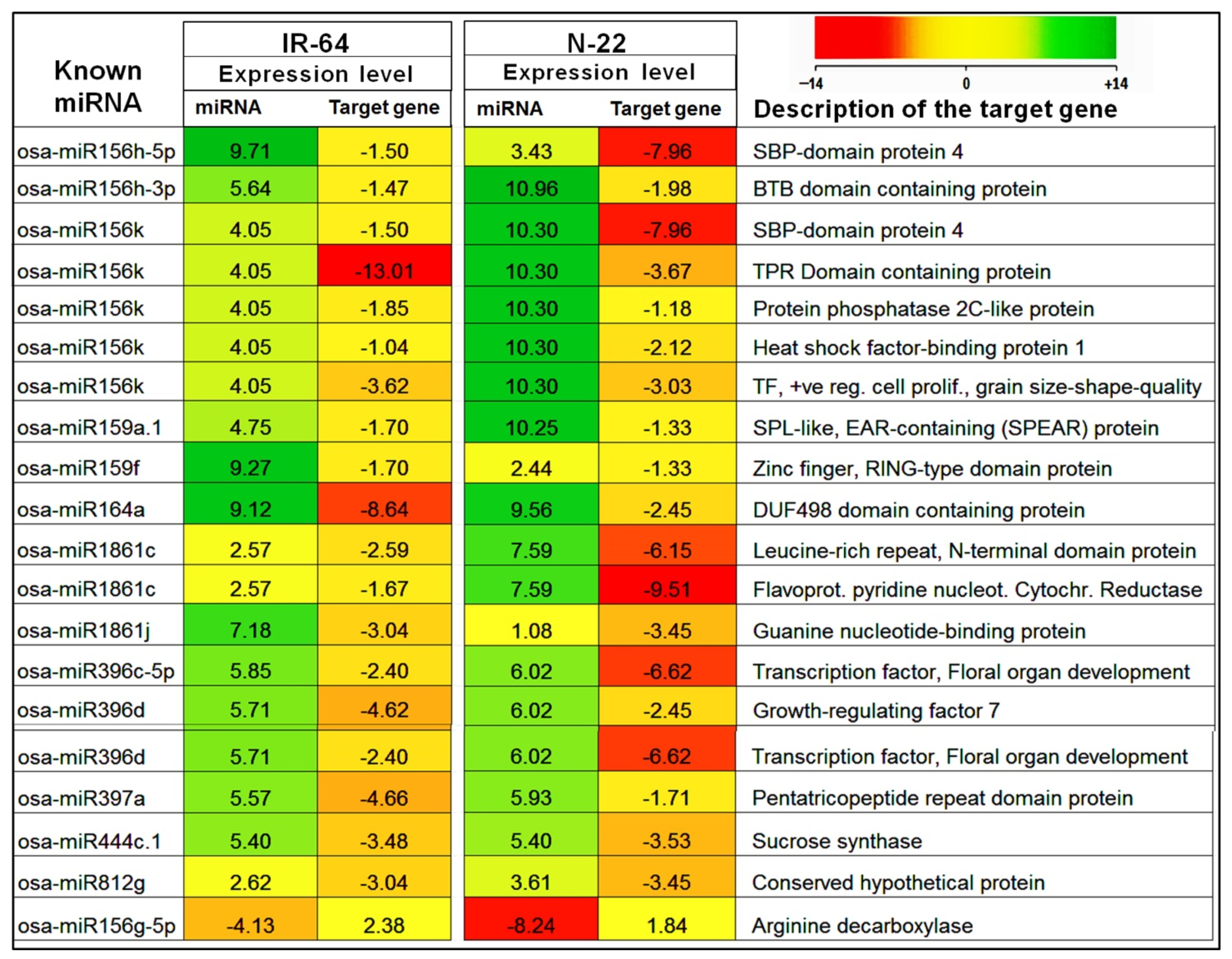
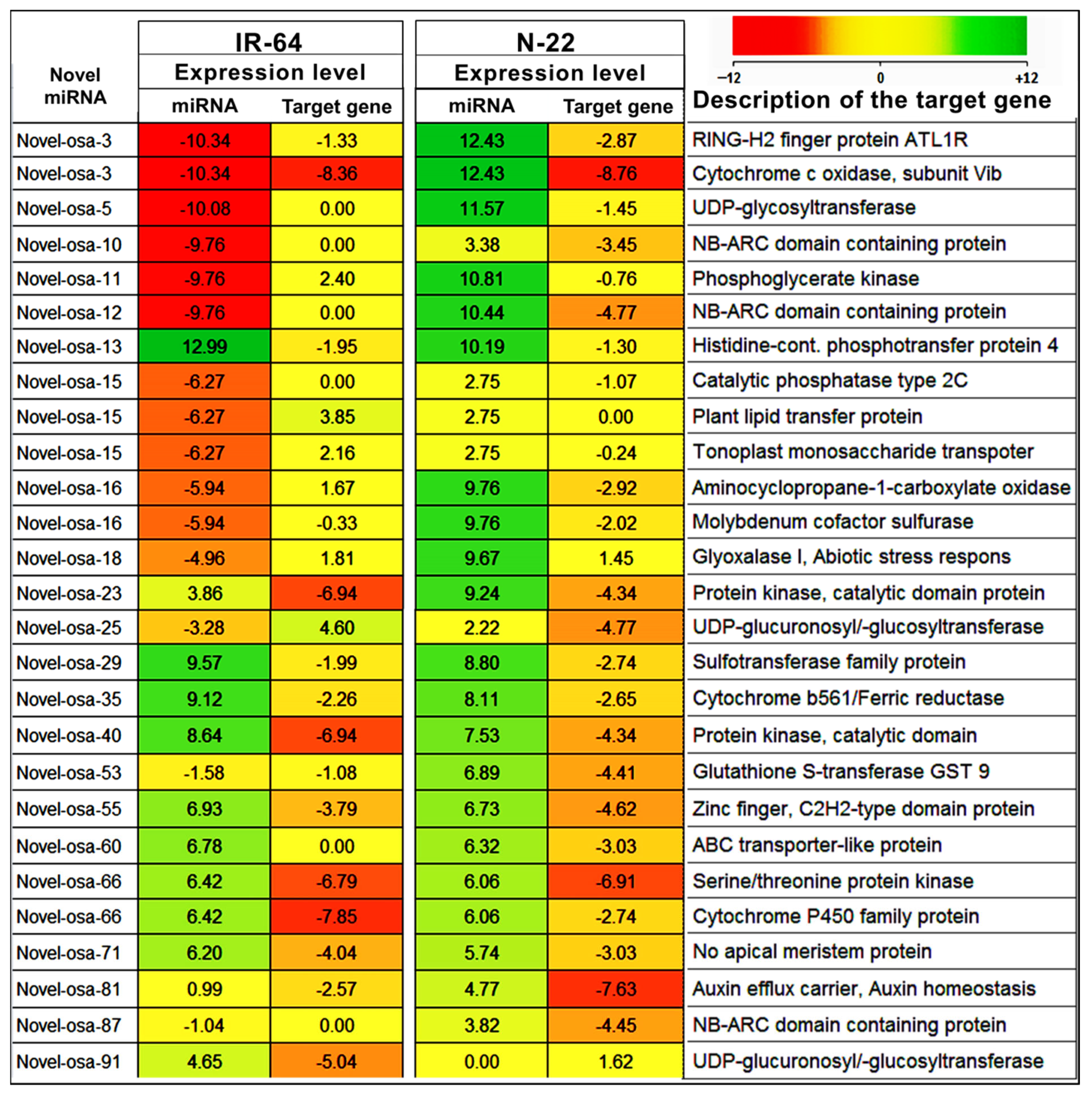
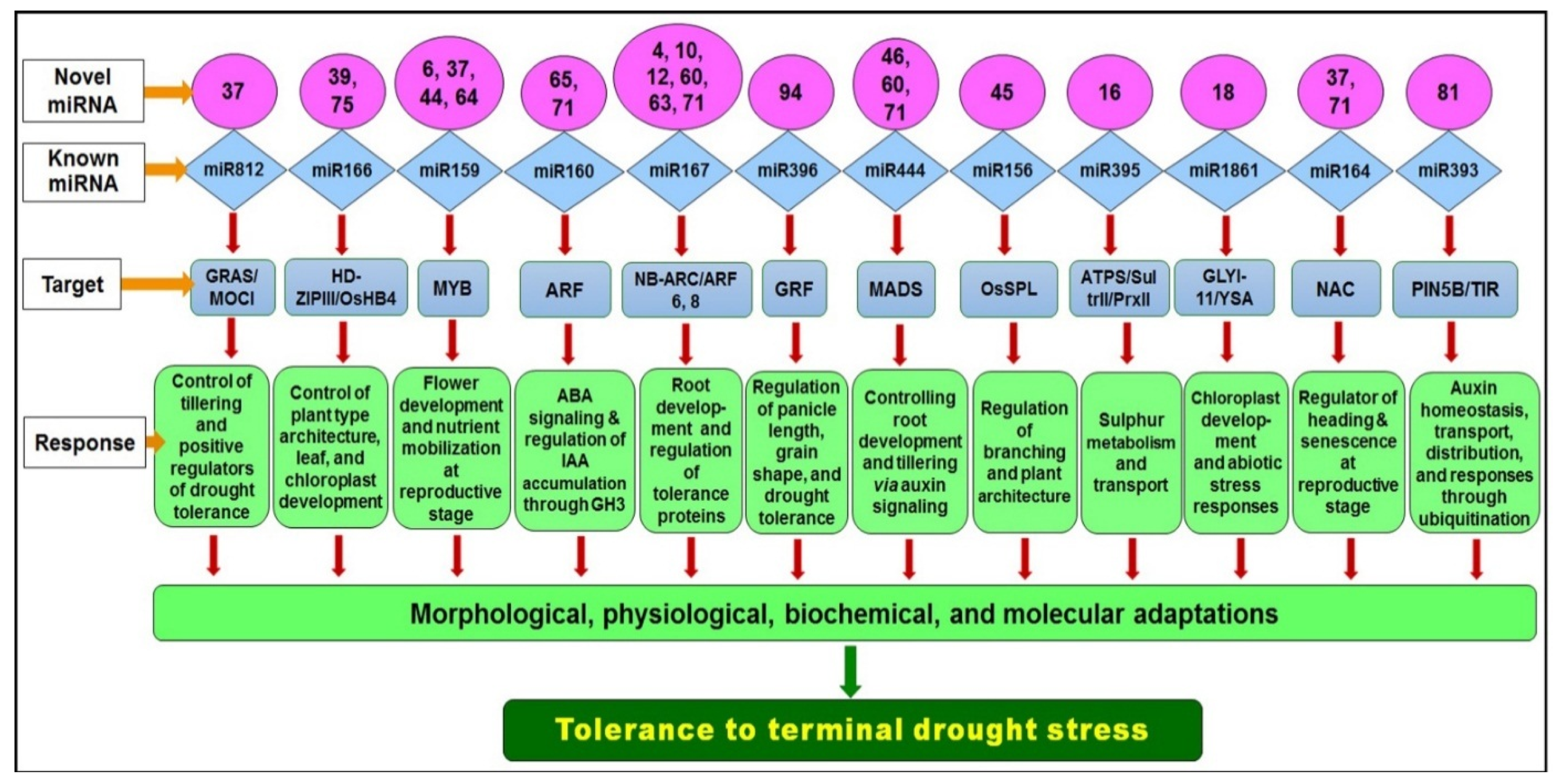
2.4. Target Prediction for Known and Novel miRNAs in Rice
2.5. Expression Analysis of miRNA Target Gene
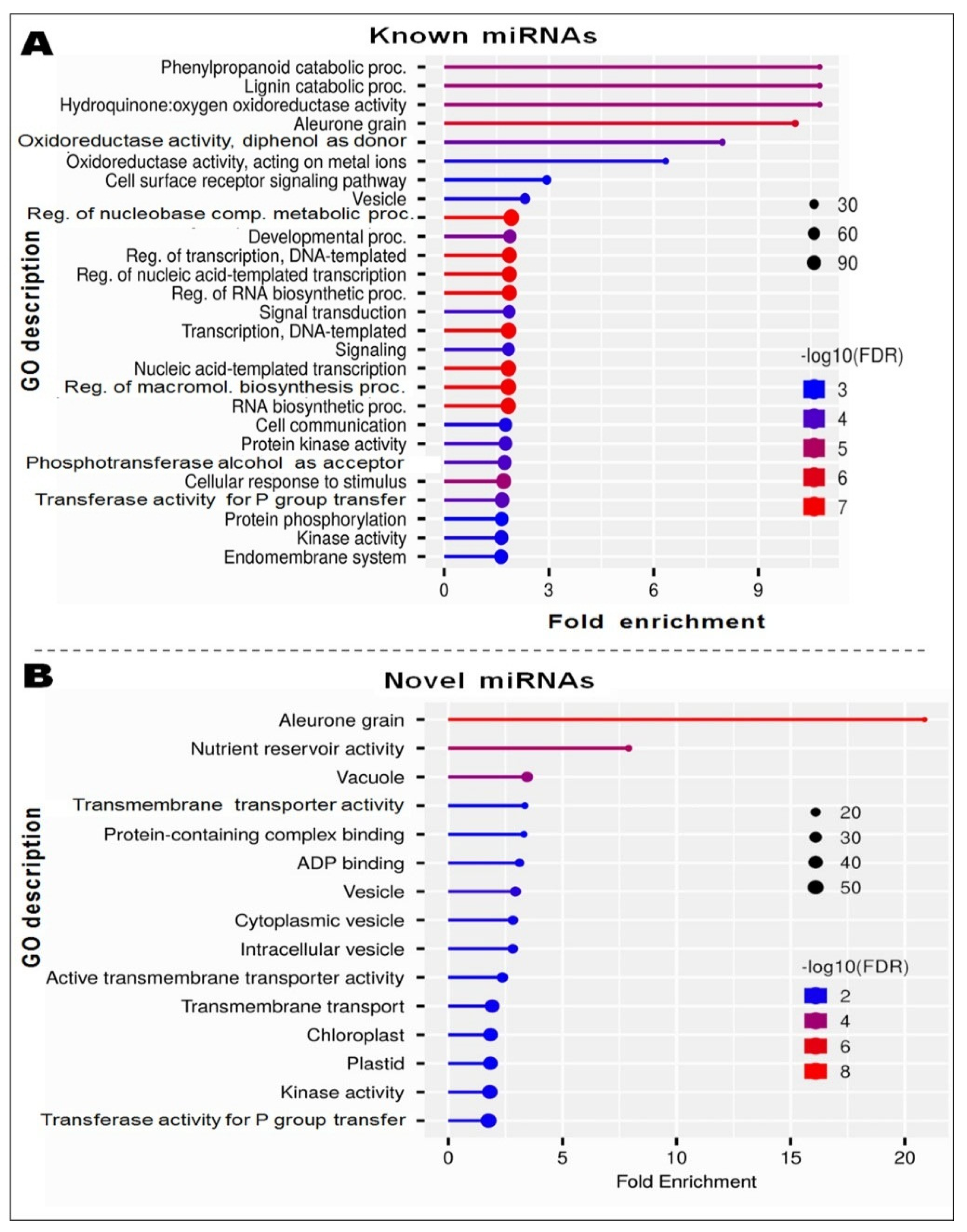
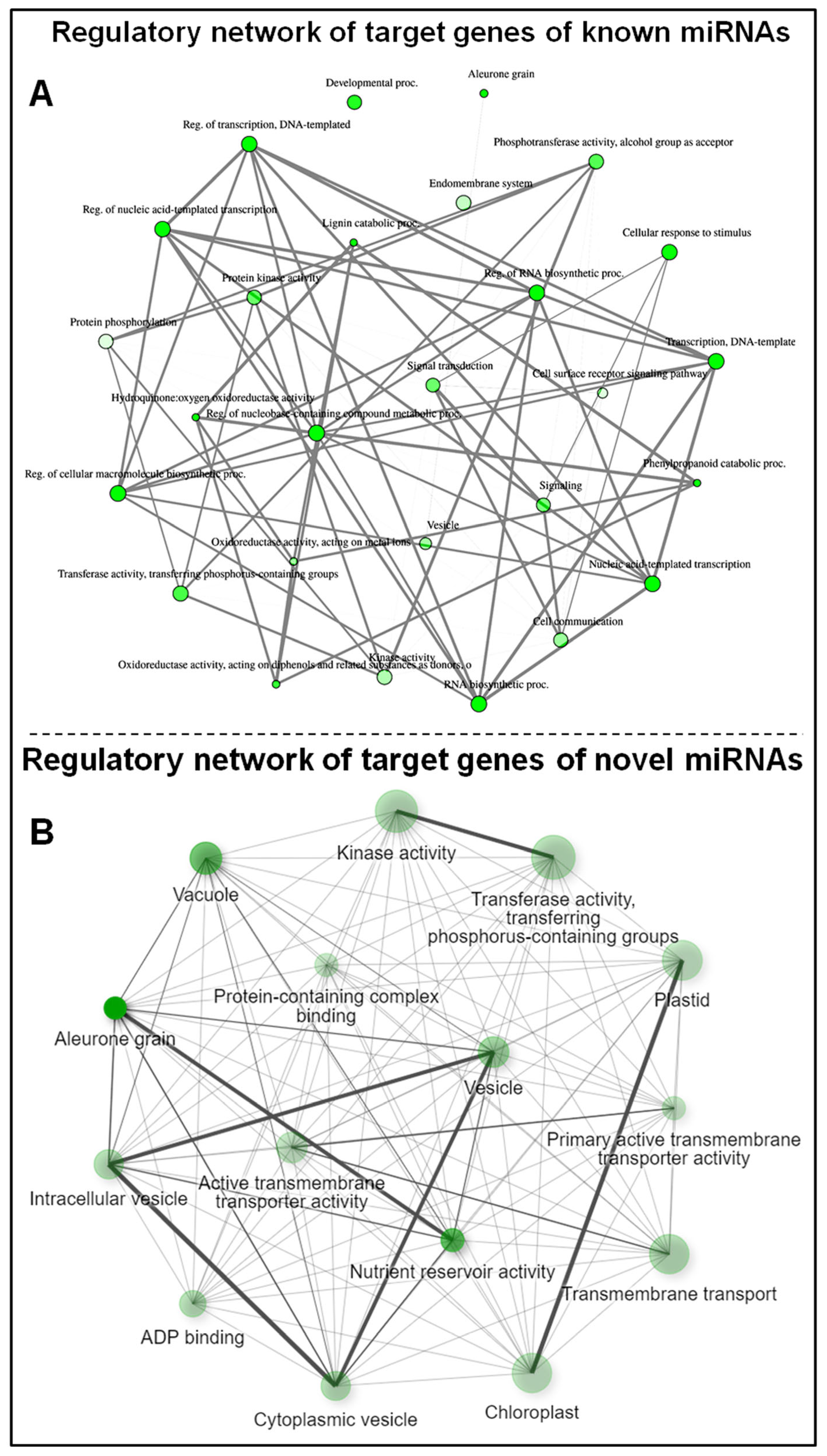
2.6. Gene Ontology Network Analysis of miRNA Targeted Genes
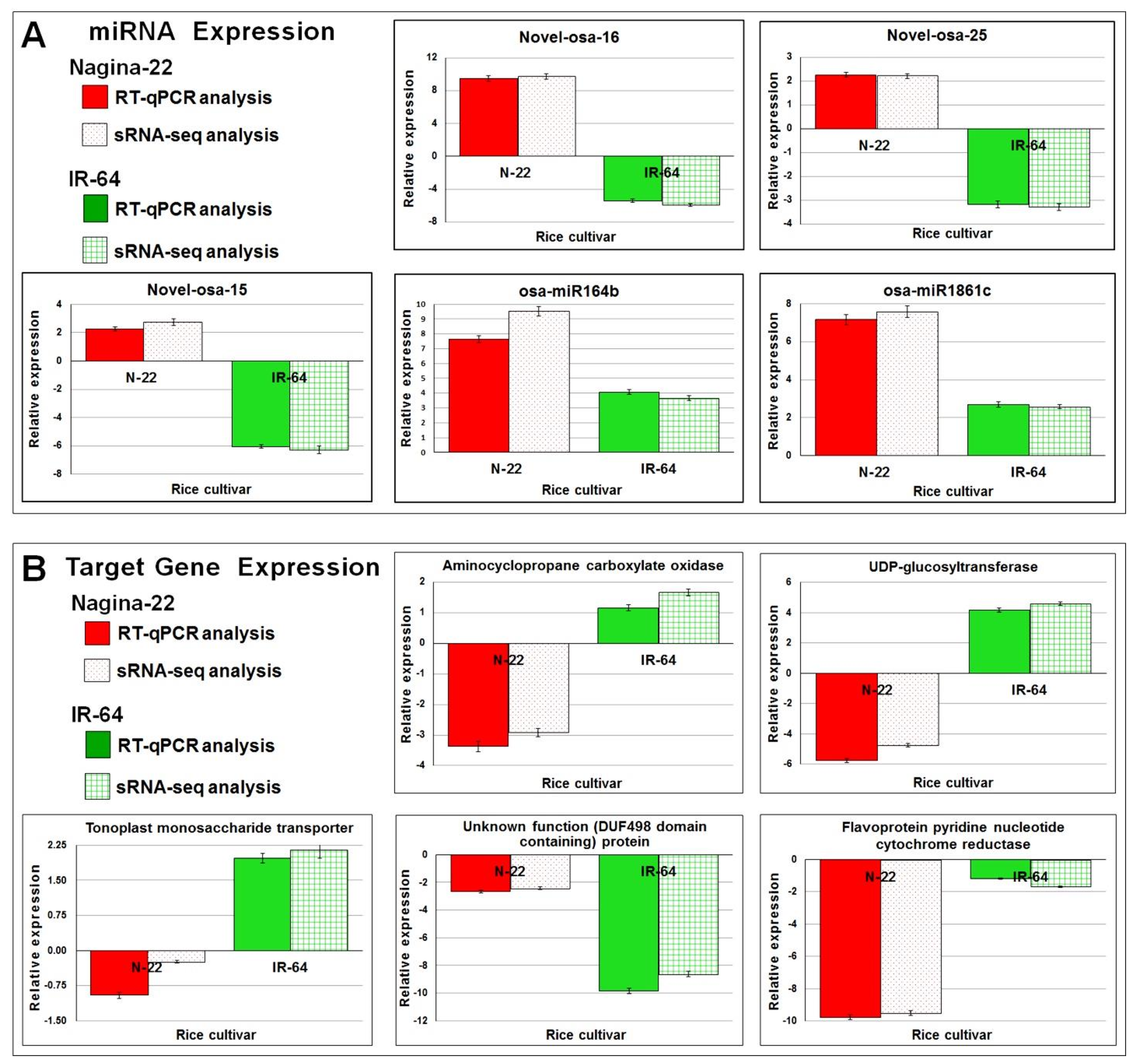
2.7. Functional Validation of miRNAs by RT-qPCR Assay
3. Discussion
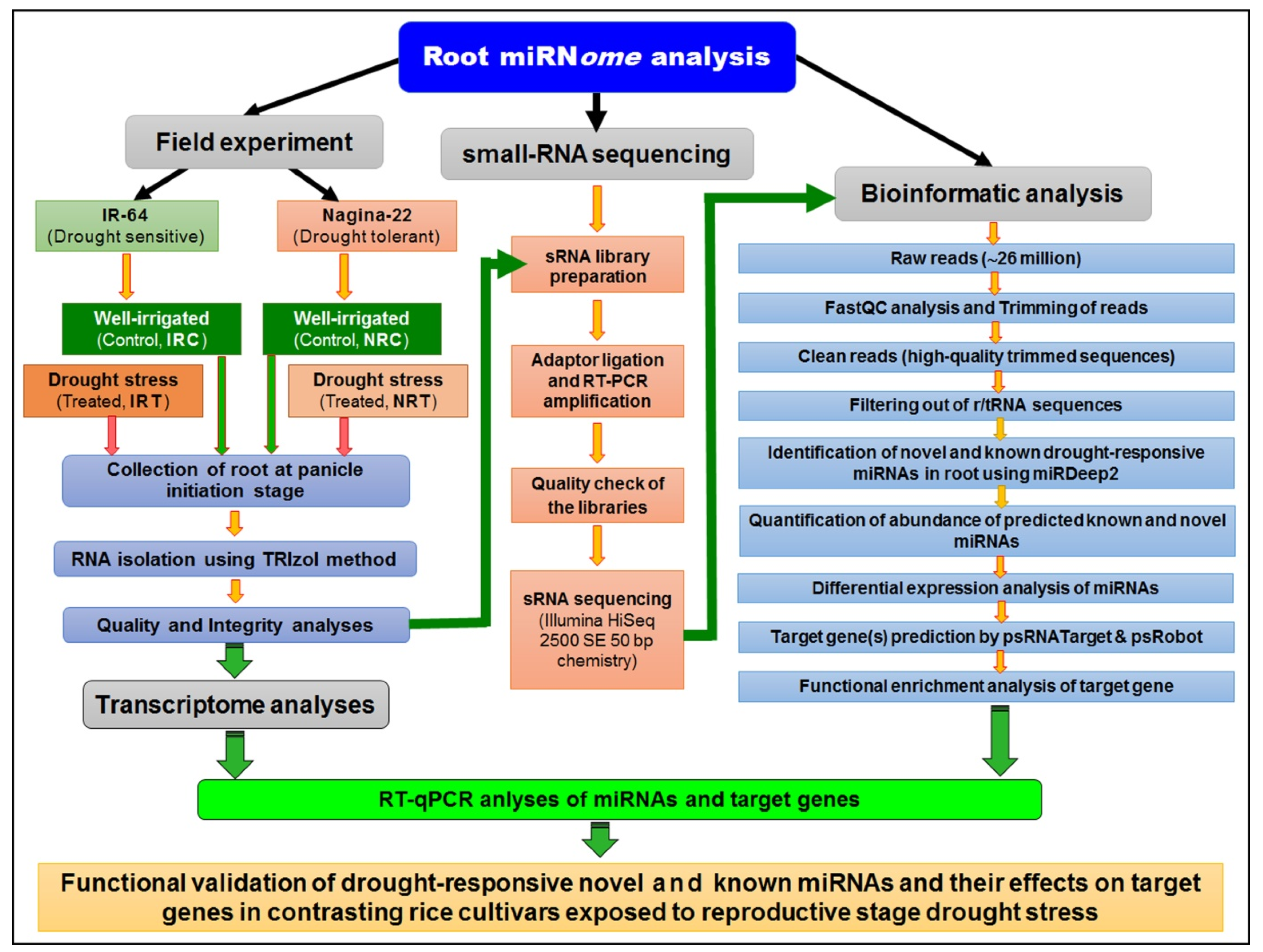
4. Materials and Methods
4.1. Plant Materials and Drought Stress Imposition
4.2. RNA Isolation, Small RNA Library Construction, and Sequencing
4.3. Small RNA Sequencing Reads Analysis
4.4. Differential Expression of Known and Novel miRNAs
4.5. Target Gene Prediction and Functional Enrichment Analysis of Targets
4.6. Whole-Genome Transcriptome Analysis for Functional Validation of miRNA
4.7. Validation of Differential Expression of miRNA by RT-qPCR
4.8. RT-qPCR Validation of Target Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shim, J.S.; Park, S.H.; Lee, D.K.; Kim, Y.S.; Park, S.C.; Redillas, M.C.F.R.; Seo, J.S.; Kim, J.K. The Rice GLYCINE-RICH PROTEIN 3 Confers Drought Tolerance by Regulating MRNA Stability of ROS Scavenging-Related Genes. Rice 2021, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Nadarajah, K.; Kumar, I.S. Drought Response in Rice: The MiRNA Story. Int. J. Mol. Sci. 2019, 20, 3766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, V.; Shukla, A. Acclimation and Tolerance Strategies of Rice under Drought Stress. Rice Sci. 2015, 22, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Bakhshi, B.; Mohseni Fard, E.; Nikpay, N.; Ali Ebrahimi, M.; Reza Bihamta, M.; Mardi, M.; Hosseini Salekdeh, G. MicroRNA Signatures of Drought Signaling in Rice Root. PLoS ONE 2016, 11, e0156814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awasthi, J.P.; Chandra, T.; Mishra, S.; Parmar, S.; Shaw, B.P.; Nilawe, P.D.; Chauhan, N.K.; Sahoo, S.; Panda, S.K. Identification and Characterization of Drought Responsive MiRNAs in a Drought Tolerant Upland Rice Cultivar KMJ 1-12-3. Plant Physiol. Biochem. 2019, 137, 62–74. [Google Scholar] [CrossRef]
- Panda, D.; Mishra, S.S.; Behera, P.K. Drought Tolerance in Rice: Focus on Recent Mechanisms and Approaches. Rice Sci. 2021, 28, 119–132. [Google Scholar] [CrossRef]
- Huang, L.; Wang, Y.; Wang, W.; Zhao, X.; Qin, Q.; Sun, F.; Hu, F.; Zhao, Y.; Li, Z.; Fu, B.; et al. Characterization of Transcription Factor Gene OSDRAP1 Conferring Drought Tolerance in Rice. Front. Plant Sci. 2018, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Kumar, S.; Krishnan, G.S.; Mohapatra, T. Molecular Basis of Genetic Plasticity to Varying Environmental Conditions on Growing Rice by Dry/Direct-Sowing and Exposure to Drought Stress: Insights for DSR Varietal Development. Front. Plant Sci. 2022, 13, 1013207. [Google Scholar] [CrossRef]
- Yue, B.; Xue, W.; Xiong, L.; Yu, X.; Luo, L.; Cui, K.; Jin, D.; Xing, Y.; Zhang, Q. Genetic Basis of Drought Resistance at Reproductive Stage in Rice: Separation of Drought Tolerance from Drought Avoidance. Genetics 2006, 172, 1213–1228. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Seem, K.; Duhan, N.; Kumar, S.; Kaundal, R.; Mohapatra, T. Transcriptome and Physio-Biochemical Profiling Reveals Differential Responses of Rice Cultivars at Reproductive-Stage Drought Stress. Int. J. Mol. Sci. 2023, 24, 1002. [Google Scholar] [CrossRef]
- Lenka, S.K.; Katiyar, A.; Chinnusamy, V.; Bansal, K.C. Comparative Analysis of Drought-Responsive Transcriptome in Indica Rice Genotypes with Contrasting Drought Tolerance. Plant Biotechnol. J. 2011, 9, 315–327. [Google Scholar] [CrossRef]
- Xia, H.; Yu, S.; Kong, D.; Xiong, J.; Ma, X.; Chen, L.; Luo, L. Temporal Responses of Conserved MiRNAs to Drought and Their Associations with Drought Tolerance and Productivity in Rice. BMC Genom. 2020, 21, 232. [Google Scholar] [CrossRef] [Green Version]
- Seem, K.; Kumar, S. Cultivation of Rice: Evolving towards Climate-Smart Crops for Precision in Resource Use Efficiency. MC Agric. Environ. Sci. 2021, 1, 41–49. [Google Scholar]
- Kumar, S.; Kaur, S.; Seem, K.; Kumar, S.; Mohapatra, T. Understanding 3D Genome Organization and Its Effect on Transcriptional Gene Regulation Under Environmental Stress in Plant: A Chromatin Perspective. Front. Cell Dev. Biol. 2021, 9, 774719. [Google Scholar] [CrossRef]
- Samota, M.K.; Sasi, M.; Awana, M.; Yadav, O.P.; AmithaMithra, S.V.; Tyagi, A.; Kumar, S.; Singh, A. Elicitor-Induced Biochemical and Molecular Manifestations to Improve Drought Tolerance in Rice (Oryza sativa L.) through Seed-Priming. Front. Plant Sci. 2017, 8, 934. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.H.; Choi, Y.D.; Kim, M.; Reuzeau, C.; Kim, J.K. Root-Specific Expression of OsNAC10 Improves Drought Tolerance and Grain Yield in Rice under Field Drought Conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Li, Z.; Xiong, L. A Plant MicroRNA Regulates the Adaptation of Roots to Drought Stress. FEBS Lett. 2012, 586, 1742–1747. [Google Scholar] [CrossRef] [Green Version]
- Shahinnia, F.; Roy, J.L.; Laborde, B.; Sznajder, B.; Kalambettu, P.; Mahjourimajd, S.; Tilbrook, J.; Fleury, D. Genetic Association of Stomatal Traits and Yield in Wheat Grown in Low Rainfall Environments. BMC Plant Biol. 2016, 16, 150. [Google Scholar] [CrossRef] [Green Version]
- Vega Riveros, C.; Villagra, P.E.; Greco, S.A. Different Root Strategies of Perennial Native Grasses under Two Contrasting Water Availability Conditions: Implications for Their Spatial Distribution in Desert Dunes. Plant Ecol. 2020, 221, 633–646. [Google Scholar] [CrossRef]
- Kulkarni, M.; Soolanayakanahally, R.; Ogawa, S.; Uga, Y.; Selvaraj, M.G.; Kagale, S. Drought Response in Wheat: Key Genes and Regulatory Mechanisms Controlling Root System Architecture and Transpiration Efficiency. Front. Chem. 2017, 5, 106. [Google Scholar] [CrossRef] [Green Version]
- Kou, X.; Han, W.; Kang, J. Responses of Root System Architecture to Water Stress at Multiple Levels: A Meta-Analysis of Trials under Controlled Conditions. Front. Plant Sci. 2022, 13, 5157. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Seem, K.; Kumar, S.; Mohapatra, T. RNA-Seq Analysis Reveals the Genes/Pathways Responsible for Genetic Plasticity of Rice to Varying Environmental Conditions on Direct-Sowing and Transplanting. Sci. Rep. 2022, 12, 2241. [Google Scholar] [CrossRef] [PubMed]
- Mutum, R.D.; Kumar, S.; Balyan, S.; Kansal, S.; Mathur, S.; Raghuvanshi, S. Identification of Novel MiRNAs from Drought Tolerant Rice Variety Nagina 22. Sci. Rep. 2016, 6, 30786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, S.; Kumar, S.; Mohapatra, T. MicroRNA: Noncoding but Still Coding, Another Example of Self-Catalysis. Funct. Integr. Genom. 2022, 23, 4. [Google Scholar] [CrossRef] [PubMed]
- Fantao, Z.; Yuan, L.; Meng, Z.; Yi, Z.; Hongping, C.; Biaolin, H.; Jiankun, X. Identification and Characterization of Drought Stress- Responsive Novel MicroRNAs in Dongxiang Wild Rice. Rice Sci. 2018, 25, 175–184. [Google Scholar] [CrossRef]
- Dong, Z.; Zhang, J.; Zhu, Q.; Zhao, L.; Sui, S.; Li, Z.; Zhang, Y.; Wang, H.; Tian, D.; Zhao, Y. Identification of MicroRNAs Involved in Drought Stress Responses in Early-Maturing Cotton by High-Throughput Sequencing. Genes Genom. 2018, 40, 305–314. [Google Scholar] [CrossRef]
- Wei, L.; Zhang, D.; Xiang, F.; Zhang, Z. Differentially Expressed MiRNAs Potentially Involved in the Regulation of Defense Mechanism to Drought Stress in Maize Seedlings. Int. J. Plant Sci. 2009, 170, 979–989. [Google Scholar] [CrossRef]
- Zhang, B. MicroRNA: A New Target for Improving Plant Tolerance to Abiotic Stress. J. Exp. Bot. 2015, 66, 1749–1761. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Y.; Liu, Z.; Kong, D.; Duan, M.; Luo, L. Genome-Wide Identification and Analysis of Drought-Responsive MicroRNAs in Oryza sativa. J. Exp. Bot. 2010, 61, 4157–4168. [Google Scholar] [CrossRef]
- Eamens, A.L.; Kim, K.W.; Curtin, S.J.; Waterhouse, P.M. DRB2 Is Required for MicroRNA Biogenesis in Arabidopsis thaliana. PLoS ONE 2012, 7, e35933. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Liang, R.; Ge, L.; Li, W.; Xiao, H.; Lin, H.; Ruan, K.; Jin, Y. Identification of Drought-Induced MicroRNAs in Rice. Biochem. Biophys. Res. Commun. 2007, 354, 585–590. [Google Scholar] [CrossRef]
- Barrera-Figueroa, B.E.; Gao, L.; Diop, N.N.; Wu, Z.; Ehlers, J.D.; Roberts, P.A.; Close, T.J.; Zhu, J.-K.; Liu, R. Identification and Comparative Analysis of Drought-Associated MicroRNAs in Two Cowpea Genotypes. BMC Plant Biol. 2011, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Shui, X.R.; Chen, Z.W.; Li, J.X. MicroRNA Prediction and Its Function in Regulating Drought-Related Genes in Cowpea. Plant Sci. 2013, 210, 25–35. [Google Scholar] [CrossRef]
- Cheah, B.H.; Nadarajah, K.; Divate, M.D.; Wickneswari, R. Identification of Four Functionally Important MicroRNA Families with Contrasting Differential Expression Profiles between Drought-Tolerant and Susceptible Rice Leaf at Vegetative Stage. BMC Genom. 2015, 16, 692. [Google Scholar] [CrossRef] [Green Version]
- Aravind, J.; Rinku, S.; Pooja, B.; Shikha, M.; Kaliyugam, S.; Mallikarjuna, M.G.; Kumar, A.; Rao, A.R.; Nepolean, T. Identification, Characterization, and Functional Validation of Drought-Responsive MicroRNAs in Subtropical Maize Inbreds. Front. Plant Sci. 2017, 8, 941. [Google Scholar] [CrossRef] [Green Version]
- Mandal, K.; Boro, P.; Chattopadhyay, S. Micro-RNA Based Gene Regulation: A Potential Way for Crop Improvements. Plant Gene 2021, 27, 100312. [Google Scholar] [CrossRef]
- Singroha, G.; Sharma, P.; Sunkur, R. Current Status of MicroRNA-Mediated Regulation of Drought Stress Responses in Cereals. Physiol. Plant 2021, 172, 1808–1821. [Google Scholar] [CrossRef]
- Jiang, D.; Zhou, L.; Chen, W.; Ye, N.; Xia, J.; Zhuang, C. Overexpression of a MicroRNA-Targeted NAC Transcription Factor Improves Drought and Salt Tolerance in Rice via ABA-Mediated Pathways. Rice 2019, 12, 76. [Google Scholar] [CrossRef]
- Goel, S.; Goswami, K.; Pandey, V.K.; Pandey, M.; Sanan-Mishra, N. Identification of MicroRNA-Target Modules from Rice Variety Pusa Basmati-1 under High Temperature and Salt Stress. Funct. Integr. Genom. 2019, 19, 867–888. [Google Scholar] [CrossRef]
- Wang, S.T.; Sun, X.L.; Hoshino, Y.; Yu, Y.; Jia, B.; Sun, Z.W.; Sun, M.Z.; Duan, X.B.; Zhu, Y.M. MicroRNA319 Positively Regulates Cold Tolerance by Targeting OsPCF6 and OsTCP21 in Rice (Oryza sativa L.). PLoS ONE 2014, 9, e91357. [Google Scholar] [CrossRef]
- Ding, Y.; Tao, Y.; Zhu, C. Emerging Roles of MicroRNAs in the Mediation of Drought Stress Response in Plants. J. Exp. Bot. 2013, 64, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Balyan, S.; Kumar, M.; Mutum, R.D.; Raghuvanshi, U.; Agarwal, P.; Mathur, S.; Raghuvanshi, S. Identification of MiRNA-Mediated Drought Responsive Multi-Tiered Regulatory Network in Drought Tolerant Rice, Nagina. Sci. Rep. 2017, 7, 15446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunkar, R.; Li, Y.F.; Jagadeeswaran, G. Functions of MicroRNAs in Plant Stress Responses. Trends Plant Sci. 2012, 17, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Shukla, L.I.; Chinnusamy, V.; Sunkar, R. The Role of MicroRNAs and Other Endogenous Small RNAs in Plant Stress Responses. Biochim. Biophys. Acta Gene Regul. Mech. 2008, 1779, 743–748. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xie, K.; Xiong, L. Conserved MiR164-Targeted NAC Genes Negatively Regulate Drought Resistance in Rice. J. Exp. Bot. 2014, 65, 2119–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, M.; Munné-Bosch, S. Ethylene Response Factors: A Key Regulatory Hub in Hormone and Stress Signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Chen, L.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of Drought-Responsive MicroRNAs in Medicago truncatula by Genome-Wide High-Throughput Sequencing. BMC Genom. 2011, 12, 367. [Google Scholar] [CrossRef] [Green Version]
- Arenas-Huertero, C.; Pérez, B.; Rabanal, F.; Blanco-Melo, D.; De La Rosa, C.; Estrada-Navarrete, G.; Sanchez, F.; Covarrubias, A.A.; Reyes, J.L. Conserved and Novel MiRNAs in the Legume Phaseolus Vulgaris in Response to Stress. Plant Mol. Biol. 2009, 70, 385–401. [Google Scholar] [CrossRef]
- Giusti, L.; Mica, E.; Bertolini, E.; De Leonardis, A.M.; Faccioli, P.; Cattivelli, L.; Crosatti, C. MicroRNAs Differentially Modulated in Response to Heat and Drought Stress in Durum Wheat Cultivars with Contrasting Water Use Efficiency. Funct. Integr. Genom. 2017, 17, 293–309. [Google Scholar] [CrossRef]
- Barrera-Figueroa, B.E.; Gao, L.; Wu, Z.; Zhou, X.; Zhu, J.; Jin, H.; Liu, R.; Zhu, J.K. High Throughput Sequencing Reveals Novel and Abiotic Stress-Regulated MicroRNAs in the Inflorescences of Rice. BMC Plant Biol. 2012, 12, 132. [Google Scholar] [CrossRef] [Green Version]
- Kulcheski, F.R.; de Oliveira, L.F.V.; Molina, L.G.; Almerão, M.P.; Rodrigues, F.A.; Marcolino, J.; Barbosa, J.F.; Stolf-Moreira, R.; Nepomuceno, A.L.; Marcelino-Guimarães, F.C.; et al. Identification of Novel Soybean MicroRNAs Involved in Abiotic and Biotic Stresses. BMC Genom. 2011, 12, 307. [Google Scholar] [CrossRef] [Green Version]
- Candar-Cakir, B.; Arican, E.; Zhang, B. Small RNA and Degradome Deep Sequencing Reveals Drought-and Tissue-Specific MicroRNAs and Their Important Roles in Drought-Sensitive and Drought-Tolerant Tomato Genotypes. Plant Biotechnol. J. 2016, 14, 1727–1746. [Google Scholar] [CrossRef] [Green Version]
- Gentile, A.; Dias, L.I.; Mattos, R.S.; Ferreira, T.H.; Menossi, M. MicroRNAs and Drought Responses in Sugarcane. Front. Plant Sci. 2015, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.K.; Mehra, S.; Chatterjee, S.; Purty, R.S. In Silico Identification and Validation of MiRNA and Their DIR Specific Targets in Oryza sativa Indica under Abiotic Stress. Noncoding RNA Res. 2020, 5, 167–177. [Google Scholar] [CrossRef]
- Seeve, C.M.; Sunkar, R.; Zheng, Y.; Liu, L.; Liu, Z.; McMullen, M.; Nelson, S.; Sharp, R.E.; Oliver, M.J. Water-Deficit Responsive MicroRNAs in the Primary Root Growth Zone of Maize. BMC Plant Biol. 2019, 19, 447. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Lv, H.; Li, Q.; Zhang, X.; Li, L.; Xu, J.; Wu, F.; Wang, Q.; Feng, X.; Lu, Y. Characteristics of MicroRNAs and Target Genes in Maize Root under Drought Stress. Int. J. Mol. Sci. 2022, 23, 4968. [Google Scholar] [CrossRef]
- Zhang, J.W.; Long, Y.; Xue, M.D.; Xiao, X.G.; Pei, X.W. Identification of MicroRNAs in Response to Drought in Common Wild Rice (Oryza rufipogon Griff.) Shoots and Roots. PLoS ONE 2017, 12, e0170330. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential Expression of MiRNAs in Response to Salt Stress in Maize Roots. Ann. Bot. 2009, 103, 29. [Google Scholar] [CrossRef] [Green Version]
- Lv, D.K.; Bai, X.; Li, Y.; Ding, X.D.; Ge, Y.; Cai, H.; Ji, W.; Wu, N.; Zhu, Y.M. Profiling of Cold-Stress-Responsive MiRNAs in Rice by Microarrays. Gene 2010, 459, 39–47. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, M.; Xu, X.; Li, X.; Li, C.; Ding, Z. System Analysis of MicroRNAs in the Development and Aluminium Stress Responses of the Maize Root System. Plant Biotechnol. J. 2014, 12, 1108–1121. [Google Scholar] [CrossRef]
- Zhai, L.; Liu, Z.; Zou, X.; Jiang, Y.; Qiu, F.; Zheng, Y.; Zhang, Z. Genome-Wide Identification and Analysis of MicroRNA Responding to Long-Term Waterlogging in Crown Roots of Maize Seedlings. Physiol. Plant 2013, 147, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yu, H.; Zhao, G.; Huang, Q.; Lu, Y.; Ouyang, B. Profiling of Drought-Responsive MicroRNA and MRNA in Tomato Using High-Throughput Sequencing. BMC Genom. 2017, 18, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Dutta, P.; Chakrabarty, D. MiRNAs Play Critical Roles in Response to Abiotic Stress by Modulating Cross-Talk of Phytohormone Signaling. Plant Cell Rep. 2021, 40, 1617–1630. [Google Scholar] [CrossRef] [PubMed]
- Baldoni, E. Improving Drought Tolerance: Can Comparative Transcriptomics Support Strategic Rice Breeding? Plant Stress 2022, 3, 100058. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, X.; Zhou, Y.; Xie, J. Genome-Wide Identification of Conserved MicroRNA and Their Response to Drought Stress in Dongxiang Wild Rice (Oryza Rufipogon Griff.). Biotechnol. Lett. 2016, 38, 711–721. [Google Scholar] [CrossRef]
- Gao, F.; Wang, K.; Liu, Y.; Chen, Y.; Chen, P.; Shi, Z.; Luo, J.; Jiang, D.; Fan, F.; Zhu, Y.; et al. Blocking MiR396 Increases Rice Yield by Shaping Inflorescence Architecture. Nat. Plants 2015, 1, 15196. [Google Scholar] [CrossRef]
- Bakhshi, B.; Salekdeh, G.H.; Bihamta, M.R.; Tohidfar, M. Characterization of Three Key MicroRNAs in Rice Root Architecture under Drought Stress Using in Silico Analysis and Quantitative Real-Time PCR. Biosci. Biotechnol. Res. Asia 2014, 11, 555–565. [Google Scholar] [CrossRef]
- Hamza, N.B.; Sharma, N.; Tripathi, A.; Sanan-Mishra, N. MicroRNA Expression Profiles in Response to Drought Stress in Sorghum bicolor. Gene Expr. Patterns 2016, 20, 88–98. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, H.; Srivastava, A.K.; Pan, Y.; Bai, J.; Fang, J.; Shi, H.; Zhu, J.K. Knockdown of Rice MicroRNA166 Confers Drought Resistance by Causing Leaf Rolling and Altering Stem Xylem Development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Uribe, L.; O’Connell, M.A. A Root-Specific BZIP Transcription Factor Is Responsive to Water Deficit Stress in Tepary Bean (Phaseolus acutifolius) and Common Bean (P. vulgaris). J. Exp. Bot. 2006, 57, 1391–1398. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Z.; Liu, D.; Zhang, K.; Li, A.; Mao, L. SQUAMOSA Promoter-Binding Protein-Like Transcription Factors: Star Players for Plant Growth and Development. J. Integr. Plant Biol. 2010, 52, 946–951. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.C.; Wang, C.Y.; Luo, Y.C.; Huang, Q.J.; Chen, S.Y.; Zhou, H.; Qu, L.H.; Chen, Y.Q. Expression Analysis of Phytohormone-Regulated MicroRNAs in Rice, Implying Their Regulation Roles in Plant Hormone Signaling. FEBS Lett. 2009, 583, 723–728. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhu, H.; Zhang, Q.; Li, M.; Yan, M.; Wang, R.; Wang, L.; Welti, R.; Zhang, W.; Wang, X. Phospholipase Dα1 and Phosphatidic Acid Regulate NADPH Oxidase Activity and Production of Reactive Oxygen Species in ABA-Mediated Stomatal Closure in Arabidopsis. Plant Cell 2009, 21, 2357–2377. [Google Scholar] [CrossRef] [Green Version]
- Sorin, C.; Bussell, J.D.; Camus, I.; Ljung, K.; Kowalczyk, M.; Geiss, G.; McKhann, H.; Garcion, C.; Vaucheret, H.; Sandberg, G.; et al. Auxin and Light Control of Adventitious Rooting in Arabidopsis Require ARGONAUTE1. Plant Cell 2005, 17, 1343. [Google Scholar] [CrossRef] [Green Version]
- Bouzroud, S.; Gouiaa, S.; Hu, N.; Bernadac, A.; Mila, I.; Bendaou, N.; Smouni, A.A.; Bouzayen, M.; Zouine, M. Auxin Response Factors (ARFs) Are Potential Mediators of Auxin Action in Tomato Response to Biotic and Abiotic Stress (Solanum lycopersicum). PLoS ONE 2018, 13, e0193517. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, Z.; Zhao, D. Deregulation of the OsmiR160 Target Gene OsARF18 Causes Growth and Developmental Defects with an Alteration of Auxin Signaling in Rice. Sci. Rep. 2016, 6, 29938. [Google Scholar] [CrossRef] [Green Version]
- Gleeson, M.; Constantin, M.; Carroll, B.J.; Mitter, N. MicroRNAs as Regulators of Adventitious Root Development. J. Plant Biochem. Biotechnol. 2014, 23, 339–347. [Google Scholar] [CrossRef]
- Ma, J.; Zhao, P.; Liu, S.; Yang, Q.; Guo, H. The Control of Developmental Phase Transitions by MicroRNAs and Their Targets in Seed Plants. Int. J. Mol. Sci. 2020, 21, 1971. [Google Scholar] [CrossRef] [Green Version]
- Xia, K.; Wang, R.; Ou, X.; Fang, Z.; Tian, C.; Duan, J.; Wang, Y.; Zhang, M. OsTIR1 and OsAFB2 Downregulation via OsmiR393 Overexpression Leads to More Tillers, Early Flowering and Less Tolerance to Salt and Drought in Rice. PLoS ONE 2012, 7, e30039. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedländer, M.R.; MacKowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. MiRDeep2 Accurately Identifies Known and Hundreds of Novel MicroRNA Genes in Seven Animal Clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From MicroRNA Sequences to Function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Pan, X.; Cannon, C.H.; Cobb, G.P.; Anderson, T.A. Conservation and Divergence of Plant MicroRNA Genes. Plant J. 2006, 46, 243–259. [Google Scholar] [CrossRef]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for Annotation of Plant MicroRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef]
- Wan, L.C.; Wang, F.; Guo, X.; Lu, S.; Qiu, Z.; Zhao, Y.; Zhang, H.; Lin, J. Identification and Characterization of Small Non-Coding RNAs from Chinese Fir by High Throughput Sequencing. BMC Plant Biol. 2012, 12, 146. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. MiRDeepFinder: A MiRNA Analysis Tool for Deep Sequencing of Plant Small RNAs. Plant Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef]
- Dai, X.; Zhuang, Z.; Zhao, P.X. PsRNATarget: A Plant Small RNA Target Analysis Server (2017 Release). Nucleic Acids Res. 2018, 46, W49–W54. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, R.; Ramachandran, G.N.; El-Manzalawy, Y.; Honavar, V. Benjamini–Hochberg Method. In Encyclopedia of Systems Biology; Springer: New York, NY, USA, 2013; p. 78. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNA Family | Known miRNAs (Total Count) | Description of Target Gene |
|---|---|---|
| miR812 | osa-miR812a, b, c, d, e, f, g, h, i, j, k, l, m, n-5p, n-3p, o-5p, o-3p, p, s, t, u, and v (22) | GRAS family nuclear protein, Control of tillering (MOCI), Similar to serine/threonine protein kinase (OsCIPK10), Leucine-rich repeat, plant-specific protein, Similar to Na+/H+ antiporter, probable CP0838, Similar to 1-aminocyclopropane-1-carboxylate oxidase (Fragment) (ACO3), Similar to PDR-like ABC transporter (PDR4 ABC transporter). |
| miR166 | osa-miR166a-5p, a-3p, b-5p, b-3p, c-5p, c-3p, d-5p, d-3p, e-5p, f, g-3p, h-5p, h-3p, i-3p, j-5p, j-3p, k-3p, l-5p, l-3p, and m (20) | Class III homeodomain Leu zipper (HD-Zip III) family member, Control of plant type architecture and leaf development (OsHB4), Leucine-rich repeat, N-terminal domain-containing protein, Similar to Rolled leaf1 (OsHox1), Serine/threonine protein kinase domain-containing protein (OsWAK105), Chloroplast-targeted Deg protease protein, Chloroplast development and maintenance of PSII function under high temperature (OsDeg10). |
| miR156 | osa-miR156a, b-5p, b-3p, c-5p, c-3p, d, e, f-5p, f-3p, g-5p, g-3p, h-5p, h-3p, i, j-5p, j-3p, k, l-5p, and l-3p (19) | Positive regulator of cell proliferation, Control of grain size, shape, and quality (GW8), Squamosa promoter-binding-like transcription activator, Regulation of branching in panicles and vegetative shoots, Semi-dominant regulator of plant architecture (OsSPL). |
| miR1861 | osa-miR1861b, c, e, f, g, h, I, j, k, l, and m (11) | Glyoxalase I, Abiotic stress response (GLYI-11), Glycoside hydrolase, carbohydrate-binding domain-containing protein, pentatricopeptide repeat protein, Chloroplast development (YSA), Similar to guanine nucleotide-binding protein α-1 subunit, Similar to GTP-binding protein-like; root hair defective 3 protein-like, Leucine-rich repeat, N-terminal domain-containing protein. |
| miR167 | osa-miR167 a-5p, b, c-5p, d-5p, e-5p, f, g, h-5p, h-3p, i-5p, and j (11) | NB-ARC domain-containing protein, Similar to glutamate-gated kainate-type ion channel receptor subunit GluR5, pentatricopeptide repeat domain-containing protein, Short-chain dehydrogenase/reductase SDR family protein. |
| miR396 | osa-miR396 a-5p, a-3p, b-5p, b-3p, c-5p, d, e-5p, f-5p, g, and h (10) | Growth regulating factor members (OsGRF), WD40/YVTN repeat-like domain-containing protein, Transcription activator, Gibberellin (GA)-induced stem elongation, Growth-regulating factor, Regulation of grain shape and panicle length, Negative regulation of seed shattering, Auxin efflux carrier protein, Auxin transport, Drought tolerance |
| miR444 | osa-miR444 a-3p.2, b.2, b.1, c.2, c.1, d.3, d.2, e, and f (9) | MADS-box transcription factor, Cold tolerance, Control of tillering (OsMADS57), MADS-box transcription factor, Homologue of the AGL17-clade MADS-box genes, Regulation of root system development via auxin signaling (OsMADS25), Zinc finger, RING/FYVE/PHD-type domain-containing protein, pentatricopeptide repeat domain-containing protein. |
| miR159 | osa-miR159a.2, a.1, b, c, d, e, and f (7) | GAMYB-like protein, Flower development and stem elongation at the reproductive stage (OsGAMYBL1), Transcriptional activator of gibberellin-dependent α-amylase expression, Regulation of nutrient mobilization in germination (GAMYB), Similar to Coatomer protein complex, β prime; β′-COP protein (OsWD40-125). |
| miR395 | osa-miR395b, d, e, g, s, and y (6) | Similar to ATP sulfurylase (OsATPS), Similar to low-affinity sulfate transporter 3 (OsSultr2), Similar to Thioredoxin peroxidase (OsPrxII), Cytochrome b5 domain-containing protein (OsMSBP2), Sulphate permease. |
| miR160 | osa-miR160a-5p, b-5p, c-5p, d-5p, and e-5p (5) | Auxin response factors (OsARF10, 13), Transcriptional factor B3 family protein (OsARF22). |
| miR164 | osa-miR164a, b, d, e, and f (5) | NAC transcription factor is a positive regulator of heading and senescence during the reproductive phase (OsY37). Sugar transporter protein, Similar to Pollen-specific kinase partner protein. |
| miR393 | osa-miR393a and b-5p (2) | Auxin receptor, Flag leaf inclination, Primary root growth, Crown root initiation, Seed development, Tillering (OsAFB2), F-Box auxin receptor protein, Nuclear protein, Flag leaf inclination, Primary root growth, Crown root initiation, Seed development, Tillering (TIR1), Protein kinase, catalytic domain-containing protein (OsWAK7/8). |
| miR397 | osa-miR397a and b (2) | Heat shock protein (HSP40), Putative tetratricopeptide repeat (TPR)-containing protein, Growth and development, salt tolerance, abiotic stress tolerance (OsHsp40), Laccase (OsLAC2, 5, 7, 9, and 29). |
| miR398 | osa-miR398a and b (2) | Similar to Superoxide dismutase [Cu-Zn] (Cu/Zn-SOD), Selenium binding protein. |
| miR408 | osa-miR408-5p and 3p (2) | Similar to Auxin-responsive protein (Aux/IAA) (Fragment) (OsIAA30), Cupredoxin domain-containing protein (OsUCL30). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaur, S.; Seem, K.; Kumar, S.; Kaundal, R.; Mohapatra, T. Comparative Genome-Wide Analysis of MicroRNAs and Their Target Genes in Roots of Contrasting Indica Rice Cultivars under Reproductive-Stage Drought. Genes 2023, 14, 1390. https://doi.org/10.3390/genes14071390
Kaur S, Seem K, Kumar S, Kaundal R, Mohapatra T. Comparative Genome-Wide Analysis of MicroRNAs and Their Target Genes in Roots of Contrasting Indica Rice Cultivars under Reproductive-Stage Drought. Genes. 2023; 14(7):1390. https://doi.org/10.3390/genes14071390
Chicago/Turabian StyleKaur, Simardeep, Karishma Seem, Suresh Kumar, Rakesh Kaundal, and Trilochan Mohapatra. 2023. "Comparative Genome-Wide Analysis of MicroRNAs and Their Target Genes in Roots of Contrasting Indica Rice Cultivars under Reproductive-Stage Drought" Genes 14, no. 7: 1390. https://doi.org/10.3390/genes14071390