
Mosquito (MS), a DD37E Family of Tc1/Mariner, Displaying a Distinct Evolution Profile from DD37E/TRT and DD37E/L18

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DD37E/MS Mining
2.2. DD37E/MS Sequence Analyses
2.3. DD37E/MS Phylogenetic Analysis
2.4. DD37E/MS HT Analyses
3. Results and Discussion
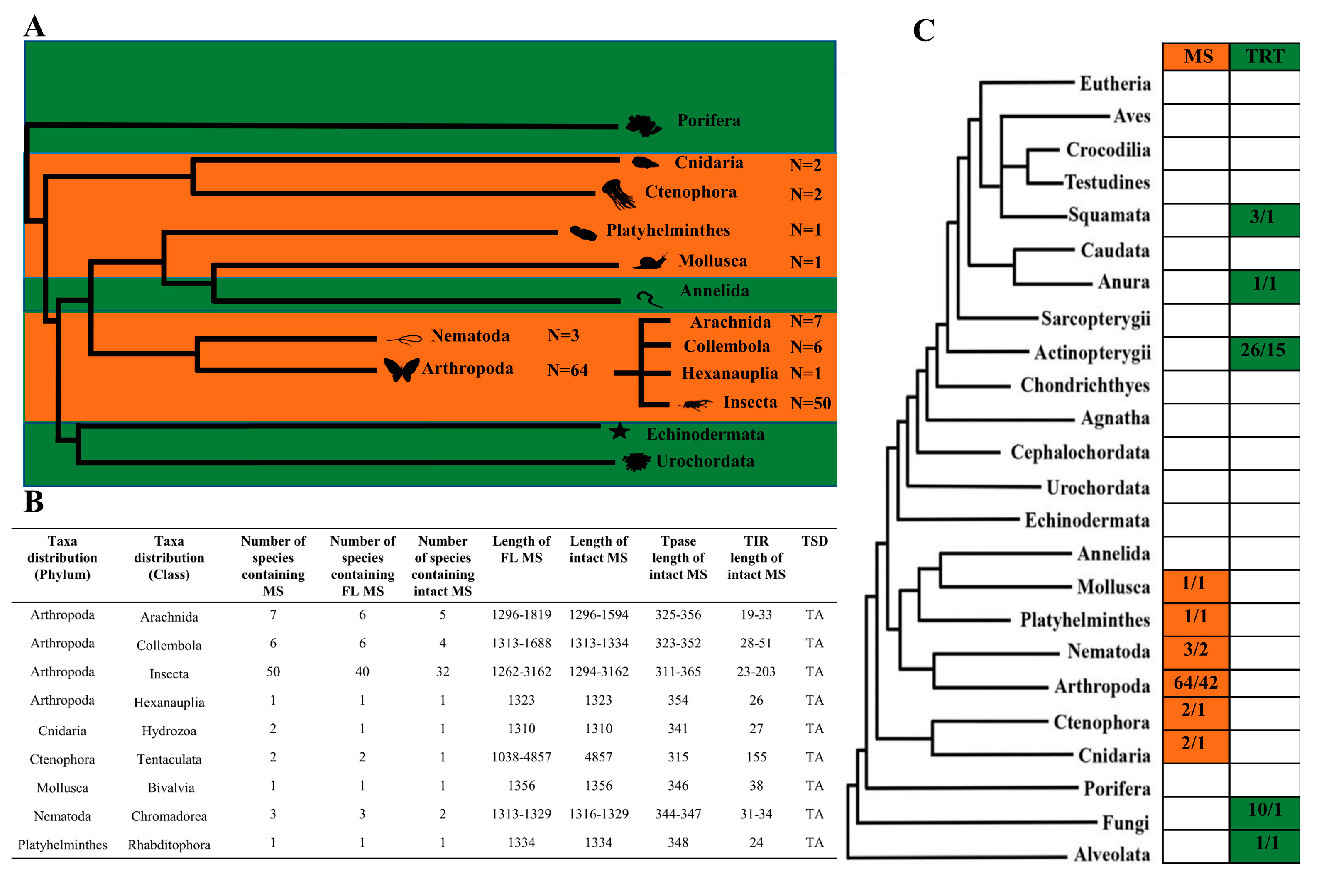
3.1. Abundance DD37E/MS Transposons
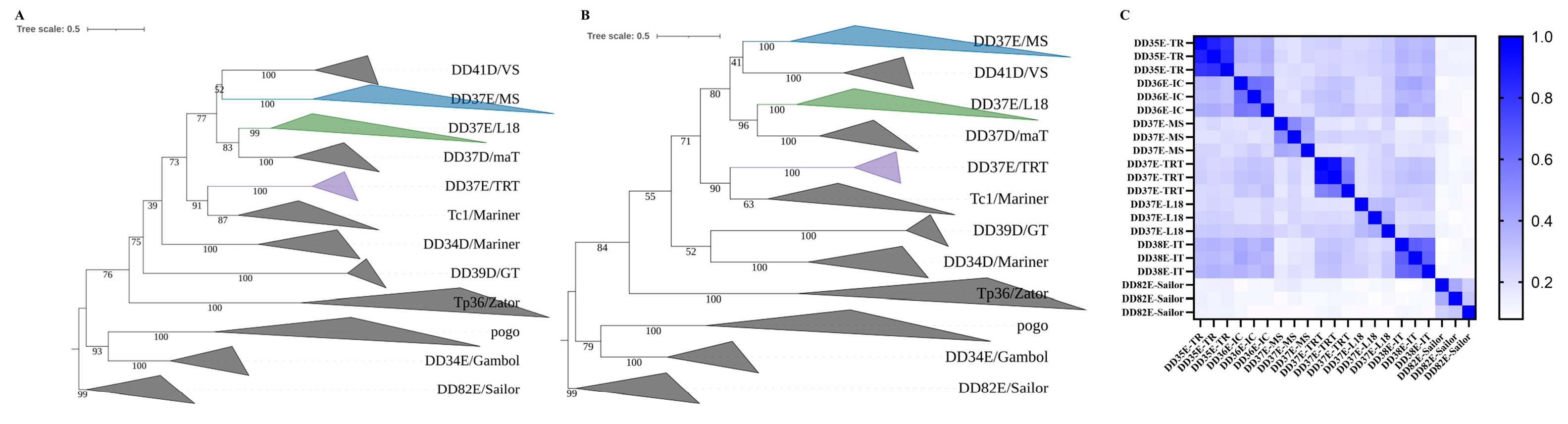
3.2. Phylogenetic Position of the DD37E/MS Family
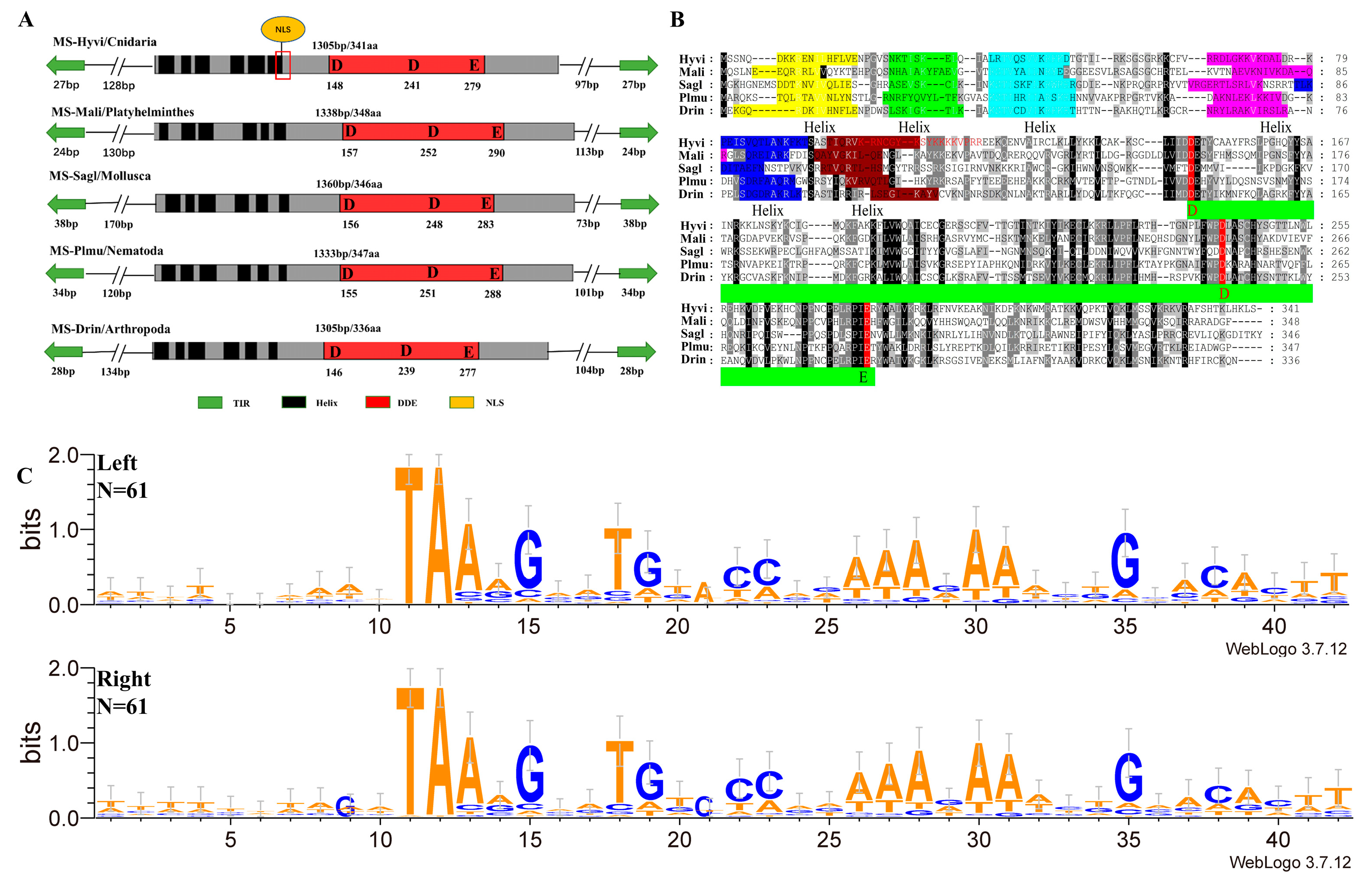
3.3. Structure of DD37E/MS Transposons
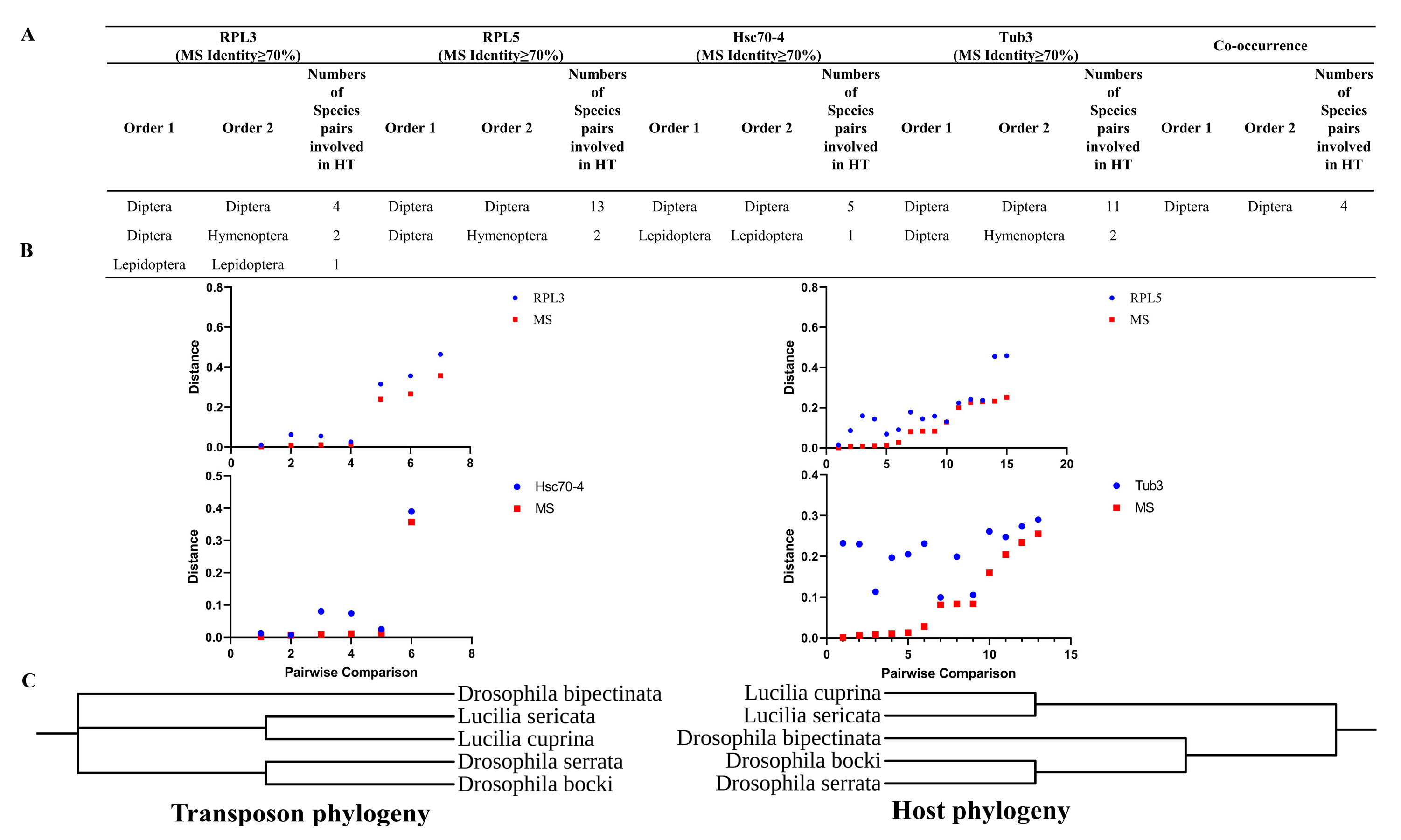
3.4. HT Analysis of DD37E/MS Transposons
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Kojima, K.K. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet. Syst. 2020, 94, 233–252. [Google Scholar] [CrossRef] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Wang, Y.; Diaby, M.; Zong, W.; Shen, D.; Wang, S.; Chen, C.; Wang, X.; Song, C. Evolution of pogo, a separate superfamily of IS630-Tc1-mariner transposons, revealing recurrent domestication events in vertebrates. Mob. DNA 2020, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Puzakov, M.; Guan, Z.; Xiang, K.; Diaby, M.; Wang, Y.; Wang, S.; Song, C.; Gao, B. Prokaryotic and Eukaryotic Horizontal Transfer of Sailor (DD82E), a New Superfamily of IS630-Tc1-Mariner DNA Transposons. Biology 2021, 10, 1005. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.W.; Wessler, S.R. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. USA 2011, 108, 7884–7889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tellier, M.; Bouuaert, C.C.; Chalmers, R. Mariner and the ITm Superfamily of Transposons. Mob. DNA III 2015, 3, 753–772. [Google Scholar] [CrossRef] [Green Version]
- Majorek, K.A.; Dunin-Horkawicz, S.; Steczkiewicz, K.; Muszewska, A.; Nowotny, M.; Ginalski, K.; Bujnicki, J.M. The RNase H-like superfamily: New members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014, 42, 4160–4179. [Google Scholar] [CrossRef]
- Doak, T.G.; Doerder, F.P.; Jahn, C.L.; Herrick, G. A proposed superfamily of transposase genes: Transposon-like elements in ciliated protozoa and a common “D35E” motif. Proc. Natl. Acad. Sci. USA 1994, 91, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Shao, H.; Tu, Z. Expanding the diversity of the IS630-Tc1-mariner superfamily: Discovery of a unique DD37E transposon and reclassification of the DD37D and DD39D transposons. Genetics 2001, 159, 1103–1115. [Google Scholar] [CrossRef]
- Shen, D.; Gao, B.; Miskey, C.; Chen, C.; Sang, Y.; Zong, W.; Wang, S.; Wang, Y.; Wang, X.; Ivics, Z.; et al. Multiple Invasions of Visitor, a DD41D Family of Tc1/mariner Transposons, throughout the Evolution of Vertebrates. Genome Biol. Evol. 2020, 12, 1060–1073. [Google Scholar] [CrossRef]
- Wang, S.; Diaby, M.; Puzakov, M.; Ullah, N.; Wang, Y.; Danley, P.; Chen, C.; Wang, X.; Gao, B.; Song, C. Divergent evolution profiles of DD37D and DD39D families of Tc1/mariner transposons in eukaryotes. Mol. Phylogenetics Evol. 2021, 161, 107143. [Google Scholar] [CrossRef]
- Puzakov, M.V.; Puzakova, L.V. Prevalence, Diversity, and Evolution of L18 (DD37E) Transposons in the Genomes of Cnidarians. Mol. Biol. 2022, 56, 476–490. [Google Scholar] [CrossRef]
- Zhang, H.H.; Li, G.Y.; Xiong, X.M.; Han, M.J.; Zhang, X.G.; Dai, F.Y. TRT, a Vertebrate and Protozoan Tc1-Like Transposon: Current Activity and Horizontal Transfer. Genome Biol. Evol. 2016, 8, 2994–3005. [Google Scholar] [CrossRef] [Green Version]
- Puzakov, M.V.; Puzakova, L.V.; Cheresiz, S.V. An Analysis of IS630/Tc1/mariner Transposons in the Genome of a Pacific Oyster, Crassostrea gigas. J. Mol. Evol. 2018, 86, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Puzakov, M.V.; Puzakova, L.V.; Cheresiz, S.V.; Sang, Y. The IS630/Tc1/mariner transposons in three ctenophore genomes. Mol. Phylogenetics Evol. 2021, 163, 107231. [Google Scholar] [CrossRef]
- Guan, Z.; Shi, S.; Diaby, M.; Danley, P.; Ullah, N.; Puzakov, M.; Gao, B.; Song, C. Horizontal transfer of Buster transposons across multiple phyla and classes of animals. Mol. Phylogenetics Evol. 2022, 173, 107506. [Google Scholar] [CrossRef] [PubMed]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumenstiel, J.P. Birth, School, Work, Death, and Resurrection: The Life Stages and Dynamics of Transposable Element Proliferation. Genes 2019, 10, 336. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, K.; Hartl, D.L. Evidence for interspecific transfer of the transposable element mariner betweenDrosophila andZaprionus. J. Mol. Evol. 1991, 33, 514–524. [Google Scholar] [CrossRef]
- Filée, J.; Rouault, J.D.; Harry, M.; Hua-Van, A. Mariner transposons are sailing in the genome of the blood-sucking bug Rhodnius prolixus. BMC Genom. 2015, 16, 1061. [Google Scholar] [CrossRef] [Green Version]
- Sanllorente, O.; Vela, J.; Mora, P.; Ruiz-Mena, A.; Torres, M.I.; Lorite, P.; Palomeque, T. Complex Evolutionary History of Mboumar, a Mariner Element Widely Represented in Ant Genomes. Sci. Rep. 2020, 10, 2610. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.L.; Lozovskaya, E.R.; Nurminsky, D.I.; Lohe, A.R. What restricts the activity of mariner-like transposable elements. Trends Genet. 1997, 13, 197–201. [Google Scholar] [CrossRef]
- Claudianos, C.; Brownlie, J.; Russell, R.; Oakeshott, J.; Whyard, S. maT—A clade of transposons intermediate between mariner and Tc1. Mol. Biol. Evol. 2002, 19, 2101–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-H.; Shen, Y.-H.; Xiong, X.-M.; Han, M.-J.; Zhang, X.-G. Identification and evolutionary history of the DD41D transposons in insects. Genes Genom. 2016, 38, 109–117. [Google Scholar] [CrossRef]
- Puzakov, M.V.; Puzakova, L.V.; Cheresiz, S.V. The Tc1-like elements with the spliceosomal introns in mollusk genomes. Mol. Genet. Genom. 2020, 295, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Izsvák, Z. Sleeping Beauty Transposition. Mob. DNA III 2015, 3, 851–872. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Plasterk, R.H.; Izsvák, Z.; Ivics, Z. Resident aliens: The Tc1/mariner superfamily of transposable elements. Trends Genet. 1999, 15, 326–332. [Google Scholar] [CrossRef]
- Zhang, H.H.; Shen, Y.H.; Xu, H.E.; Liang, H.Y.; Han, M.J.; Zhang, Z. A novel hAT element in Bombyx mori and Rhodnius prolixus: Its relationship with miniature inverted repeat transposable elements (MITEs) and horizontal transfer. Insect Mol. Biol. 2013, 22, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.O. Lateral gene transfer in eukaryotes. Cell. Mol. Life Sci. 2005, 62, 1182–1197. [Google Scholar] [CrossRef] [PubMed]
- Cummings, M.P. Transmission patterns of eukaryotic transposable elements: Arguments for and against horizontal transfer. Trends Ecol. Evol. 1994, 9, 141–145. [Google Scholar] [CrossRef]
- Wallau, G.L.; Ortiz, M.F.; Loreto, E.L. Horizontal transposon transfer in eukarya: Detection, bias, and perspectives. Genome Biol. Evol. 2012, 4, 689–699. [Google Scholar] [CrossRef] [Green Version]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, K.; Puzakov, M.; Shi, S.; Diaby, M.; Ullah, N.; Gao, B.; Song, C. Mosquito (MS), a DD37E Family of Tc1/Mariner, Displaying a Distinct Evolution Profile from DD37E/TRT and DD37E/L18. Genes 2023, 14, 1379. https://doi.org/10.3390/genes14071379
Xiang K, Puzakov M, Shi S, Diaby M, Ullah N, Gao B, Song C. Mosquito (MS), a DD37E Family of Tc1/Mariner, Displaying a Distinct Evolution Profile from DD37E/TRT and DD37E/L18. Genes. 2023; 14(7):1379. https://doi.org/10.3390/genes14071379
Chicago/Turabian StyleXiang, Kuilin, Mikhail Puzakov, Shasha Shi, Mohamed Diaby, Numan Ullah, Bo Gao, and Chengyi Song. 2023. "Mosquito (MS), a DD37E Family of Tc1/Mariner, Displaying a Distinct Evolution Profile from DD37E/TRT and DD37E/L18" Genes 14, no. 7: 1379. https://doi.org/10.3390/genes14071379