
Insights into the Adaptation to High Altitudes from Transcriptome Profiling: A Case Study of an Endangered Species, Kingdonia uniflora
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction Library Construction and Sequencing
2.3. Sequencing of All Samples and Filtering of Clean Reads
- (1)
- Removing reads containing adapters;
- (2)
- Removing reads containing polyA and polyG;
- (3)
- Removing reads containing more than 5% of unknown nucleotides (N);
- (4)
- Removing low quality reads containing more than 20% of low-quality (q-value ≤ 20) bases. Then, sequence quality was verified using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, 0.11.9), including the Q20, Q30, and GC-content of the clean data. After that, a total of approximately 6G bp of cleaned, paired-end reads were produced for each sample; detailed information is given in Supplemental Table S1. We submitted the raw sequence data to the NCBI Sequence Read Archive (SRA) database with accession number PRJNA971146.
2.4. Alignment with the Reference Genome
2.5. Quantification of Gene Abundance
2.6. Differentially Expressed Gene (DEG) Analysis
2.7. Relationship Analysis of Samples
2.8. GO Enrichment Analysis
2.9. Pathway Enrichment Analysis (KEGG)
2.10. Gene Set Enrichment Analysis (GSEA)
3. Results
3.1. Transcriptome Data of 18 Samples and Mapping Information
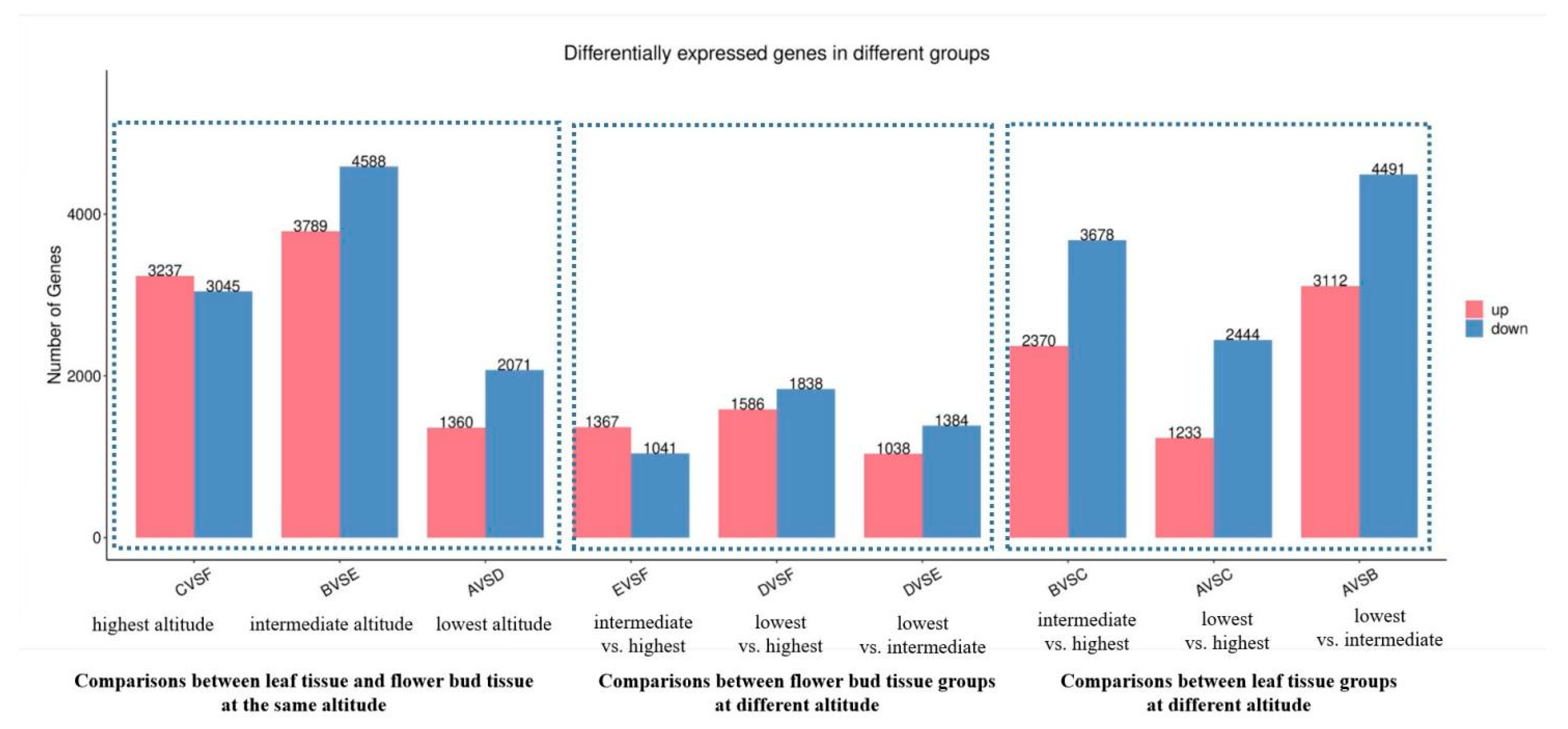
3.2. Differentially Expressed Genes (DEGs) Detected from Leaf or Flower Bud Tissue at Different Altitudes
3.3. Differentially Expressed Genes (DEGs) Detected from Pairwise Comparison of Leaf and Flower Bud Tissue at the Same Altitude
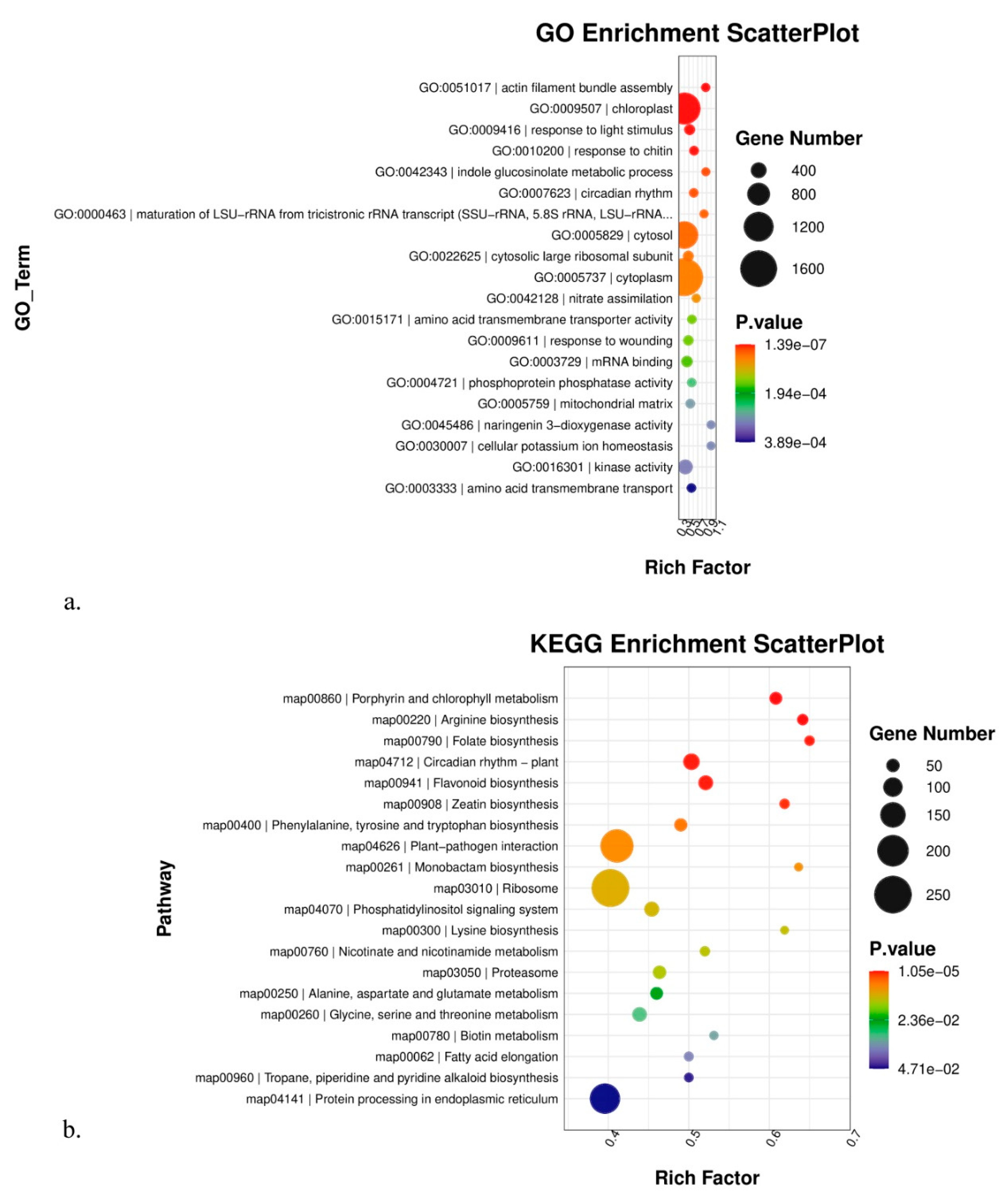
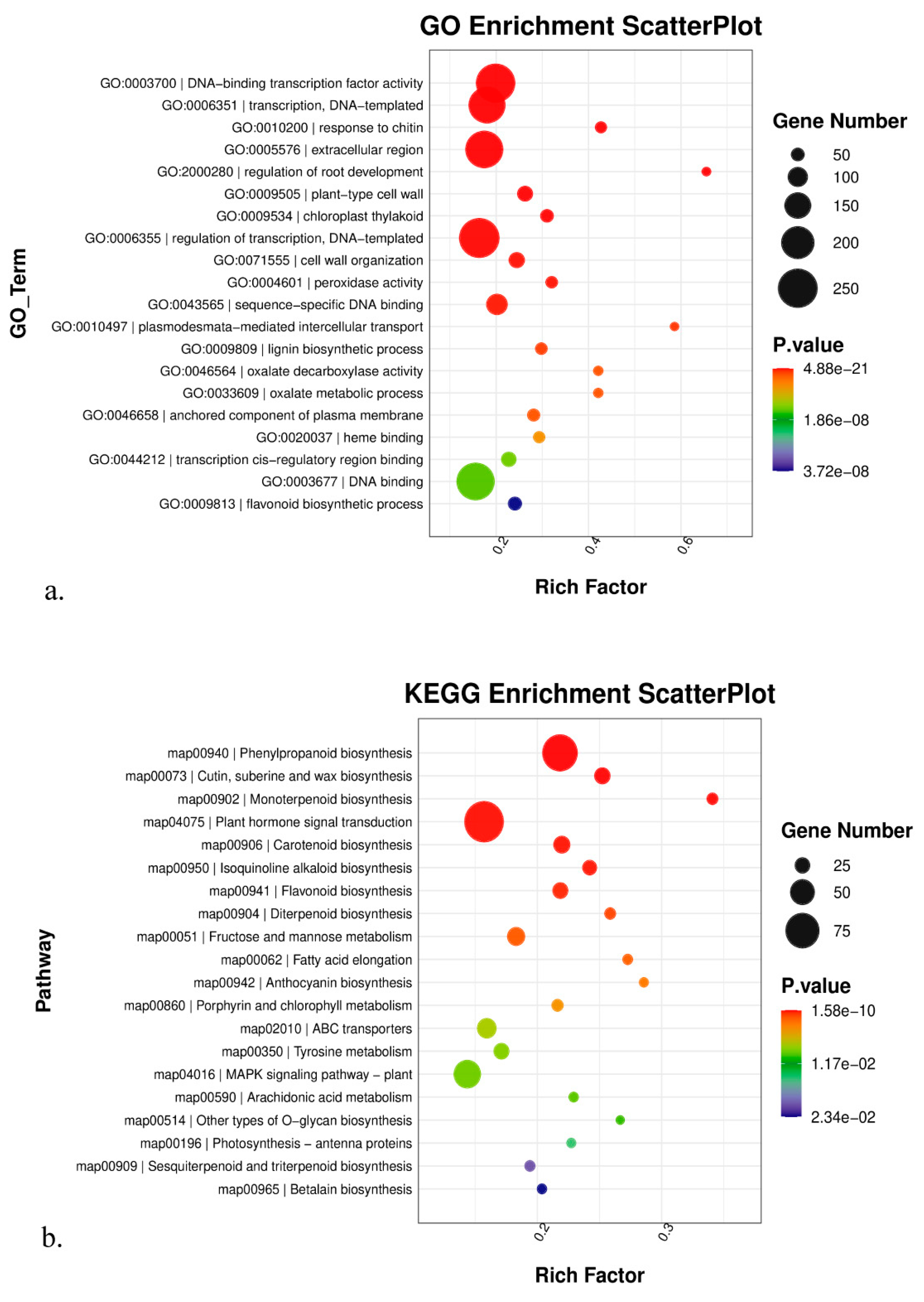
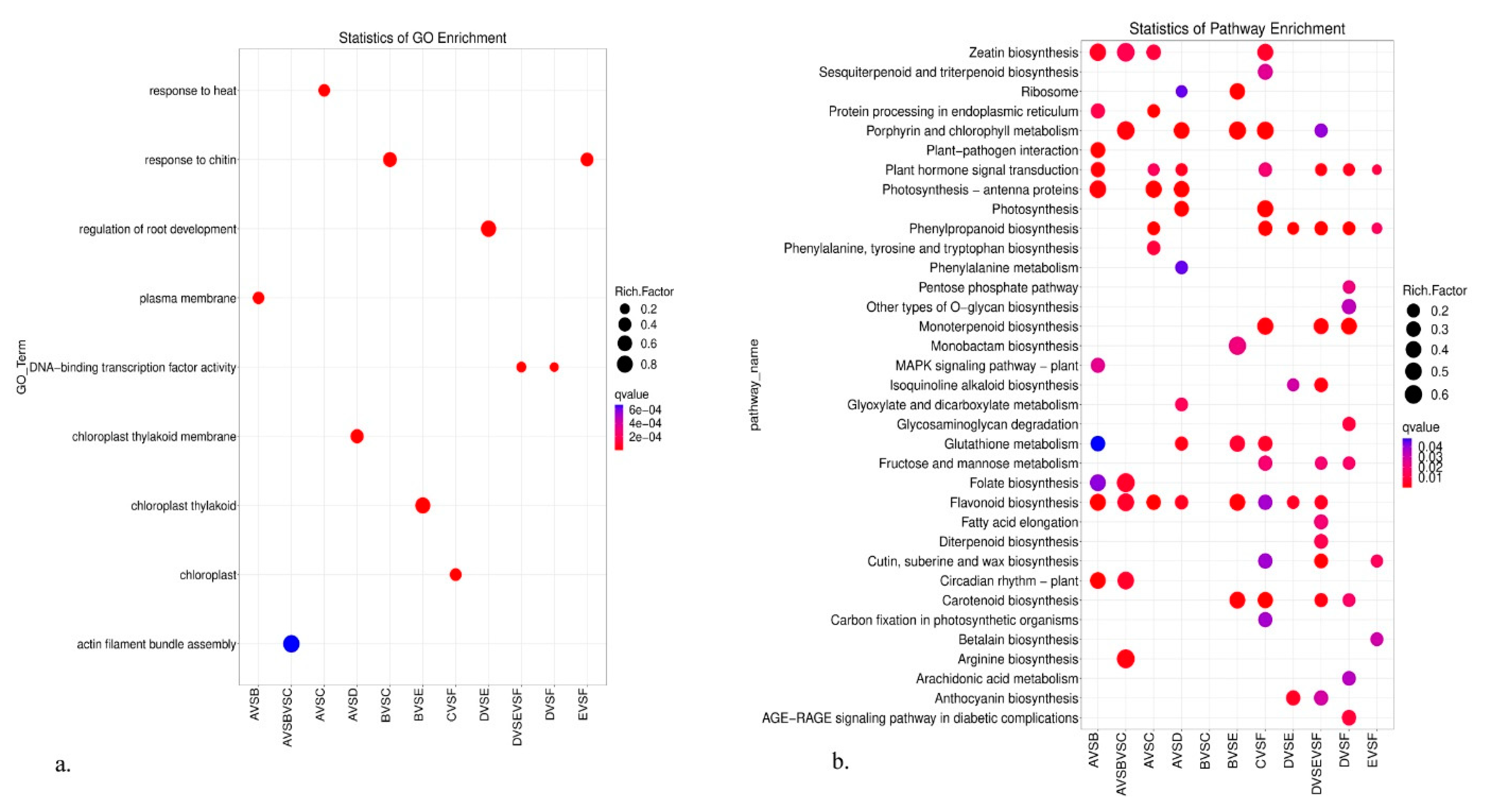
3.4. GO, KEGG, and Gsea Enrichment Analyses of DEGs
4. Discussion
4.1. Molecular Mechanism Underlying the High-Altitude Adaptation of K. uniflora
4.2. Response to Altitude Variation in Different Tissues of K. uniflora
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Tian, Y.; Yan, L.; Zhang, G.; Wang, X.; Zeng, Y.; Zhang, J.; Ma, X.; Tan, Y.; Long, N.; et al. Genome of Plant Maca (Lepidium meyenii) Illuminates genomic basis for high-altitude adaptation in the central Andes. Mol. Plant. 2016, 9, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.B.; Fu, T.T.; Jin, J.Q.; Murphy, R.W.; Hillis, D.M.; Zhang, Y.P.; Che, J. Species groups distributed across elevational gradients reveal convergent and continuous genetic adaptation to high elevations. Proc. Natl. Acad. Sci. USA 2018, 115, E10634–E10641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.Y.; Hu, Q.J.; Hao, G.Q.; Wang, X.; Zhang, D.; Ma, T.; Liu, J. The genomes of two eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 2018, 25, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Qiao, Q.; Novikova, P.Y.; Wang, Q.; Yue, J.; Guan, Y.; Ming, S.; Liu, T.; De, J.; Liu, Y.; et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc. Natl. Acad. Sci. USA 2019, 116, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Li, M.L.; Yue, M.; Zhao, Z.; Zhao, G.F.; Li, Z.H. Comparative transcriptome analysis reveals adaptive evolution of Notopterygium incisum and Notopterygium franchetii, two high-alpine herbal species endemic to China. Molecules 2017, 22, 1158. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Sun, Y.X.; Landis, J.B.; Shen, J.; Zhang, H.; Juang, T.; Sun, W.; Sun, J.; Tiamiyu, B.B.; Deng, T.; et al. Transcriptomes of Saussurea (Asteraceae) provide insights into high-altitude adaptation. Plants 2021, 10, 1715. [Google Scholar] [CrossRef]
- Angiosperm Phylogeny Group. An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 2016, 181, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Deng, T.; Zhang, A.; Moore, M.J.; Landis, J.B.; Lin, N.; Zhang, H.; Zhang, X.; Huang, J.; Zhang, X.; et al. Genome sequencing of the endangered Kingdonia uniflora (circaeasteraceae, ranunculales) reveals potential mechanisms of evolutionary specialization. IScience 2020, 23, 101124. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBNET J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Thompson, O.; von Meyenn, F.; Hewitt, Z.; Alexander, J.; Wood, A.; Weightman, R.; Gregory, S.; Krueger, F.; Andrews, S.; Barbaric, I.; et al. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nat. Commun. 2020, 11, 1528. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol 2015, 33, 290–295. [Google Scholar]
- Sahraeian, S.M.E.; Mohiyuddin, M.; Sebra, R.; Tilgner, H.; Afshar, P.T.; Au, K.F.; Lam, H.Y. Gaining comprehensive biological insight into the transcriptome by performing a broad-spectrum RNA-seq analysis. Nat. Commun. 2017, 8, 59. [Google Scholar] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, 545–551. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheviron, Z.A.; Brumfield, R.T. Genomic insights into adaptation to high- altitude environments. Heredity 2012, 108, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.K.; Lin, W.D.; Yu, B.B.; Zhu, J.; Li, J. Integrated metabolomic and transcriptomic analysis of the flavonoid accumulation in the leaves of Cyclocarya paliurus at different altitudes. Front. Plant Sci. 2022, 12, 794137. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, J.P.; Liu, L.K.; Jing, H.; Zuo, W.; Zeng, Y. Transcriptome analysis provides insights into Potentilla bifurca adaptation to high altitude. Life 2022, 12, 1337. [Google Scholar] [CrossRef] [PubMed]
- Creux, N.; Harmer, S. Circadian rhythms in plants. Cold Spring Harb. Perspect. Biol. 2019, 11, a034611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, L. Transport barriers made of cutin, suberin and associated waxes. Trends Plant Sci. 2010, 15, 546–553. [Google Scholar] [CrossRef]
- Franke, R.; Schreiber, L. Suberin—A biopolyester forming apoplastic plant interfaces. Curr. Opin. Plant Biol. 2007, 10, 252–259. [Google Scholar] [CrossRef]
- Guo, N.; Gao, J.; He, Y.; Guo, Y. Compositae plants differed in leaf cuticular waxes between high and low altitudes. Chem. Biodivers. 2016, 13, 710–718. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, L.; Zhang, M.; Wei, Y.; Lin, W. Recent advances on feasible strategies for monoterpenoid production in saccharomyces cerevisiae. Front. Bioeng. Biotechnol. 2020, 8, 609800. [Google Scholar] [CrossRef]
- Shi, Y.; Su, Z.; Yang, H.; Wang, W.; Jin, G.; He, G.; Siddique, A.N.; Zhang, L.; Zhu, A.; Xue, R.; et al. Alternative splicing coupled to nonsense-mediated mrna decay contributes to the high-altitude adaptation of maca (Lepidium meyenii). Gene 2019, 694, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.D.; Upadhyay, A.K.; Bhardwaj, P.K.; Sowdhamini, R.; Ramakrishnan, U. Transcriptome analysis reveals plasticity in gene regulation due to environmental cues in Primula sikkimensis, a high altitude plant species. BMC Genom. 2019, 20, 989. [Google Scholar] [CrossRef] [PubMed]
- Rathore, N.; Kumar, P.; Mehta, N.; Swarnkar, M.K.; Shankar, R.; Chawla, A. Time-series RNA-seq transcriptome profiling reveals novel insights about cold acclimation and de-acclimation processes in an evergreen shrub of high altitude. Sci. Rep. 2022, 12, 15553. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group Name | Location | Latitude | Longitude | Altitude (m) | Tissue Type |
|---|---|---|---|---|---|
| A | Honghegu, Meixian, Shaanxi province | 34°0′46″ | 107°47′26″ | 2346 | leaf |
| D | Honghegu, Meixian, Shaanxi province | 34°0′46″ | 107°47′26″ | 2346 | flower bud |
| B | Xiabansi, Meixian, Shaanxi province | 33°42′11″ | 107°46′53″ | 2771 | leaf |
| E | Xiabansi, Meixian, Shaanxi province | 33°42′11″ | 107°46′53″ | 2771 | flower bud |
| C | Fangyangsi, Meixian, Shaanxi province | 33°58′28″ | 107°46′15″ | 3294 | leaf |
| F | Fangyangsi, Meixian, Shaanxi province | 33°58′28″ | 107°46′15″ | 3294 | flower bud |
| Gene Name | p-Value | q-Value | Putative Function |
|---|---|---|---|
| GIB67_027134 | 0 | 0 | Response to heat, 17.3 kDa class II heat shock protein |
| GIB67_005997 | 0 | 0 | Response to heat, Small heat shock protein HSP |
| GIB67_007978 | 0 | 0 | Response to heat, 17.1 kDa class II heat shock protein-like |
| GIB67_028015 | 0 | 0 | Response to heat, HSP20 domain-containing protein |
| GIB67_035570 | 0 | 0 | Response to heat, 17.3 kDa class II heat shock protein |
| GIB67_023343 | 0 | 0 | Response to cold, ACT domain-containing protein DS12, chloroplastic-like |
| GIB67_027084 | 0 | 0 | Response to cold, cold-inducible protein |
| GIB67_027089 | 0 | 0 | Response to cold, early light induced protein 2 |
| GIB67_027141 | 0 | 0 | Response to cold, Chlorophyll A-B binding protein |
| GIB67_033329 | 0 | 0 | Response to cold, photosystem I chlorophyll a/b-binding protein 3-1, chloroplastic |
| GIB67_042678 | 0 | 0 | Response to water deprivation, hypothetical protein AQUCO_00200416v1 |
| GIB67_023028 | 0 | 0 | Response to water deprivation, Stress-related protein |
| GIB67_035316 | 0 | 0 | Response to water deprivation, plasma membrane-associated cation-binding protein 1 |
| GIB67_037972 | 0 | 0 | Response to water deprivation, hypothetical protein AQUCO_08400041v1 |
| GIB67_017844 | 0 | 0 | Cellular response to hypoxia, lignin-forming anionic peroxidase |
| GIB67_017843 | 0 | 0 | Cellular response to hypoxia, lignin-forming anionic peroxidase |
| GIB67_007868 | 0 | 0 | Cellular response to hypoxia, lignin-forming anionic peroxidase |
| GIB67_035929 | 0 | 0 | Cellular response to hypoxia, lignin-forming anionic peroxidase |
| GIB67_035933 | 0 | 0 | Cellular response to hypoxia, lignin-forming anionic peroxidase |
| GIB67_016048 | 0 | 0 | Response to light stimulus, PREDICTED: metacaspase-4 |
| GIB67_025148 | 0 | 0 | Response to light stimulus, Chlorophyll A-B binding protein |
| GIB67_026789 | 0 | 0 | Response to light stimulus, PREDICTED: chlorophyll a-b binding protein of LHCII type 1-like |
| GIB67_026779 | 0 | 0 | Response to light stimulus, glyceraldehyde-3-phosphate dehydrogenase B, chloroplastic |
| GIB67_000124 | 0 | 0 | Response to light stimulus, β tubulin1 |
| GIB67_034645 | 0 | 0 | Response to high light intensity, heat shock 70 kDa protein |
| GIB67_031347 | 0 | 0 | Response to high light intensity, hypothetical protein AQUCO_00800081v1 |
| GIB67_025134 | 0 | 0 | Response to high light intensity, Heat shock protein 70 family |
| GIB67_040467 | 0 | 0 | Response to high light intensity, Heat shock protein 70 family |
| GIB67_019867 | 0 | 0 | Response to high light intensity, small heat shock protein, chloroplastic-like |
| GIB67_001312 | 0 | 0 | Circadian rhythm—plant, phytochrome B |
| GIB67_007364 | 0 | 0 | Circadian rhythm—plant, Cyclic dof factor 2 |
| GIB67_001069 | 0 | 0 | Circadian rhythm—plant, zinc finger protein |
| GIB67_035301 | 0 | 0 | Circadian rhythm—plant, Chal_sti_synt_N domain-containing protein |
| GIB67_029338 | 0 | 0 | Circadian rhythm—plant, Basic-leucine zipper domain |
| GIB67_008159 | 0.02 | 0.04 | Cutin, suberine and wax biosynthesis, fatty acyl-CoA reductase 3-like |
| GIB67_019375 | 0.02 | 0.03 | Cutin, suberine and wax biosynthesis, omega- hydroxypalmitate O-feruloyl transferase |
| GIB67_042139 | 0 | 0 | Cutin, suberine and wax biosynthesis, Fatty acid hydroxylase |
| GIB67_038019 | 0 | 0 | Cellular response to nitrogen starvation, |
| GIB67_011552 | 0 | 0 | Cellular response to nitrogen starvation, |
| GIB67_026985 | 0 | 0 | Cellular response to nitrogen starvation, |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nong, M.-L.; Luo, X.-H.; Zhu, L.-X.; Zhang, Y.-N.; Dun, X.-Y.; Huang, L. Insights into the Adaptation to High Altitudes from Transcriptome Profiling: A Case Study of an Endangered Species, Kingdonia uniflora. Genes 2023, 14, 1291. https://doi.org/10.3390/genes14061291
Nong M-L, Luo X-H, Zhu L-X, Zhang Y-N, Dun X-Y, Huang L. Insights into the Adaptation to High Altitudes from Transcriptome Profiling: A Case Study of an Endangered Species, Kingdonia uniflora. Genes. 2023; 14(6):1291. https://doi.org/10.3390/genes14061291
Chicago/Turabian StyleNong, Man-Li, Xiao-Hui Luo, Li-Xin Zhu, Ya-Nan Zhang, Xue-Yi Dun, and Lei Huang. 2023. "Insights into the Adaptation to High Altitudes from Transcriptome Profiling: A Case Study of an Endangered Species, Kingdonia uniflora" Genes 14, no. 6: 1291. https://doi.org/10.3390/genes14061291