
Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genetic Analysis
2.2. Clinical Analysis
3. Results
3.1. Molecular Results
3.2. Review of Known WDR19 Variants and Their Associated Phenotypes
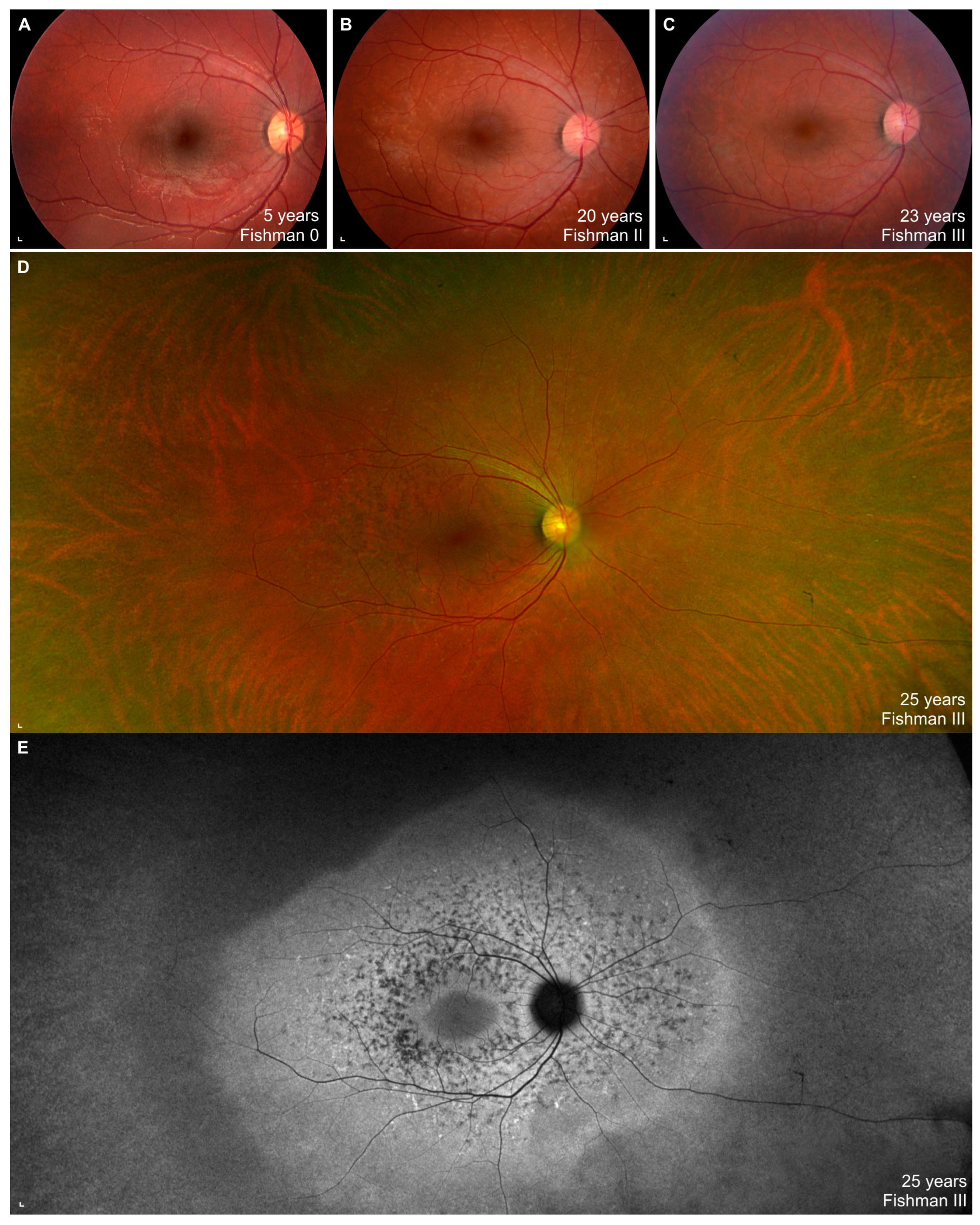
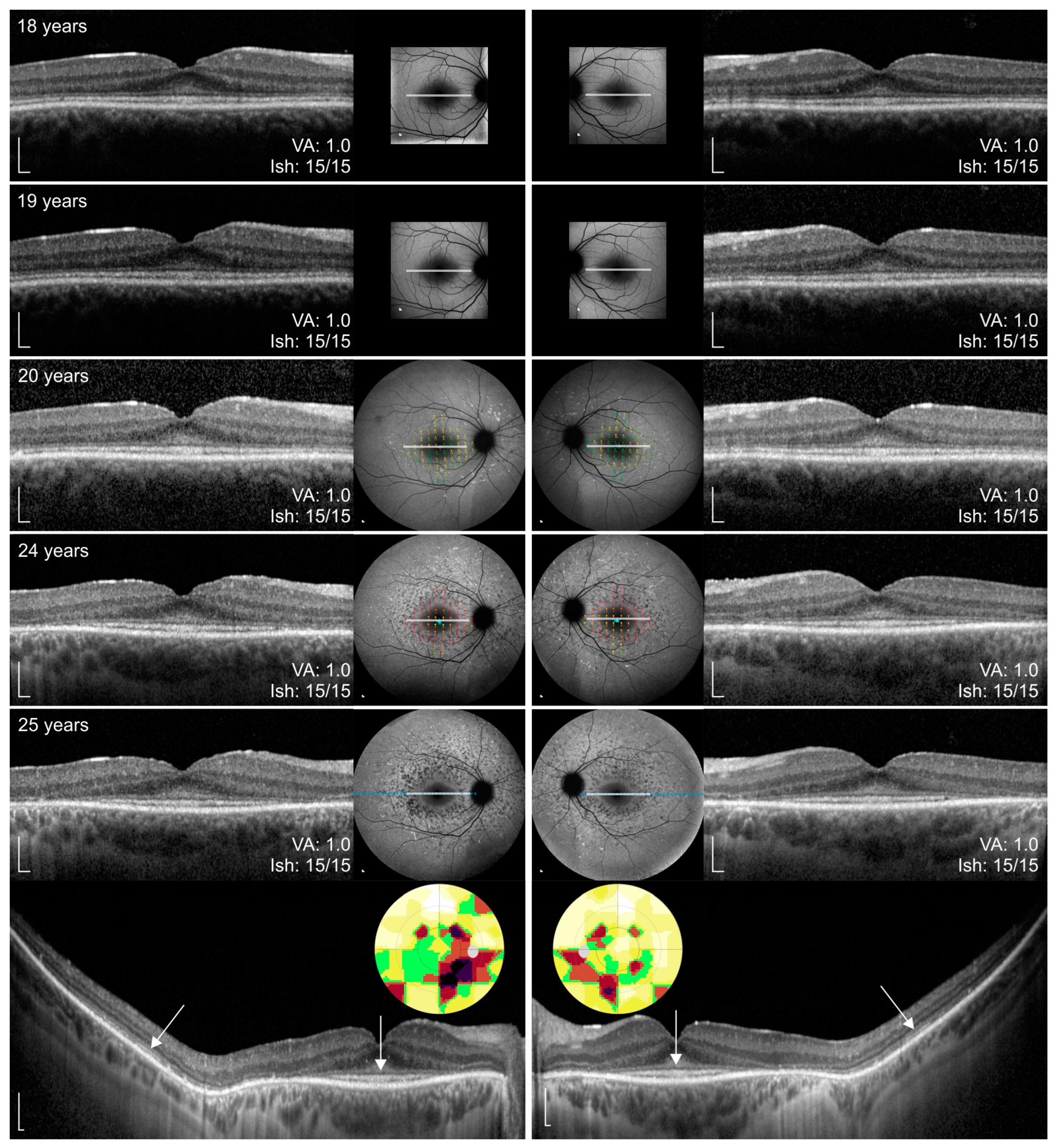
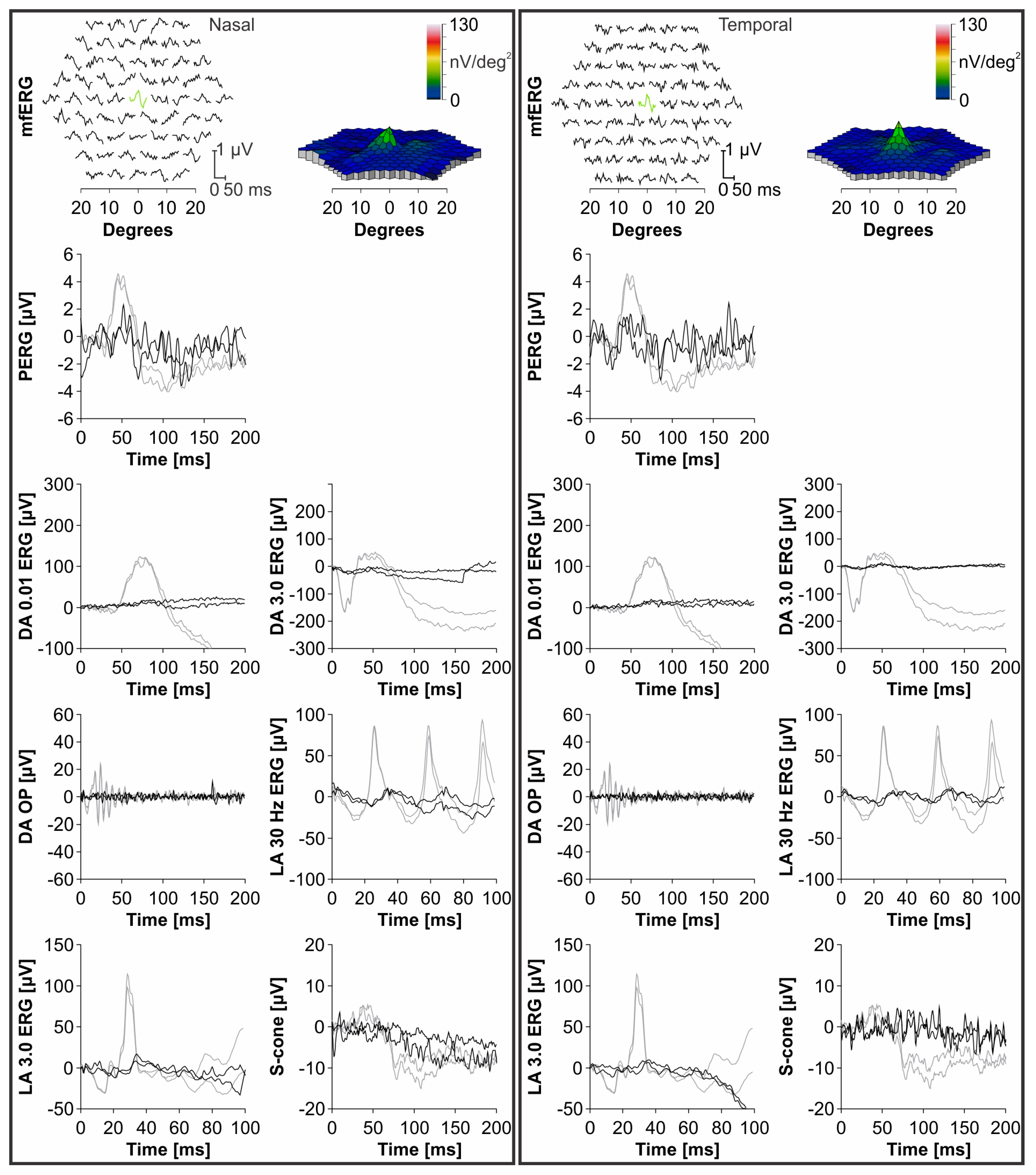
3.3. Clinical Data
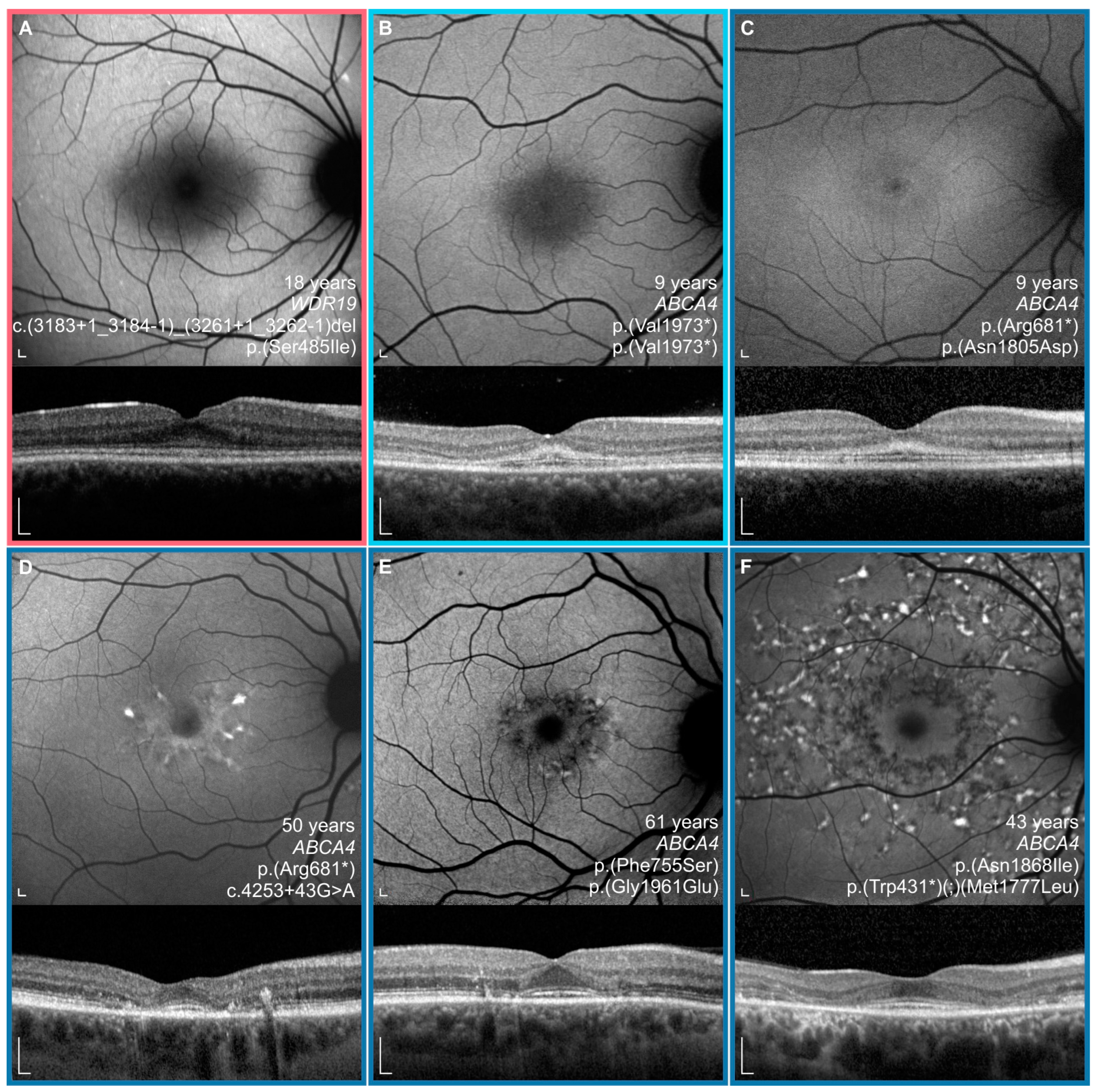
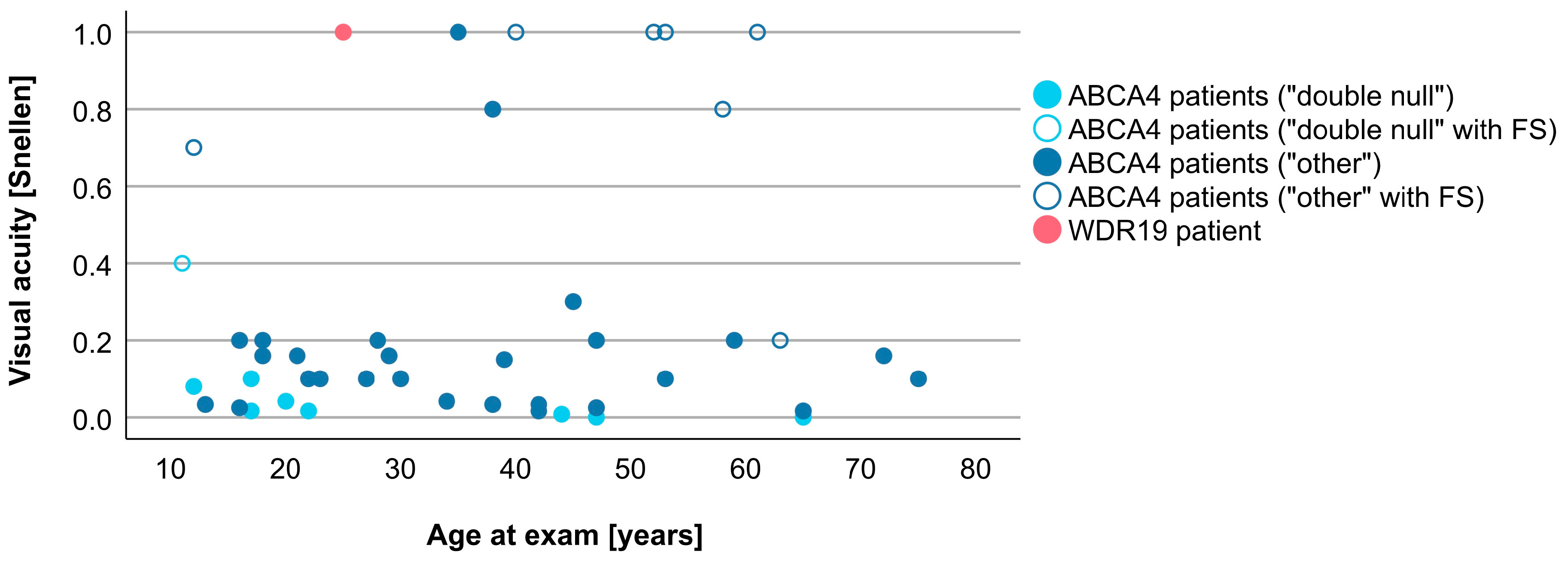
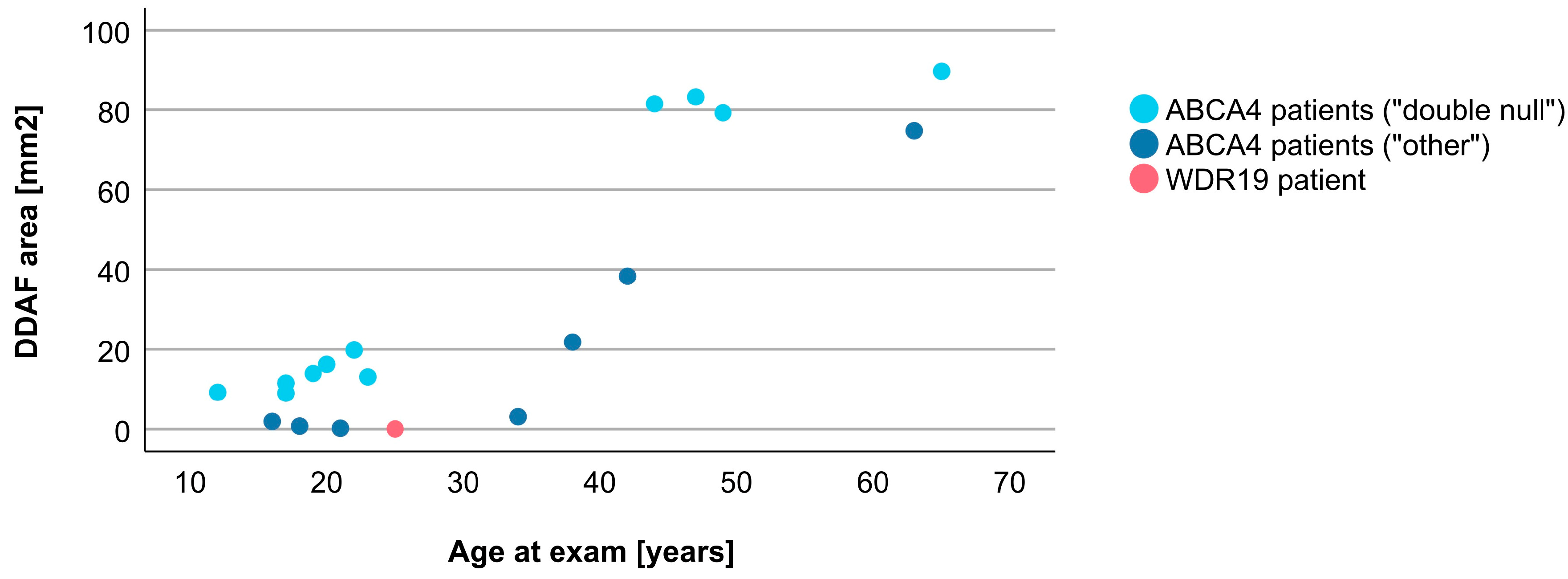
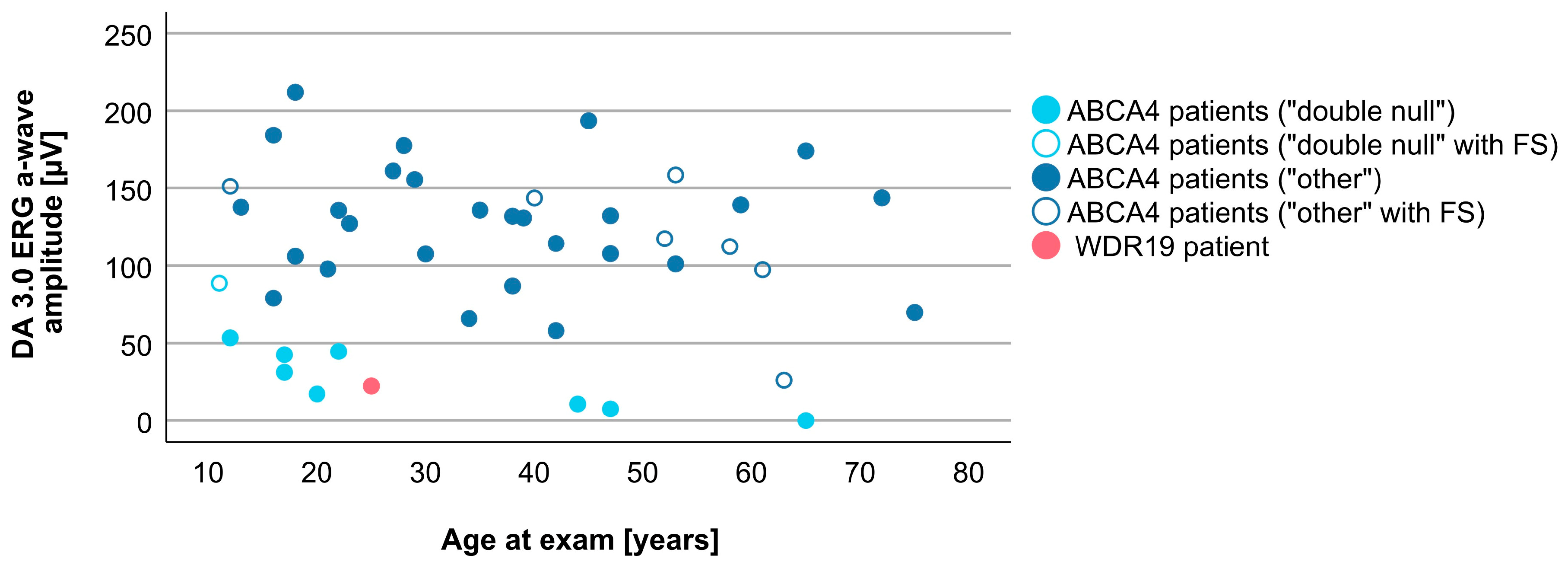
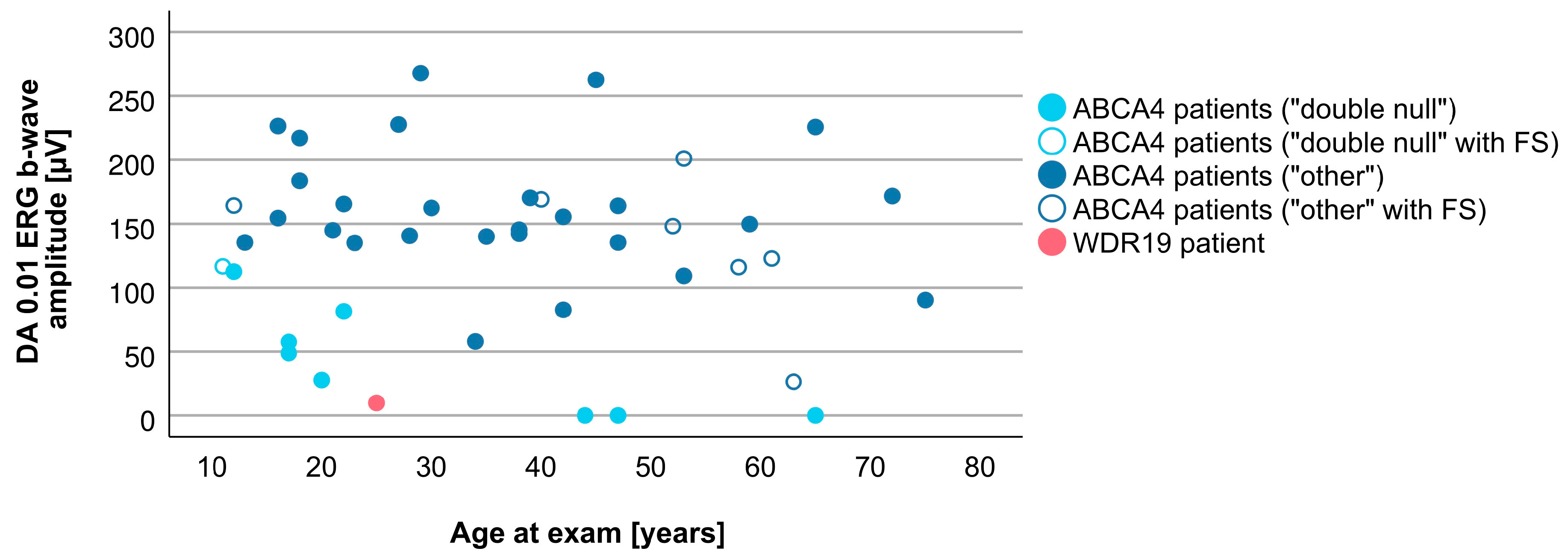
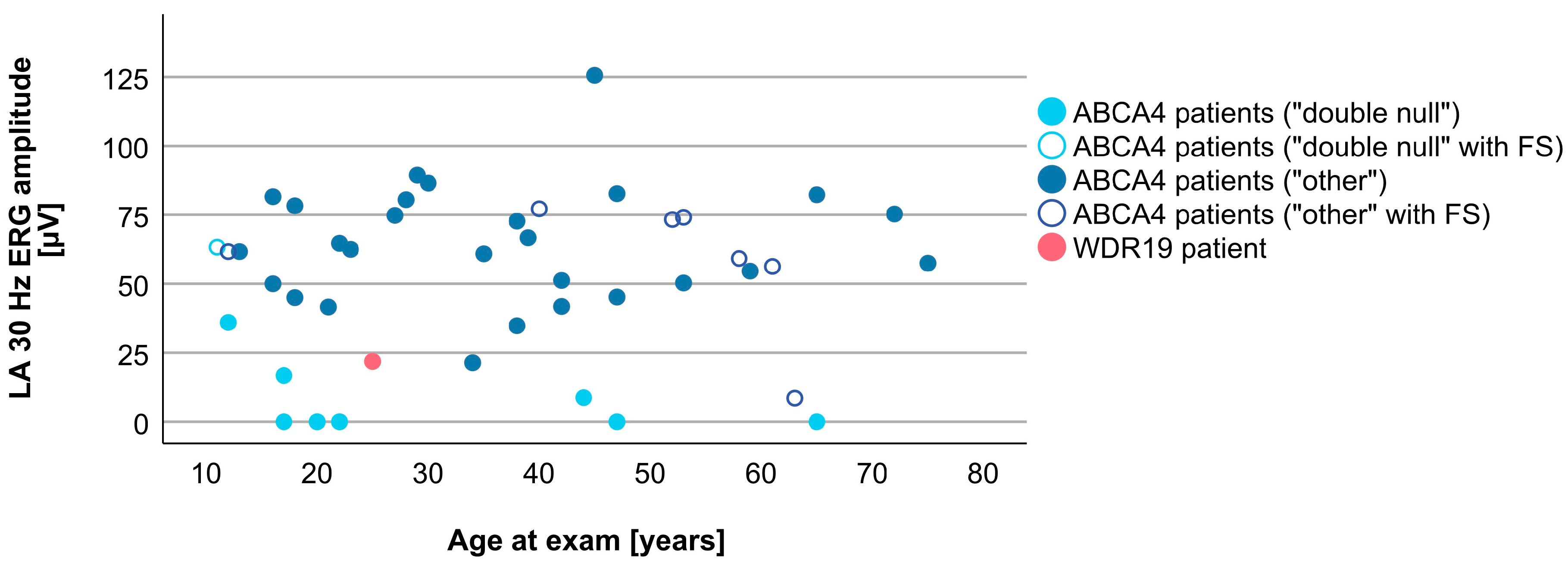
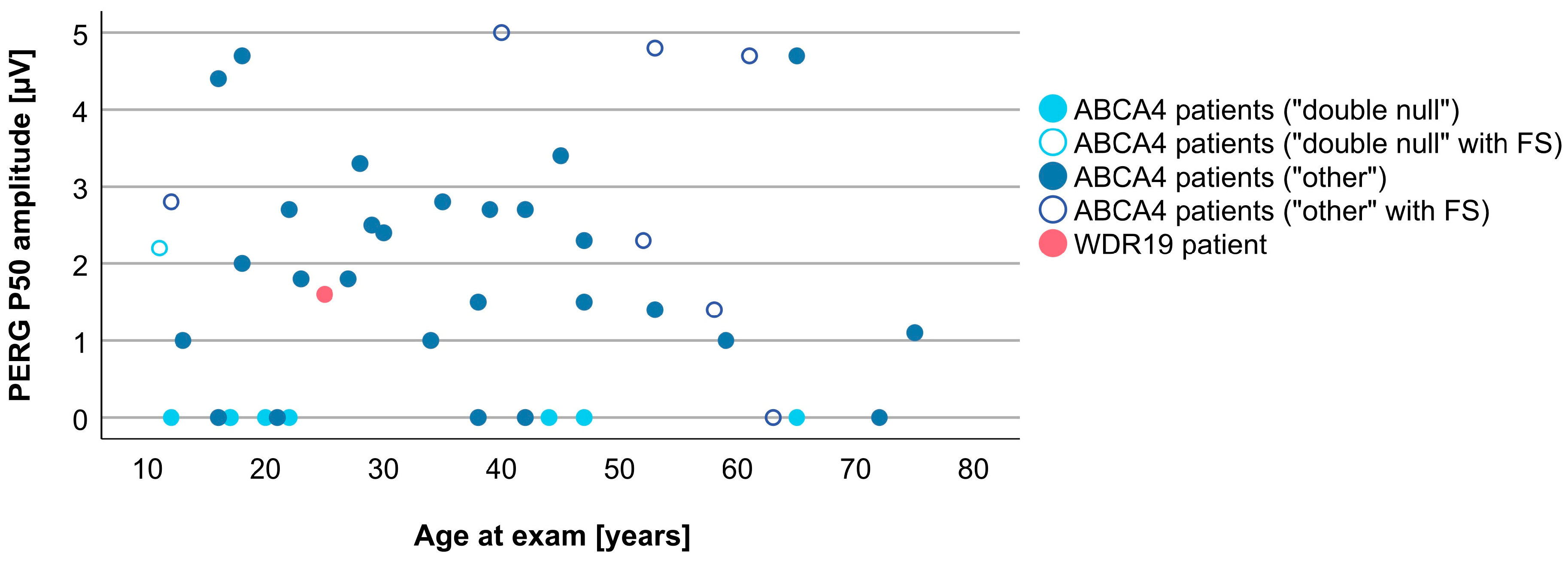
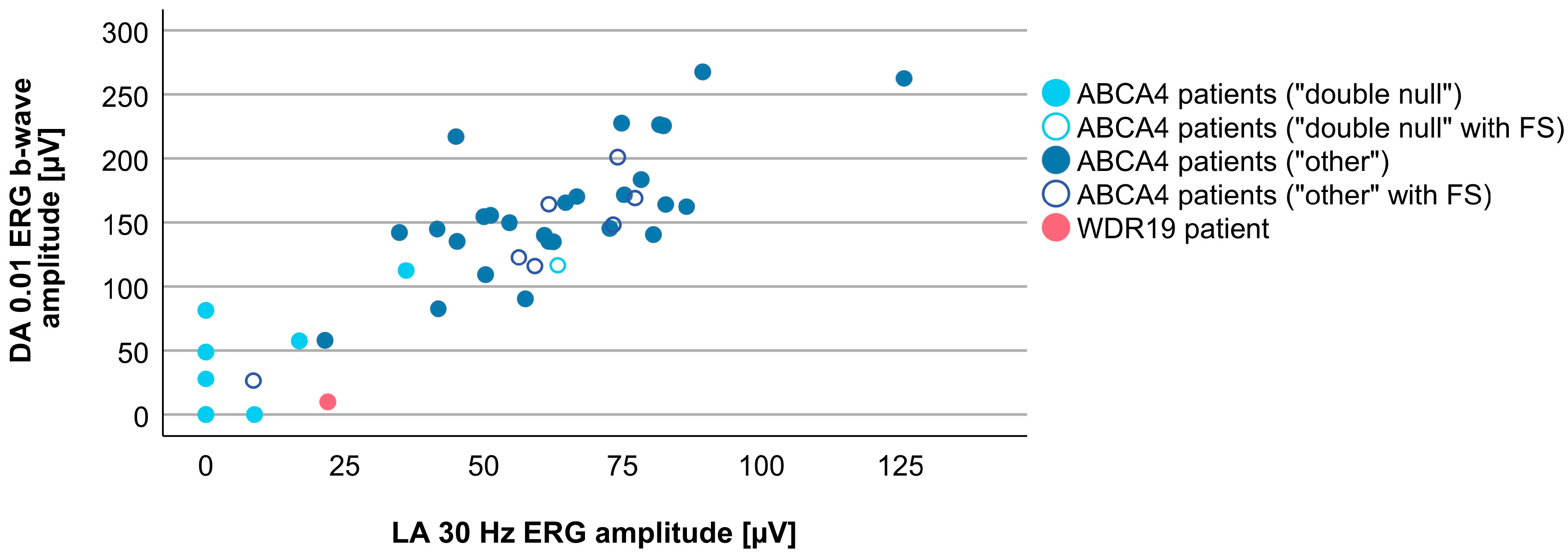
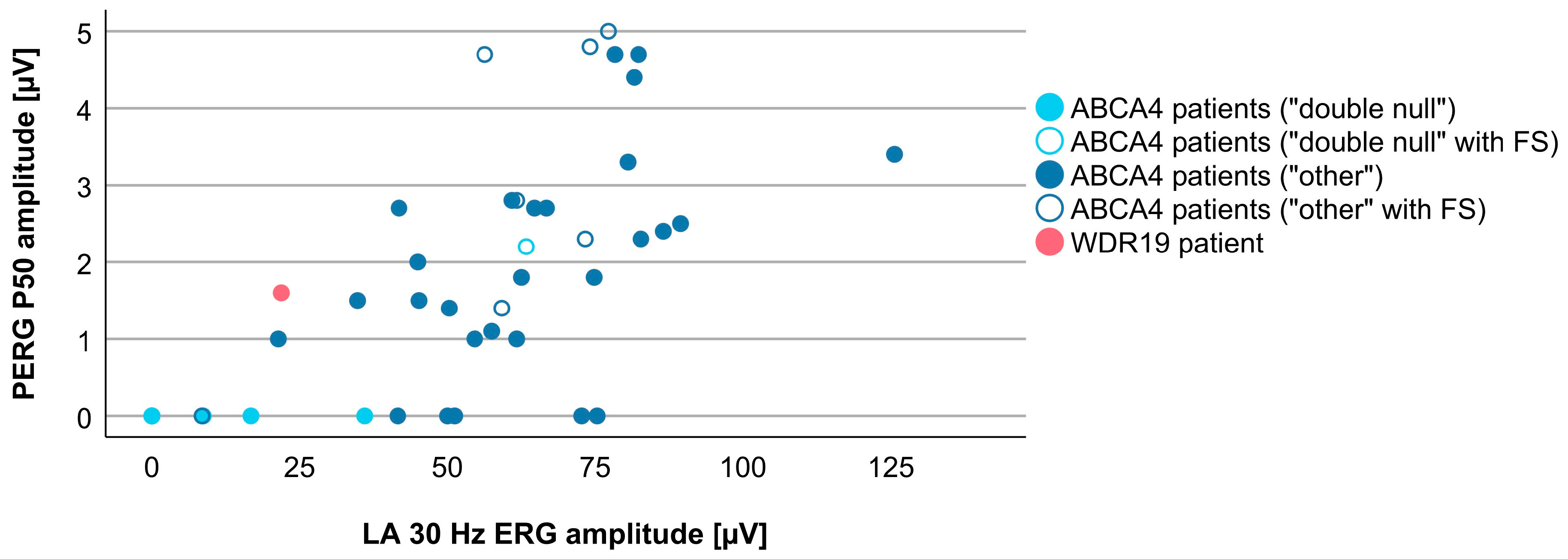
3.4. Comparison between WDR19 and ABCA4 Stargardt Patients
4. Discussion
4.1. Genetic Considerations
4.2. Clinical Findings and Comparison with ABCA4-Stargardt Phenotype
4.3. WDR19 and Other Genes Producing Phenocopies of Stargardt Disease
4.4. Advantages and Disadvantages of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations and Acronyms
| RPE | retinal pigment epithelium |
| ERG | electroretinography |
| IFT144 | intraflagellar transport 144 protein |
| RP | retinitis pigmentosa |
| FAF | fundus autofluorescence |
| SD-OCT | spectral-domain optical coherence tomography |
| DDAF | definitely decreased autofluorescence |
| BCVA | best-corrected visual acuity |
| VA | visual acuity |
| PERG | pattern electroretinography |
| mfERG | multifocal electroretinography |
| ffERG | full-field ERG |
| ISCEV | International Society of Clinical Electrophysiology of Vision |
| VUS | variants of uncertain significance |
| ELM | external limiting membrane |
| ONL | outer nuclear layer |
| DA | dark-adapted |
| LA | light-adapted |
| Ish | Ishihara plates |
| OP | oscillatory potentials |
| Ise band | inner segment ellipsoid band |
| FS | foveal sparing |
References
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A. Fundus Flavimaculatus. A Clinical Classification. Arch. Ophthalmol. 1976, 94, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Tanna, P.; Strauss, R.W.; Fujinami, K.; Michaelides, M. Stargardt disease: Clinical features, molecular genetics, animal models and therapeutic options. Br. J. Ophthalmol. 2017, 101, 25–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molday, L.L.; Rabin, A.R.; Molday, R.S. ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nat. Genet. 2000, 25, 257–258. [Google Scholar] [CrossRef]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa Rim Protein of Retinal Rod Outer Segments Is a Member of the ABC Transporter Superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenis, T.L.; Hu, J.; Ng, S.Y.; Jiang, Z.; Sarfare, S.; Lloyd, M.B.; Esposito, N.J.; Samuel, W.; Jaworski, C.; Bok, D.; et al. Expression of ABCA4 in the retinal pigment epithelium and its implications for Stargardt macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, e11120–e11127. [Google Scholar] [CrossRef] [Green Version]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor ABC transporter ABCA4 in lipid transport and Stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Insights into autofluorescence patterns in Stargardt macular dystrophy using ultra-wide-field imaging. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 1917–1922. [Google Scholar] [CrossRef]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic Subtypes of Stargardt Macular Dystrophy-Fundus Flavimaculatus. Arch. Ophthalmol. 2001, 119, 359–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Huet, R.A.; Bax, N.M.; Westeneng-Van Haaften, S.C.; Muhamad, M.; Zonneveld-Vrieling, M.N.; Hoefsloot, L.H.; Cremers, F.P.; Boon, C.J.; Klevering, B.J.; Hoyng, C.B. Foveal Sparing in Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7467–7478. [Google Scholar] [CrossRef]
- Fujinami, K.; Sergouniotis, P.I.; Davidson, A.E.; Wright, G.; Chana, R.K.; Tsunoda, K.; Tsubota, K.; Egan, C.A.; Robson, A.G.; Moore, A.T.; et al. Clinical and Molecular Analysis of Stargardt Disease With Preserved Foveal Structure and Function. Am. J. Ophthalmol. 2013, 156, 487–501.e1. [Google Scholar] [CrossRef] [PubMed]
- Lambertus, S.; Lindner, M.; Bax, N.M.; Mauschitz, M.M.; Nadal, J.; Schmid, M.; Schmitz-Valckenberg, S.; den Hollander, A.I.; Weber, B.H.F.; Holz, F.G.; et al. Progression of Late-Onset Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5186–5191. [Google Scholar] [CrossRef]
- Kong, X.; West, S.K.; Strauss, R.W.; Munoz, B.; Cideciyan, A.V.; Michaelides, M.; Ho, A.; Ahmed, M.; Schönbach, E.M.; Cheetham, J.K.; et al. Progression of Visual Acuity and Fundus Autofluorescence in Recent-Onset Stargardt Disease: ProgStar Study Report #4. Ophthalmol. Retina 2017, 1, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Miedziak, A.; Vrabec, T.; Verhoeven, J.; Acott, T.S.; Weleber, R.G.; Donoso, L.A. Autosomal dominant Stargardt-like macular dystrophy: I. Clinical characterization, longitudinal follow-up, and evidence for a common ancestry in families linked to chromosome 6q14. Am. J. Ophthalmol. 1999, 127, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Nichols, B.E.; Kimura, A.E.; Weingeist, T.A.; Drack, A.; Sheffield, V.C. Clinical Features of a Stargardt-Like Dominant Progressive Macular Dystrophy With Genetic Linkage to Chromosome 6q. Arch. Ophthalmol. 1994, 112, 765–772. [Google Scholar] [CrossRef]
- Maugeri, A.; Meire, F.; Hoyng, C.B.; Vink, C.; Van Regemorter, N.; Karan, G.; Yang, Z.; Cremers, F.P.M.; Zhang, K. A Novel Mutation in the ELOVL4 Gene Causes Autosomal Dominant Stargardt-like Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4263–4267. [Google Scholar] [CrossRef] [Green Version]
- Palejwala, N.V.; Gale, M.J.; Clark, R.F.; Schlechter, C.; Weleber, R.G.; Pennesi, M.E. Insights into Autosomal Dominant Stargardt-Like Macular Dystrophy Through Multimodality Diagnostic Imaging. Retina 2016, 36, 119–130. [Google Scholar] [CrossRef]
- Del Pozo-Valero, M.; Martin-Merida, I.; Jimenez-Rolando, B.; Arteche, A.; Avila-Fernandez, A.; Blanco-Kelly, F.; Riveiro-Alvarez, R.; Van Cauwenbergh, C.; De Baere, E.; Rivolta, C.; et al. Expanded Phenotypic Spectrum of Retinopathies Associated with Autosomal Recessive and Dominant Mutations in PROM1. Am. J. Ophthalmol. 2019, 207, 204–214. [Google Scholar] [CrossRef]
- Zhang, K.; Kniazeva, M.; Han, M.; Li, W.; Yu, Z.; Yang, Z.; Li, Y.; Metzker, M.L.; Allikmets, R.; Zack, D.J.; et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat. Genet. 2001, 27, 89–93. [Google Scholar] [CrossRef]
- Agbaga, M.P.; Brush, R.S.; Mandal, M.N.; Henry, K.; Elliott, M.H.; Anderson, R.E. Role of Stargardt-3 macular dystrophy protein (ELOVL4) in the biosynthesis of very long chain fatty acids. Proc. Natl. Acad. Sci. USA 2008, 105, 12843–12848. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Investig. 2008, 118, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Yin, J.; Winborn, C.S.; Zhang, Q.; Yue, J.; Chaum, E. Prominin-1 Is a Novel Regulator of Autophagy in the Human Retinal Pigment Epithelium. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2366–2387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cehajic-Kapetanovic, J.; Birtel, J.; McClements, M.E.; Shanks, M.E.; Clouston, P.; Downes, S.M.; Charbel Issa, P.; MacLaren, R.E. Clinical and Molecular Characterization of PROM1-Related Retinal Degeneration. JAMA Netw. Open 2019, 2, e195752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kniazeva, M.; Chiang, M.F.; Morgan, B.; Anduze, A.L.; Zack, D.J.; Han, M.; Zhang, K. A New Locus for Autosomal Dominant Stargardt-Like Disease Maps to Chromosome 4. Am. J. Hum. Genet. 1999, 64, 1394–1399. [Google Scholar] [CrossRef] [Green Version]
- Michaelides, M.; Gaillard, M.C.; Escher, P.; Tiab, L.; Bedell, M.; Borruat, F.X.; Barthelmes, D.; Carmona, R.; Zhang, K.; White, E.; et al. The PROM1 Mutation p.R373C Causes an Autosomal Dominant Bull’s Eye Maculopathy Associated with Rod, Rod-Cone, and Macular Dystrophy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4771–4780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, G.H.; Sutcliffe, J.G.; Bok, D. The retinal degeneration slow (rds) gene product is a photoreceptor disc membrane-associated glycoprotein. Neuron 1991, 6, 61–70. [Google Scholar] [CrossRef]
- Connell, G.; Bascom, R.; Molday, L.; Reid, D.; McInnes, R.R.; Molday, R.S. Photoreceptor peripherin is the normal product of the gene responsible for retinal degeneration in the rds mouse. Proc. Natl. Acad. Sci. USA 1991, 88, 723–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, G.; Parravano, M.; Barbano, L.; Costanzo, E.; Bertelli, M.; Medori, M.C.; Parisi, V.; Ziccardi, L. Multimodal Study of PRPH2 Gene-Related Retinal Phenotypes. Diagnostics 2022, 12, 1851. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.; van Schooneveld, M.J.; den Hollander, A.I.; van Lith-Verhoeven, J.J.; Zonneveld-Vrieling, M.N.; Theelen, T.; Cremers, F.P.; Hoyng, C.B.; Klevering, B.J. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Br. J. Ophthalmol. 2007, 91, 1504–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolock, C.J.; Stong, N.; Ma, C.J.; Nagasaki, T.; Lee, W.; Tsang, S.H.; Kamalakaran, S.; Goldstein, D.B.; Allikmets, R. A case-control collapsing analysis identifies retinal dystrophy genes associated with ophthalmic disease in patients with no pathogenic ABCA4 variants. Genet. Med. 2019, 21, 2336–2344. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Broadgate, S.; Shanks, M.; Clouston, P.; Yu, J.; MacLaren, R.E.; Németh, A.H.; Halford, S.; Downes, S.M. Association of Clinical and Genetic Heterogeneity With BEST1 Sequence Variations. JAMA Ophthalmol. 2020, 138, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Arno, G.; Robson, A.G.; Broadgate, S.; Plagnol, V.; McKibbin, M.; Halford, S.; Michaelides, M.; Holder, G.E.; Moore, A.T.; et al. Characterization of CDH3-Related Congenital Hypotrichosis With Juvenile Macular Dystrophy. JAMA Ophthalmol. 2016, 134, 992–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamseldin, H.E.; Shaheen, R.; Ewida, N.; Bubshait, D.K.; Alkuraya, H.; Almardawi, E.; Howaidi, A.; Sabr, Y.; Abdalla, E.M.; Alfaifi, A.Y.; et al. The morbid genome of ciliopathies: An update. Genet. Med. 2020, 22, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically Focused Molecular Investigation of 1000 Consecutive Families with Inherited Retinal Disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Bredrup, C.; Saunier, S.; Oud, M.M.; Fiskerstrand, T.; Hoischen, A.; Brackman, D.; Leh, S.M.; Midtbø, M.; Filhol, E.; Bole-Feysot, C.; et al. Ciliopathies with Skeletal Anomalies and Renal Insufficiency due to Mutations in the IFT-A Gene WDR19. Am. J. Hum. Genet. 2011, 89, 634–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, C.; Cormier-Daire, V. Ciliary disorder of the skeleton. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 165–174. [Google Scholar] [CrossRef]
- Halbritter, J.; Porath, J.D.; Diaz, K.A.; Braun, D.A.; Kohl, S.; Chaki, M.; Allen, S.J.; Soliman, N.A.; Hildebrandt, F.; Otto, E.A. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum. Genet. 2013, 132, 865–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coussa, R.G.; Otto, E.A.; Gee, H.Y.; Arthurs, P.; Ren, H.; Lopez, I.; Keser, V.; Fu, Q.; Faingold, R.; Khan, A.; et al. WDR19: An ancient, retrograde, intraflagellar ciliary protein is mutated in autosomal recessive retinitis pigmentosa and in Senior-Loken syndrome. Clin. Genet. 2013, 84, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Perlman, I.; Kondo, M.; Chelva, E.; Robson, A.G.; Holder, G.E. ISCEV extended protocol for the S-cone ERG. Doc. Ophthalmol. 2020, 140, 95–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robson, A.G.; Frishman, L.J.; Grigg, J.; Hamilton, R.; Jeffrey, B.G.; Kondo, M.; Li, S.; McCulloch, D.L. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc. Ophthalmol. 2022, 144, 165–177. [Google Scholar] [CrossRef]
- Bach, M.; Brigell, M.G.; Hawlina, M.; Holder, G.E.; Johnson, M.A.; McCulloch, D.L.; Meigen, T.; Viswanathan, S. ISCEV standard for clinical pattern electroretinography (PERG): 2012 update. Doc. Ophthalmol. 2013, 126, 1–7. [Google Scholar] [CrossRef]
- Hoffmann, M.B.; Bach, M.; Kondo, M.; Li, S.; Walker, S.; Holopigian, K.; Viswanathan, S.; Robson, A.G. ISCEV standard for clinical multifocal electroretinography (mfERG) (2021 update). Doc. Ophthalmol. 2021, 142, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Hawlina, M.; Konec, B. New noncorneal HK-loop electrode for clinical electroretinography. Doc Ophthalmol 1992, 81, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Abou Tayoun, A.N.; Pesaran, T.; DiStefano, M.T.; Oza, A.; Rehm, H.L.; Biesecker, L.G.; Harrison, S.M. Recommendations for interpreting the loss of function PVS1 ACMG/AMP variant criterion. Hum. Mutat. 2018, 39, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Dharmadhikari, A.V.; Ghosh, R.; Yuan, B.; Liu, P.; Dai, H.; Al Masri, S.; Scull, J.; Posey, J.E.; Jiang, A.H.; He, W.; et al. Copy number variant and runs of homozygosity detection by microarrays enabled more precise molecular diagnoses in 11,020 clinical exome cases. Genome Med. 2019, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Maddirevula, S.; Alsahli, S.; Alhabeeb, L.; Patel, N.; Alzahrani, F.; Shamseldin, H.E.; Anazi, S.; Ewida, N.; Alsaif, H.S.; Mohamed, J.Y.; et al. Expanding the phenome and variome of skeletal dysplasia. Genet. Med. 2018, 20, 1609–1616. [Google Scholar] [CrossRef]
- Bax, N.M.; Valkenburg, D.; Lambertus, S.; Klevering, B.J.; Boon, C.J.F.; Holz, F.G.; Cremers, F.P.M.; Fleckenstein, M.; Hoyng, C.B.; Lindner, M. Foveal Sparing in Central Retinal Dystrophies. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3456–3467. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.N.; Kasilian, M.; Mahroo, O.A.R.; Tanna, P.; Kalitzeos, A.; Robson, A.G.; Tsunoda, K.; Iwata, T.; Moore, A.T.; Fujinami, K.; et al. Early Patterns of Macular Degeneration in ABCA4-Associated Retinopathy. Ophthalmology 2018, 125, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Fakin, A.; Robson, A.G.; Chiang, J.P.; Fujinami, K.; Moore, A.T.; Michaelides, M.; Holder, G.E.; Webster, A.R. The Effect on Retinal Structure and Function of 15 Specific ABCA4 Mutations: A Detailed Examination of 82 Hemizygous Patients. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5963–5973. [Google Scholar] [CrossRef]
- Schulz, H.L.; Grassmann, F.; Kellner, U.; Spital, G.; Rüther, K.; Jägle, H.; Hufendiek, K.; Rating, P.; Huchzermeyer, C.; Baier, M.J.; et al. Mutation Spectrum of the ABCA4 Gene in 335 Stargardt Disease Patients From a Multicenter German Cohort-Impact of Selected Deep Intronic Variants and Common SNPs. Investig. Ophthalmol. Vis. Sci. 2017, 58, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boubakri, M.; Chaya, T.; Hirata, H.; Kajimura, N.; Kuwahara, R.; Ueno, A.; Malicki, J.; Furukawa, T.; Omori, Y. Loss of ift122, a Retrograde Intraflagellar Transport (IFT) Complex Component, Leads to Slow, Progressive Photoreceptor Degeneration Due to Inefficient Opsin Transport*. J. Biol. Chem. 2016, 291, 24465–24474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldt, K.; Mans, D.A.; Won, J.; van Reeuwijk, J.; Vogt, A.; Kinkl, N.; Letteboer, S.J.F.; Hicks, W.L.; Hurd, R.E.; Naggert, J.K.; et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J. Clin. Investig. 2011, 121, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Rivolta, C.; Ayyagari, R.; Sieving, P.A.; Berson, E.L.; Dryja, T.P. Evaluation of the ELOVL4 gene in patients with autosomal recessive retinitis pigmentosa and Leber congenital amaurosis. Mol. Vis. 2003, 9, 49–51. [Google Scholar]
- Ruiz, A.; Borrego, S.; Marcos, I.; Antiñolo, G. A Major Locus for Autosomal Recessive Retinitis Pigmentosa on 6q, Determined by Homozygosity Mapping of Chromosomal Regions That Contain Gamma-Aminobutyric Acid-Receptor Clusters. Am. J. Hum. Genet. 1998, 62, 1452–1459. [Google Scholar] [CrossRef] [Green Version]
- Khaliq, S.; Hameed, A.; Ismail, M.; Mehdi, S.Q.; Bessant, D.A.; Payne, A.M.; Bhattacharya, S.S. Refinement of the Locus for Autosomal Recessive Retinitis pigmentosa (RP25) Linked to Chromosome 6q in a Family of Pakistani Origin. Am. J. Hum. Genet. 1999, 65, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.; Binkley, E.M.; Rexach, J.; Knight-Johnson, A.; Khemani, P.; Fogel, B.L.; Das, S.; Stone, E.M.; Gomez, C.M. A family with spinocerebellar ataxia and retinitis pigmentosa attributed to an ELOVL4 mutation. Neurol. Genet. 2019, 5, e357. [Google Scholar] [CrossRef] [Green Version]
- Dharmaraj, S.; Li, Y.; Robitaille, J.M.; Silva, E.; Zhu, D.; Mitchell, T.N.; Maltby, L.P.; Baffoe-Bonnie, A.B.; Maumenee, I.H. A Novel Locus for Leber Congenital Amaurosis Maps to Chromosome 6q. Am. J. Hum. Genet. 2000, 66, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zulfiqar, F.; Xiao, X.; Riazuddin, S.A.; Ahmad, Z.; Caruso, R.; MacDonald, I.; Sieving, P.; Riazuddin, S.; Hejtmancik, J.F. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum. Genet. 2007, 122, 293–299. [Google Scholar] [CrossRef]
- Permanyer, J.; Navarro, R.; Friedman, J.; Pomares, E.; Castro-Navarro, J.; Marfany, G.; Swaroop, A.; Gonzàlez-Duarte, R. Autosomal Recessive Retinitis Pigmentosa with Early Macular Affectation Caused by Premature Truncation in PROM1. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2656–2663. [Google Scholar] [CrossRef] [Green Version]
- Imani, S.; Cheng, J.; Shasaltaneh, M.D.; Wei, C.; Yang, L.; Fu, S.; Zou, H.; Khan, M.A.; Zhang, X.; Chen, H.; et al. Genetic identification and molecular modeling characterization reveal a novel PROM1 mutation in Stargardt4-like macular dystrophy. Oncotarget 2018, 9, 122–141. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.M.; Naash, M.I. Gene Therapy for PRPH2-Associated Ocular Disease: Challenges and Prospects. Cold Spring Harb. Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manes, G.; Guillaumie, T.; Vos, W.L.; Devos, A.; Audo, I.; Zeitz, C.; Marquette, V.; Zanlonghi, X.; Defoort-Dhellemmes, S.; Puech, B.; et al. High Prevalence of PRPH2 in Autosomal Dominant Retinitis Pigmentosa in France and Characterization of Biochemical and Clinical Features. Am. J. Ophthalmol. 2015, 159, 302–314. [Google Scholar] [CrossRef]
- Boon, C.J.F.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.M.; Klevering, B.J.; Keunen, J.E.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype–phenotype associations in a large PRPH2-related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef] [PubMed]
- Harkewicz, R.; Du, H.; Tong, Z.; Alkuraya, H.; Bedell, M.; Sun, W.; Wang, X.; Hsu, Y.H.; Esteve-Rudd, J.; Hughes, G.; et al. Essential Role of ELOVL4 Protein in Very Long Chain Fatty Acid Synthesis and Retinal Function. J. Biol. Chem. 2012, 287, 11469–11480. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
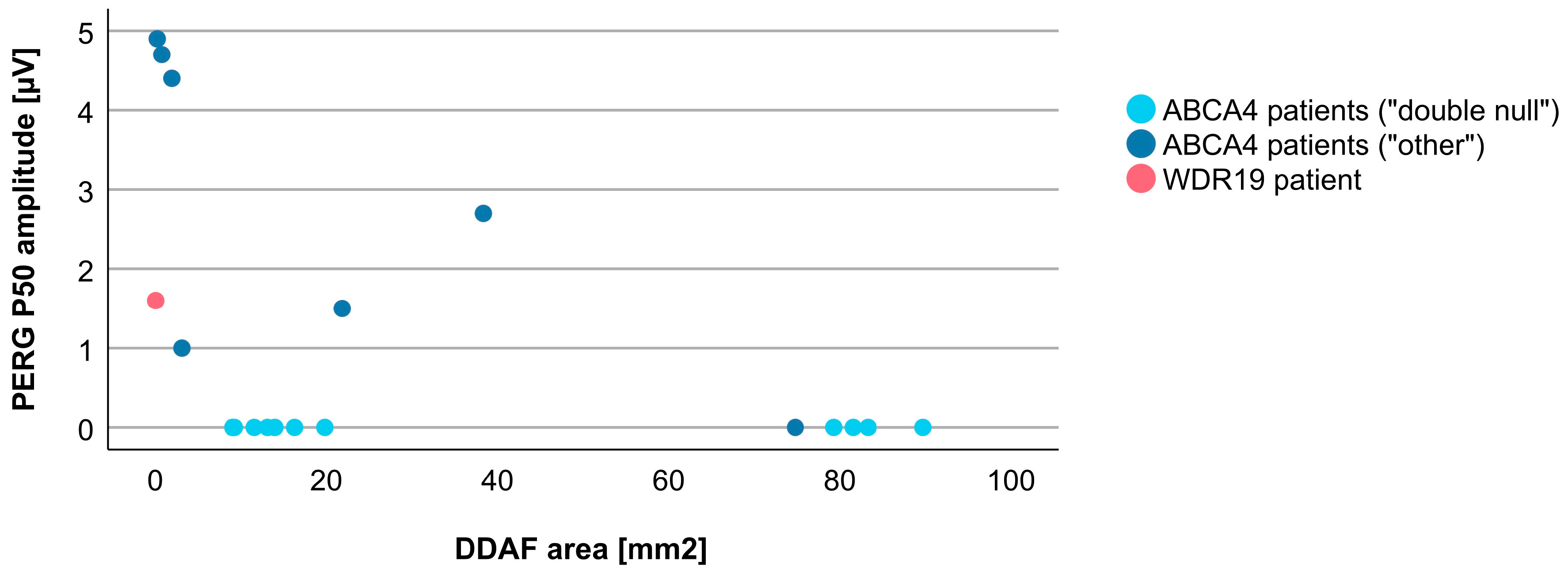{kind=link}
| Parameter | WDR19 Patient | ABCA4 Patients (Median, Range) |
|---|---|---|
| Age at exam | 24 years | 38 years (11–75 years) |
| Age at onset | 5 years | 16 years (5–60 years) |
| Disease duration | 21 years | 12 years (0–55 years) |
| Visual acuity (Snellen decimal) | 1.0 | 0.1 (0.0–1.0) |
| PERG P50 amplitude | 1.6 µV | 1.5 µV (0.5–5.0 µV) |
| S-cone ERG amplitude | 0.0 µV | 3.4 µV (0.0–8.9 µV) |
| DA 0.01 ERG b-wave amplitude | 9.8 µV | 143.6 (0.0–267.8 µV) |
| DA 3.0 ERG a-wave amplitude | 22.3 µV | 113.3 µV (0.0–211.9 µV) |
| Oscillatory potentials | 0.0 µV | 9.3 µV (0.0–53.9 µV) |
| LA 30 Hz ERG amplitude | 21.9 µV | 58.4 µV (0.0–125.6 µV) |
| LA 3.0 ERG b-wave amplitude | 16.7 µV | 60.5 µV (0.0–128.7 µV) |
| DDAF area | 0.04 mm2 | 15.09 mm2 (0.22–89.67 mm2), N = 18 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajovic, J.; Meglič, A.; Volk, M.; Maver, A.; Jarc-Vidmar, M.; Hawlina, M.; Fakin, A. Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene. Genes 2023, 14, 291. https://doi.org/10.3390/genes14020291
Sajovic J, Meglič A, Volk M, Maver A, Jarc-Vidmar M, Hawlina M, Fakin A. Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene. Genes. 2023; 14(2):291. https://doi.org/10.3390/genes14020291
Chicago/Turabian StyleSajovic, Jana, Andrej Meglič, Marija Volk, Aleš Maver, Martina Jarc-Vidmar, Marko Hawlina, and Ana Fakin. 2023. "Stargardt-like Clinical Characteristics and Disease Course Associated with Variants in the WDR19 Gene" Genes 14, no. 2: 291. https://doi.org/10.3390/genes14020291