
Molecular Genetic Characteristics of FANCI, a Proposed New Ovarian Cancer Predisposing Gene
 , ,
, ,  , , , , , , , , , and
, , , , , , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Study Subjects
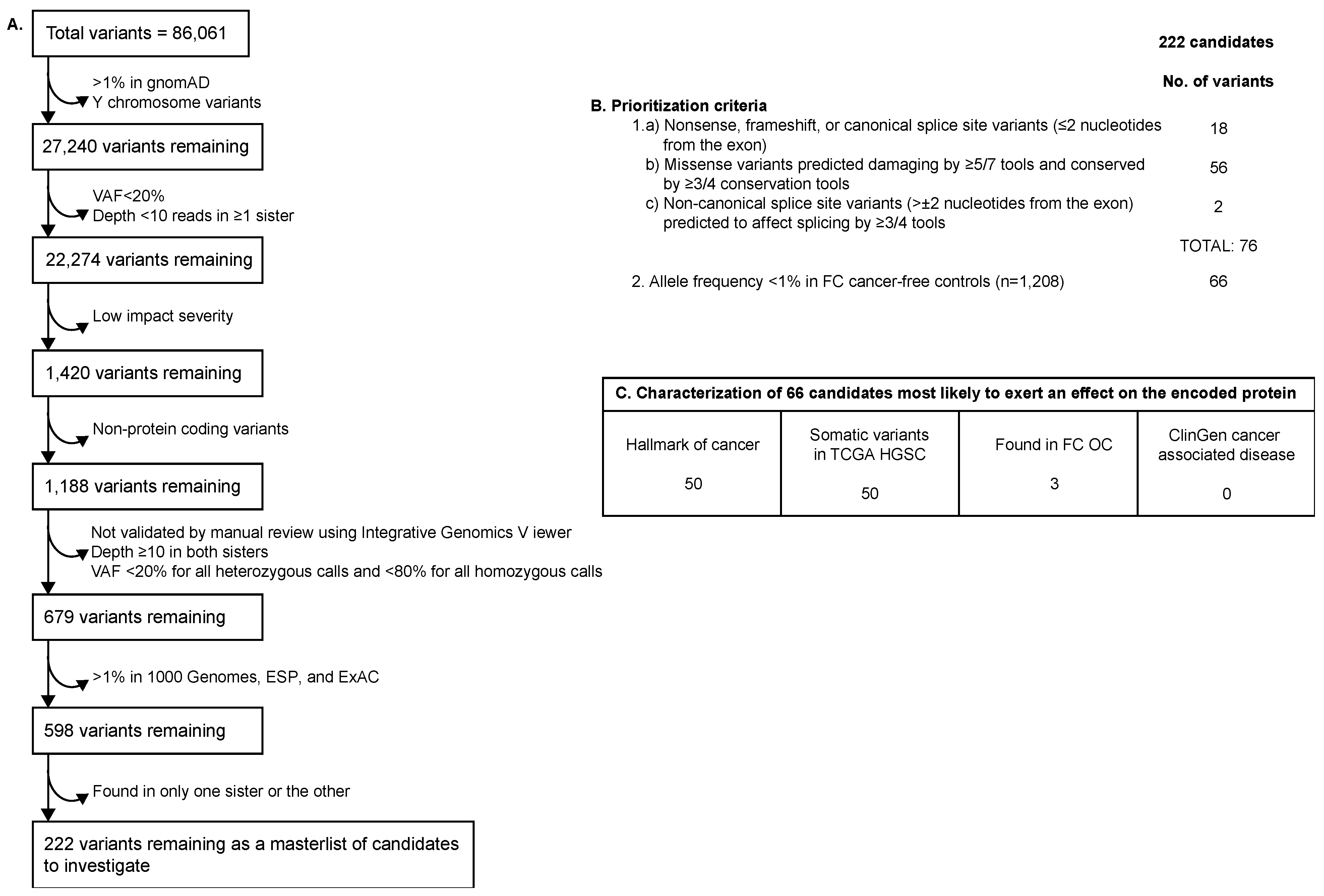
2.2. WES Filtering and Prioritization of Variants Identified in Family F1528
2.3. Investigation of Genetic Landscape Variants
2.4. Loss of Heterozygosity Analyses of FANCI c.1813C>T in OC Tumour DNA from Candidate Variant Carriers
2.5. Somatic Genetic Landscape of FANCI c.1813C>T Carriers
2.6. FANCI c.1813C>T Germline Carrier Frequency across Different Cancer Types from TCGA PanCancer Atlas
2.7. Identification of Somatic FANCI Variants in Different Cancer Types from TCGA PanCancer Atlas
2.8. Investigation of Missense Variants in FANCI Reported in Public Databases
2.9. Identification of Variants in the FANCI Protein Interactome
3. Results
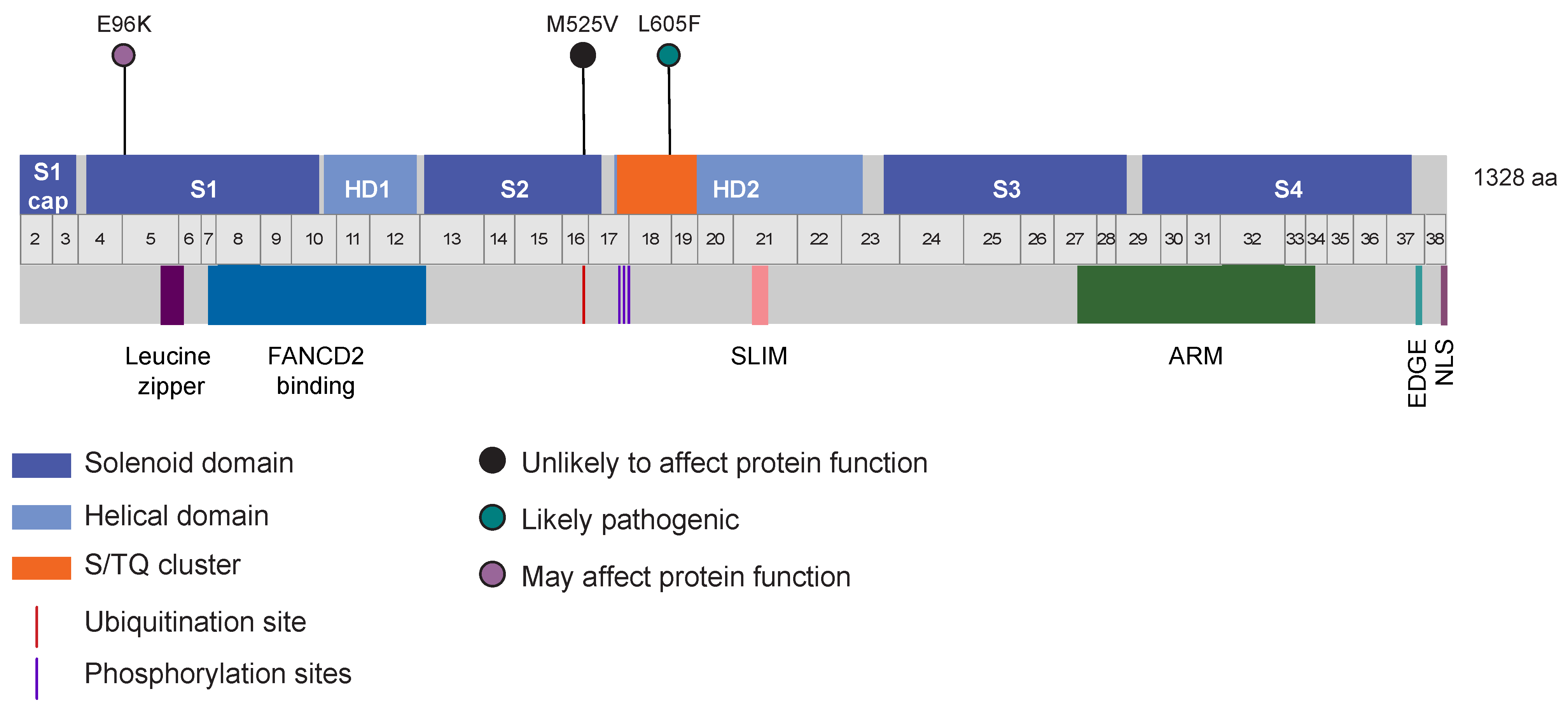
3.1. Candidate Variants Identified in Family F1528
3.2. Genetic Analyses of Variants Identified in FANCI Carrier Siblings in FC Study Groups
3.3. Genetic Analyses of Variants Identified in FANCI Carrier Siblings in Non-FC Study Groups
3.4. Genetic Analyses of Germline FANCI Interactome Variants Identified in FC OC Cases
3.5. Identification of Other Germline Potentially Deleterious Variants in FANCI
3.6. Loss of Heterozygosity Analyses of FANCI c.1813C>T in OC Tumour DNA from Carriers
3.7. Somatic Genetic Analyses of OC Tumours from FANCI c.1813C>T Carriers
3.8. Germline FANCI c.1813C>T Carriers Identified in Other Cancer Types
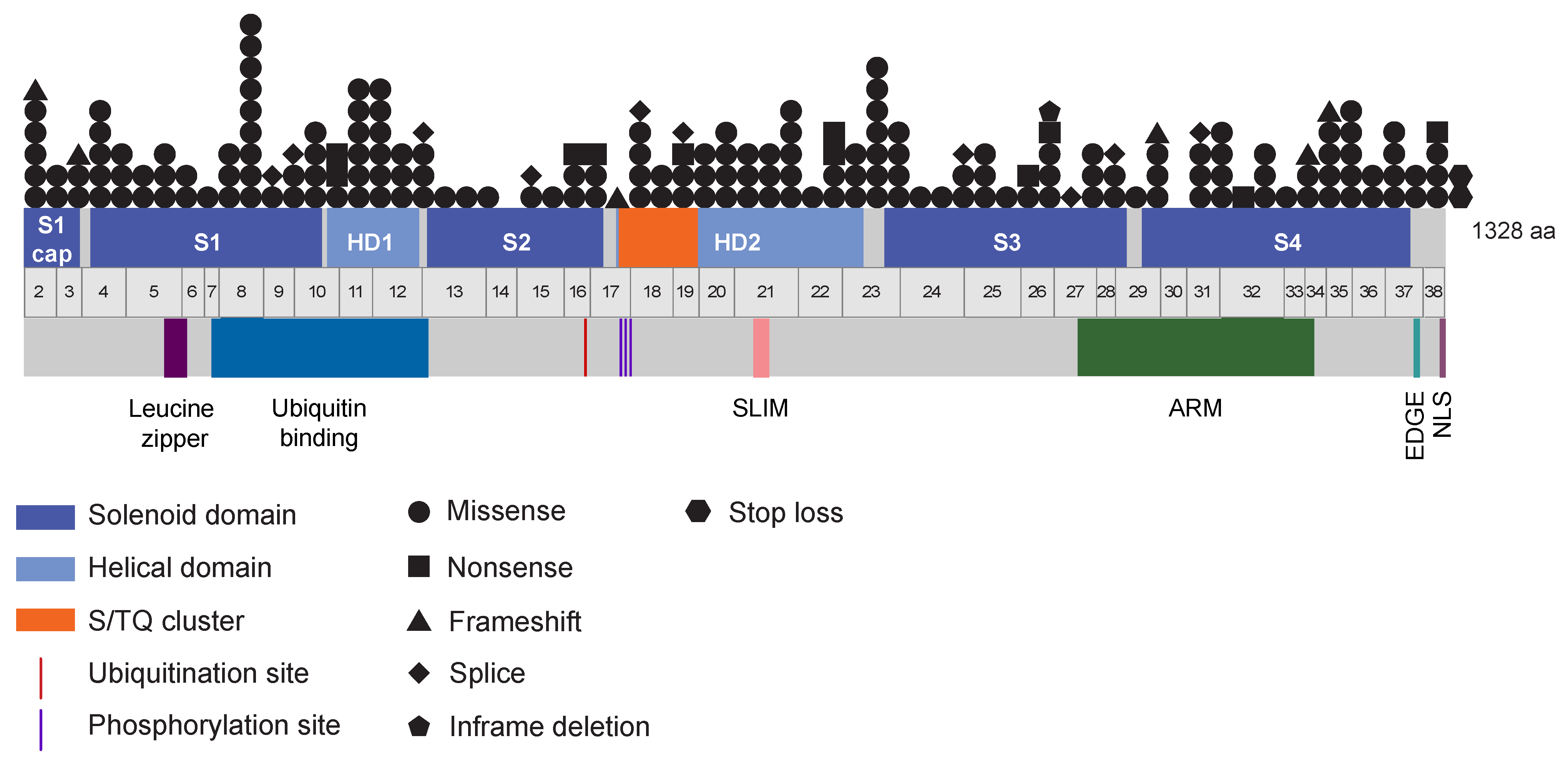
3.9. A Wide Spectrum of Somatic FANCI Variants Identified in a Variety of Cancer Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miki, Y.; Swensen, J.; Shattuck-eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. Strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Ramus, S.J.; Kjaer, S.K.; Hogdall, E.; Dicioccio, R.A.; Whittemore, A.S.; McGuire, V.; Hogdall, C.; Jacobs, I.J.; Easton, D.F.; et al. Tagging single nucleotide polymorphisms in the BRIP1 gene and susceptibility to breast and ovarian cancer. PLoS ONE 2007, 2, e268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, J.; Hope, K.; Niu, Q.; Huo, D.; Olopade, O.I. Screening RAD51C nucleotide alterations in patients with a family history of breast and ovarian cancer. Breast Cancer Res. Treat. 2010, 124, 857–861. [Google Scholar] [CrossRef]
- Somyajit, K.; Subramanya, S.; Nagaraju, G. RAD51C: A novel cancer susceptibility gene is linked to Fanconi anemia and breast cancer. Carcinogenesis 2010, 31, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Pavanello, M.; Chan, I.H.Y.; Ariff, A.; Pharoah, P.D.P.; Gayther, S.A.; Ramus, S.J. Rare germline genetic variants and the risks of epithelial ovarian cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; Adams, M.D. Mutation of a mutL homolog in hereditary colon cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef] [PubMed]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Miyaki, M.; Konishi, M.; Tanaka, K.; Kikuchi-Yanoshita, R.; Muraoka, M.; Yasuno, M.; Igari, T.; Koike, M.; Chiba, M.; Mori, T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1997, 17, 271–272. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Wei, Y.F.; Carter, K.C.; Ruben, S.M.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371, 75–80. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous REcombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol. 2019, 5, 51–57. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Weber-Lassalle, N.; Borde, J.; Weber-Lassalle, K.; Horváth, J.; Niederacher, D.; Arnold, N.; Kaulfuß, S.; Ernst, C.; Paul, V.G.; Honisch, E.; et al. Germline loss-of-function variants in the BARD1 gene are associated with early-onset familial breast cancer but not ovarian cancer. Breast Cancer Res. BCR 2019, 21, 55. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature review of BARD1 as a cancer predisposing gene with a focus on breast and ovarian cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef]
- Kanchi, K.L.; Johnson, K.J.; Lu, C.; Mclellan, M.D.; Leiserson, D.M.; Wendl, M.C.; Zhang, Q.; Koboldt, D.C.; Kandoth, C.; Mcmichael, J.F.; et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat. Commun. 2014, 5, 3156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicks, E.; Song, H.; Ramus, S.J.; Van Oudenhove, E.; Tyrer, J.P.; Intermaggio, M.P.; Kar, S.; Harrington, P.; Bowtell, D.D.; Study Group, A.; et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017, 8, 50930–50940. [Google Scholar] [CrossRef] [PubMed]
- Fierheller, C.T.; Alenezi, W.M.; Tonin, P.N. The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers 2021, 13, 3406. [Google Scholar] [CrossRef]
- Tonin, P.N.; Mes-Masson, A.M.; Futreal, P.A.; Morgan, K.; Mahon, M.; Foulkes, W.D.; Cole, D.E.; Provencher, D.; Ghadirian, P.; Narod, S.A. Founder BRCA1 and BRCA2 mutations in French Canadian breast and ovarian cancer families. Am. J. Hum. Genet. 1998, 63, 1341–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oros, K.K.; Ghadirian, P.; Greenwood, C.M.T.; Perret, C.; Shen, Z.; Paredes, Y.; Arcand, S.L.; Mes-Masson, A.M.; Narod, S.A.; Foulkes, W.D.; et al. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5 BRCA1 and BRCA2 mutations. Int. J. Cancer 2004, 112, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Ghadirian, P.; Akbari, M.R.; Hamel, N.; Giroux, S.; Sabbaghian, N.; Darnel, A.; Royer, R.; Poll, A.; Fafard, E.; et al. Identification of a novel truncating PALB2 mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007, 9, R83. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.; Xia, B.; Sabbaghian, N.; Reis-filho, J.S.; Hamel, N.; Li, G.; Beers, E.H.V.; Li, L.; Khalil, T.; Quenneville, L.A.; et al. Analysis of PALB2/FANCN-associated breast cancer families. Proc. Natl. Acad. Sci. USA 2007, 104, 6788–6793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenezi, W.M.; Milano, L.; Fierheller, C.T.; Serruya, C.; Revil, T.; Oros, K.K.; Behl, S.; Arcand, S.L.; Nayar, P.; Spiegelman, D.; et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers 2022, 14, 2251. [Google Scholar] [CrossRef]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally null RAD51D missense mutation associates strongly with ovarian carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef]
- Fierheller, C.T.; Guitton-Sert, L.; Alenezi, W.M.; Revil, T.; Oros, K.K.; Gao, Y.; Bedard, K.; Arcand, S.L.; Serruya, C.; Behl, S.; et al. A functionally impaired missense variant identified in French Canadian families implicates FANCI as a candidate ovarian cancer-predisposing gene. Genome Med. 2021, 13, 186. [Google Scholar] [CrossRef] [PubMed]
- Callahan, M.J.; Crum, C.P.; Medeiros, F.; Kindelberger, D.W.; Elvin, J.A.; Garber, J.E.; Feltmate, C.M.; Berkowitz, R.S.; Muto, M.G. Primary fallopian tube malignancies in BRCA-positive women undergoing surgery for ovarian cancer risk reduction. J. Clin. Oncol. 2007, 25, 3985–3990. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.W.; Miron, A.; Jarboe, E.A.; Parast, M.M.; Hirsch, M.S.; Lee, Y.; Muto, M.G.; Kindelberger, D.; Crum, C.P. Serous tubal intraepithelial carcinoma: Its potential role in primary peritoneal serous carcinoma and serous cancer prevention. J. Clin. Oncol. 2008, 26, 4160–4165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, A.; Shaw, P.; Rosen, B.; Murphy, J.; Narod, S.A.; Colgan, T.J. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers. Gynecol. Oncol. 2006, 100, 58–64. [Google Scholar] [CrossRef]
- Labidi-galy, S.I.; Papp, E.; Hallberg, D.; Niknafs, N.; Adleff, V.; Noe, M.; Bhattacharya, R.; Novak, M.; Jones, S.; Phallen, J.; et al. High grade serous ovarian carcinomas originiate in the fallopian tube. Nat. Commun. 2017, 8, 1093. [Google Scholar] [CrossRef] [Green Version]
- Leeper, K.; Garcia, R.; Swisher, E.; Goff, B.; Greer, B.; Paley, P. Pathologic findings in prophylactic oophorectomy specimens in high-risk women. Gynecol. Oncol. 2002, 87, 52–56. [Google Scholar] [CrossRef]
- Powell, C.B.; Kenley, E.; Chen, L.; Crawford, B.; Mclennan, J.; Zaloudek, C.; Komaromy, M.; Beattie, M.; Ziegler, J. Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: Role of seial sectioning in the setection of occult malignancy. J. Clin. Oncol. 2005, 23, 127–132. [Google Scholar] [CrossRef]
- Tonin, P.N.; Maugard, C.M.; Perret, C.; Mes-Masson, A.M.; Provencher, D.M. A review of histopathological subtypes of ovarian cancer in BRCA-related French Canadian cancer families. Fam. Cancer 2007, 6, 491–497. [Google Scholar] [CrossRef]
- Tonin, P.N.; Mes-Masson, A.-M.; Narod, S.A.; Ghadirian, P.; Provencher, D. Founder BRCA1 and BRCA2 mutations in French Canadian ovarian cancer cases unselected for family history. Clin. Genet. 1999, 55, 318–324. [Google Scholar] [CrossRef]
- Hodgkinson, A.; Idaghdour, Y.; Gbeha, E.; Grenier, J.-C.; Hip-Ki, E.; Bruat, V.; Goulet, J.-P.; de Malliard, T.; Awadalla, P. High-resolution genomic analysis of human mitochondrial RNA sequence variation. Science 2014, 344, 413–415. [Google Scholar] [CrossRef]
- Hussin, J.G.; Hodgkinson, A.; Idaghdour, Y.; Grenier, J.-C.; Goulet, J.-P.; Gbeha, E.; Hip-Ki, E.; Awadalla, P. Recombination affects accumulation of damaging and disease-associated mutations in human populations. Nat. Genet. 2015, 47, 400–404. [Google Scholar] [CrossRef]
- Peischl, S.; Dupanloup, I.; Foucal, A.; Jomphe, M.; Bruat, V.; Grenier, J.-C.; Gouy, A.; Gilbert, K.J.; Gbeha, E.; Bosshard, L.; et al. Relaxed Selection During a Recent Human Expansion. Genetics 2018, 208, 763–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemette, L.; Allard, C.; Lacroix, M.; Patenaude, J.; Battista, M.-C.; Doyon, M.; Moreau, J.; Ménard, J.; Bouchard, L.; Ardilouze, J.-L.; et al. Genetics of Glucose regulation in Gestation and Growth (Gen3G): A prospective prebirth cohort of mother–child pairs in Sherbrooke, Canada. BMJ Open 2016, 6, e010031. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.N.; Zethoven, M.; Mcinerny, S.; Morgan, J.A.; Rowley, S.M.; Lee, J.E.A.; Li, N.; Gorringe, K.L.; James, P.A.; Campbell, I.G. Exome sequencing of familial high-grade serous ovarian carcinoma reveals heterogeneity for rare candidate susceptibility genes. Nat. Commun. 2020, 11, 1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.-L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic germline variants in 10,389 adult cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Onur, S.; Larsson, E.; Antipin, Y.; Reva, B.; Goldberg, A.P.; Sander, C. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2014, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, M.; Guttman, M.; Clamp, M.; Zody, M.C.; Friedman, N.; Xie, X. Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 2009, 25, i54–i62. [Google Scholar] [CrossRef] [Green Version]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.; Bejerano, G.; Pedersen, J.S.; Hinrichs, A.S.; Hou, M.; Rosenbloom, K.; Clawson, H.; Spieth, J.; Hillier, L.W.; Richards, S.; et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005, 15, 1034–1050. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Ionita-Laza, I.; McCallum, K.; Xu, B.; Buxbaum, J.D. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 2016, 48, 214–220. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douville, C.; Masica, D.L.; Stenson, P.D.; Cooper, D.N.; Gygax, D.M.; Kim, R.; Ryan, M.; Karchin, R. Assessing the Pathogenicity of Insertion and Deletion Variants with the Variant Effect Scoring Tool (VEST-Indel). Hum. Mutat. 2016, 37, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.M.; McLaren, W.; Parsons, M.T.; Thompson, B.A.; O’Mara, T.A.; Hunt, S.E.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Kyriazopoulou Panagiotopoulou, S.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G. Rare and common variants: Twenty arguments. Nat. Rev. Genet. 2012, 13, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Baker, S.; Ali, I.; Silins, I.; Pyysalo, S.; Guo, Y.; Högberg, J.; Stenius, U.; Korhonen, A. Cancer Hallmarks Analytics Tool (CHAT): A text mining approach to organize and evaluate scientific literature on cancer. Bioinformatics 2017, 33, 3973–3981. [Google Scholar] [CrossRef] [Green Version]
- Menyhárt, O.; Harami-Papp, H.; Sukumar, S.; Schäfer, R.; Magnani, L.; de Barrios, O.; Győrffy, B. Guidelines for the selection of functional assays to evaluate the hallmarks of cancer. Biochim. Biophys. Acta 2016, 1866, 300–319. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Rehm, H.L.; Berg, J.S.; Brooks, L.D.; Bustamante, C.D.; Evans, J.P.; Landrum, M.J.; Ledbetter, D.H.; Maglott, D.R.; Martin, C.L.; Nussbaum, R.L.; et al. ClinGen—The Clinical Genome Resource. N. Engl. J. Med. 2015, 372, 2235–2242. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Shen, R.; Seshan, V.E. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016, 44, e131. [Google Scholar] [CrossRef] [Green Version]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [Green Version]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 2022, 376, 6591. [Google Scholar] [CrossRef]
- Kautto, E.A.; Bonneville, R.; Miya, J.; Yu, L.; Krook, M.A.; Reeser, J.W.; Roychowdhury, S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget 2017, 8, 7452–7463. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, 1062–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Stark, C.; Breitkreutz, B.-J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [Green Version]
- Tomkins, J.E.; Ferrari, R.; Vavouraki, N.; Hardy, J.; Lovering, R.C.; Lewis, P.A.; McGuffin, L.J.; Manzoni, C. PINOT: An intuitive resource for integrating protein-protein interactions. Cell Commun. Signal. 2020, 18, 92. [Google Scholar] [CrossRef]
- Licata, L.; Lo Surdo, P.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2020, 48, D504–D510. [Google Scholar] [CrossRef] [PubMed]
- Calderone, A.; Iannuccelli, M.; Peluso, D.; Licata, L. Using the MINT Database to Search Protein Interactions. Curr. Protoc. Bioinform. 2020, 69, e93. [Google Scholar] [CrossRef]
- Gioutlakis, A.; Klapa, M.I.; Moschonas, N.K. PICKLE 2.0: A human protein-protein interaction meta-database employing data integration via genetic information ontology. PLoS ONE 2017, 12, e0186039. [Google Scholar] [CrossRef] [Green Version]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004, 32, D449–D451. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Cubuk, C.; Garrett, A.; Choi, S.; King, L.; Loveday, C.; Torr, B.; Burghel, G.J.; Durkie, M.; Callaway, A.; Robinson, R.; et al. Clinical likelihood ratios and balanced accuracy for 44 in silico tools against multiple large-scale functional assays of cancer susceptibility genes. Genet. Med. 2021, 23, 2096–2104. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Sabbaghian, N.; Hamel, N.; Pouchet, C.; Foulkes, W.D.; Mes-Masson, A.-M.; Provencher, D.M.; Tonin, P.N. Contribution of the PALB2 c.2323C>T [p. Q775X] Founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med. Genet. 2013, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oros, K.K.; Ghadirian, P.; Maugard, C.M.; Perret, C.; Paredes, Y.; Mes-Masson, A.-M.; Foulkes, W.D.; Provencher, D.; Tonin, P.N. Application of BRCA1 and BRCA2 mutation carrier prediction models in breast and/or ovarian cancer families of French Canadian descent. Clin. Genet. 2006, 70, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Tonin, P.N.; Perret, C.; Lambert, J.A.; Paradis, A.J.; Kantemiroff, T.; Benoît, M.H.; Martin, G.; Foulkes, W.D.; Ghadirian, P. Founder BRCA1 and BRCA2 mutations in early-onset french Canadian breast cancer cases unselected for family history. Int. J. Cancer 2001, 95, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Arcand, S.L.; Akbari, M.R.; Provencher, D.; Foulkes, W.D.; Narod, S.A.; Tonin, P.N. Germline TP53 mutational spectrum in French Canadians with breast cancer. BMC Med. Genet. 2015, 16, 24. [Google Scholar] [CrossRef] [Green Version]
- Cote, S.; Arcand, S.L.; Royer, R.; Nolet, S.; Mes-Masson, A.; Ghadirian, P.; Foulkes, W.D.; Tischkowitz, M.; Narod, S.A.; Provencher, D.; et al. The BRCA2 c.9004G>A (E2002K) [corrected] variant is likely pathogenic and recurs in breast and/or ovarian cancer families of French Canadian descent. Breast Cancer Res. Treat. 2012, 131, 333–340. [Google Scholar] [CrossRef]
- Osher, D.J.; Leeneer, K.D.; Michils, G.; Hamel, N.; Tomiak, E.; Poppe, B.; Leunen, K.; Legius, E.; Shuen, A.; Smith, E.; et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br. J. Cancer 2012, 106, 1460–1463. [Google Scholar] [CrossRef] [Green Version]
- Arcand, S.L.; Maugard, C.M.; Ghadirian, P.; Robidoux, A.; Perret, C.; Zhang, P.; Fafard, E.; Mes-Masson, A.M.; Foulkes, W.D.; Provencher, D.; et al. Germline TP53 mutations in BRCA1 and BRCA2 mutation-negative French Canadian breast cancer families. Breast Cancer Res. Treat. 2008, 108, 399–408. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Revil, T.; Serruya, C.; Mes-Masson, A.-M.; Foulkes, W.D.; Provencher, D.; El Haffaf, Z.; Ragoussis, J.; Tonin, P.N. Case Review: Whole-Exome Sequencing Analyses Identify Carriers of a Known Likely Pathogenic Intronic BRCA1 Variant in Ovarian Cancer Cases Clinically Negative for Pathogenic BRCA1 and BRCA2 Variants. Genes 2022, 13, 697. [Google Scholar] [CrossRef]
- Sims, A.E.; Spiteri, E.; Sims, R.J.; Arita, A.G.; Lach, F.P.; Landers, T.; Wurm, M.; Freund, M.; Neveling, K.; Hanenberg, H.; et al. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2007, 14, 564–567. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald III, E.R.; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the Fanconi anemia (FANC) I protein, a monoubiquitinated FANCD2 paralog required for crosslink repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef]
- Ishiai, M.; Kitao, H.; Smogorzewska, A.; Tomida, J.; Kinomura, A.; Uchida, E.; Saberi, A.; Kinoshita, E.; Kinoshita-Kikuta, E.; Koike, T.; et al. FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat. Struct. Mol. Biol. 2008, 15, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; van Twest, S.; Murphy, V.J.; Deans, A.J. ATR-Mediated FANCI Phosphorylation Regulates Both Ubiquitination and Deubiquitination of FANCD2. Front. Cell Dev. Biol. 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; El Hokayem, J.; Zhou, W.; Zhang, Y. FANCI protein binds to DNA and interacts with FANCD2 to recognize branched structures. J. Biol. Chem. 2009, 284, 24443–24452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Wang, S.; Dhar, A.; Peralta, C.; Pavletich, N.P. DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 2020, 580, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Moldovan, G.-L.; Vinciguerra, P.; Murai, J.; Takeda, S.; D’Andrea, A.D. Regulation of the Fanconi anemia pathway by a SUMO-like delivery network. Genes Dev. 2011, 25, 1847–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colnaghi, L.; Jones, M.J.K.; Cotto-Rios, X.M.; Schindler, D.; Hanenberg, H.; Huang, T.T. Patient-derived C-terminal mutation of FANCI causes protein mislocalization and reveals putative EDGE motif function in DNA repair. Blood 2011, 117, 2247–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; DeFazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Wojnarowicz, P.M.; Oros, K.K.; Quinn, M.C.J.; Arcand, S.L.; Gambaro, K.; Madore, J.; Birch, A.H.; de Ladurantaye, M.; Rahimi, K.; Provencher, D.M.; et al. The genomic landscape of TP53 and p53 annotated high grade ovarian serous carcinomas from a defined founder population associated with patient outcome. PLoS ONE 2012, 7, e45484. [Google Scholar] [CrossRef]
- PDQ Cancer Genetics Editorial Board. Genetics of Breast and Gynecologic Cancers (PDQ®): Health Professional Version; National Cancer Institute: Bethesda, MD, USA, 2022. [Google Scholar]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, D.G.; Balasubramanian, S.; Frankish, A.; Huang, N.; Morris, J.; Walter, K.; Jostins, L.; Habegger, L.; Pickrell, J.K.; Montgomery, S.B.; et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science 2012, 335, 823–828. [Google Scholar] [CrossRef]
- Halldorsson, B.V.; Eggertsson, H.P.; Moore, K.H.S.; Hauswedell, H.; Eiriksson, O.; Ulfarsson, M.O.; Palsson, G.; Hardarson, M.T.; Oddsson, A.; Jensson, B.O.; et al. The sequences of 150,119 genomes in the UK Biobank. Nature 2022, 607, 732–740. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Tyler-Smith, C. Loss-of-function variants in the genomes of healthy humans. Hum. Mol. Genet. 2010, 19, R125–R130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sulem, P.; Helgason, H.; Oddson, A.; Stefansson, H.; Gudjonsson, S.A.; Zink, F.; Hjartarson, E.; Sigurdsson, G.T.; Jonasdottir, A.; Jonasdottir, A.; et al. Identification of a large set of rare complete human knockouts. Nat. Genet. 2015, 47, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.-H.; Kuo, W.-H.; Huang, A.-C.; Lu, Y.-S.; Lin, C.-H.; Kuo, S.-H.; Wang, M.-Y.; Liu, C.-Y.; Cheng, F.T.-F.; Yeh, M.-H.; et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget 2016, 7, 8310–8320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Dicks, E.M.; Tyrer, J.; Intermaggio, M.; Chenevix-Trench, G.; Bowtell, D.D.; Traficante, N.; Group, A.; Brenton, J.; Goranova, T.; et al. Population-based targeted sequencing of 54 candidate genes identifies PALB2 as a susceptibility gene for high-grade serous ovarian cancer. J. Med. Genet. 2021, 58, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Volkova, N.V.; Meier, B.; González-Huici, V.; Bertolini, S.; Gonzalez, S.; Vöhringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 2169. [Google Scholar] [CrossRef]
- Degasperi, A.; Amarante, T.D.; Czarnecki, J.; Shooter, S.; Zou, X.; Glodzik, D.; Morganella, S.; Nanda, A.S.; Badja, C.; Koh, G.; et al. A practical framework and online tool for mutational signature analyses show inter-tissue variation and driver dependencies. Nat. Cancer 2020, 1, 249–263. [Google Scholar] [CrossRef]
- Sondka, Z.; Bamford, S.; Cole, C.G.; Ward, S.A.; Dunham, I.; Forbes, S.A. The COSMIC Cancer Gene Census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 2018, 18, 696–705. [Google Scholar] [CrossRef]
- Webster, A.L.H.; Sanders, M.A.; Patel, K.; Dietrich, R.; Noonan, R.J.; Lach, F.P.; White, R.R.; Goldfarb, A.; Hadi, K.; Edwards, M.M.; et al. Fanconi Anemia Pathway Deficiency Drives Copy Number Variation in Squamous Cell Carcinomas. Biorxiv 2021, 82, 6196. [Google Scholar] [CrossRef]
- Dorsman, J.C.; Levitus, M.; Rockx, D.; Rooimans, M.A.; Oostra, A.B.; Haitjema, A.; Bakker, S.T.; Steltenpool, J.; Schuler, D.; Mohan, S.; et al. Identification of the Fanconi anemia complementation group I gene, FANCI. Cell. Oncol. 2007, 29, 211–218. [Google Scholar] [CrossRef]
- Mehta, P.A.; Tolar, J. Fanconi Anemia. In GeneReviews(®) [Internet]; Pagon, R., Adam, M., Ardinger, H., Eds.; University of Washington: Seattle, WA, USA, 2002. [Google Scholar]
- Savage, S.A.; Ballew, B.J.; Giri, N.; Dceg, N.C.I.; Genomics, C.; Chandrasekharappa, S.C.; Ameziane, N.; Winter, J.D.; Alter, B.P.; Dceg, N.C.I.; et al. Novel FANCI Mutations in Fanconi Anemia with VACTERL Association. Am. J. Med. Genet. Part A 2015, 170A, 386–391. [Google Scholar] [CrossRef]
- George, M.; Solanki, A.; Chavan, N.; Rajendran, A.; Raj, R.; Mohan, S.; Nemani, S.; Kanvinde, S.; Munirathnam, D.; Rao, S.; et al. A comprehensive molecular study identified 12 complementation groups with 56 novel FANC gene variants in Indian Fanconi anemia subjects. Hum. Mutat. 2021, 42, 1648–1665. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jones, M.J.K.; Yin, Y.; Crist, S.B.; Colnaghi, L.; Sims III, R.J.; Rothenberg, E.; Jallepalli, P.V.; Huang, T.T. ATR-mediated phosphorylation of FANCI regulates dormant origin firing in response to replication stress. Mol. Cell 2016, 58, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lu, X.; Akhter, S.; Georgescu, M.; Legerski, R.J. FANCI is a negative regulator of Akt activation. Cell Cycle 2016, 15, 1134–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sondalle, S.B.; Longerich, S.; Ogawa, L.M.; Sung, P.; Baserga, S.J. Fanconi anemia protein FANCI functions in ribosome biogenesis. Proc. Natl. Acad. Sci. USA 2019, 116, 2561–2570. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Giri, N.; Leathwood, L.; Risch, M.O.; Carr, A.G.; Alter, B.P. Risk of cancer in heterozygous relatives of patients with Fanconi anemia. Genet. Med. 2022, 24, 245–250. [Google Scholar] [CrossRef]
- Berwick, M.; Satagopan, J.M.; Ben-Porat, L.; Carlson, A.; Mah, K.; Henry, R.; Diotti, R.; Milton, K.; Pujara, K.; Landers, T.; et al. Genetic heterogeneity among fanconi anemia heterozygotes and risk of cancer. Cancer Res. 2007, 67, 9591–9596. [Google Scholar] [CrossRef] [Green Version]
- Swift, M.; Caldwell, R.J.; Chase, C. Reassessment of cancer predisposition of Fanconi anemia heterozygotes. J. Natl. Cancer Inst. 1980, 65, 863–867. [Google Scholar]
- Maxwell, K.N.; Wubbenhorst, B.; Wenz, B.M.; Sloover, D.D.; Pluta, J.; Emery, L.; Barrett, A.; Kraya, A.A.; Anastopoulos, I.N.; Yu, S.; et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nat. Commun. 2017, 8, 319. [Google Scholar] [CrossRef] [Green Version]
- Ovarian, Fallopian Tube, and Primary Peritoneal Cancer Prevention (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute: Bethesda, MD, USA, 2002.
- Guidi, S.; Berghella, V.; Scambia, G.; Fagotti, A.; Vidiri, A.; Restaino, S.; Vizzielli, G.; Inzani, F.; Cavaliere, A.F. Adult Granulosa Cell Tumor in Pregnancy: A New Case and a Review of the Literature. Healthcare 2021, 9, 1455. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, A.F.; Perelli, F.; Zaami, S.; D’Indinosante, M.; Turrini, I.; Giusti, M.; Gullo, G.; Vizzielli, G.; Mattei, A.; Scambia, G.; et al. Fertility Sparing Treatments in Endometrial Cancer Patients: The Potential Role of the New Molecular Classification. Int. J. Mol. Sci. 2021, 22, 12248. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2019. Nucleic Acids Res. 2019, 47, D948–D954. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | Coding Change | Protein Change | F1528-PT0056 1 | F1528-PT0057 1 | F1085-PT0134 2 | F1085-PT0135 2 | F1601-PT0138 | F845-PT0196 | F1490-PT0047 | F1620-PT0100 | F1506-PT0136 | F1543-PT0137 | F1288-PT0158 | F1617-PT0090 | F694-PT0128 | F439-PT0184 | F1650-PT0142 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FANCI | c.1813C>T | p.L605F | x | x | x | x | |||||||||||
| PTPN22 | c.993-1G>A | NA | x | x | x | x | |||||||||||
| GPD1 | c.431T>C | p.M144T | x | x | x | ||||||||||||
| SEC14L4 | c.364C>T | p.R122W | x | x | x | ||||||||||||
| PIWIL3 | c.2023T>G | p.C675R | x | x | |||||||||||||
| PIWIL3 | c.1932+1G>A | NA | x | ||||||||||||||
| CACNA1S | c.4340G>A | p.R1447Q | x | x | |||||||||||||
| CACNA1S | c.773G>A | p.G258D | x | ||||||||||||||
| MYO7A | c.5866G>A | p.V1956I | x | x | |||||||||||||
| MYO7A | c.1078G>T | p.E360 * | x | ||||||||||||||
| SCN10A | c.3776G>A | p.R1259Q | x | x | |||||||||||||
| SCN10A | c.2972C>T | p.P991L | x | ||||||||||||||
| PCDH15 | c.3127C>T | p.P1043S | x | x | |||||||||||||
| PCDH15 | c.2581G>A | p.V861M | x | ||||||||||||||
| TEX2 | c.73G>T | p.V25L | x | x | |||||||||||||
| TEX2 | c.3040G>A | p.E1014K | x | ||||||||||||||
| DNAH3 | c.10382C>G | p.P3461R | x | x | |||||||||||||
| DNAH3 | c.5368A>T | p.I1790F | x | ||||||||||||||
| DNAH1 | c.1941_1944del | p.N648Afs * 36 | x | x | |||||||||||||
| DNAH1 | c.2717A>G | p.D906G | x | ||||||||||||||
| DNAH1 | c.10216G>A | p.V3406I | x | ||||||||||||||
| IQCA1 | c.29G>A | p.W10 * | x | x | |||||||||||||
| IQCA1 | c.979G>C | p.A327P | x |
| Gene | Coding Change | Protein Change | No. of Carriers (%) |
|---|---|---|---|
| CACNA1S | c.4340G>A | p.R1447Q | 1 (0.2) |
| NBAS | c.3217C>T | p.R1073C | 6 (1.2) |
| ANKAR | c.3815G>A | p.R1272H | 2 (0.4) |
| PARD3B | c.365T>C | p.I122T | 1 (0.2) |
| TNS1 | c.1333G>C | p.G445R | 4 (0.8) |
| IQCA1 | c.29G>A | p.W10 * | 3 (0.6) |
| CXCL6 | c.239dup | p.V81Gfs * 44 | 9 (1.7) |
| CEP120 | c.2134C>T | p.L712F | 8 (1.6) |
| KCNU1 | c.2731G>A | p.A911T | 1 (0.2) |
| NUP188 | c.3974G>A | p.R1325H | 4 (0.8) |
| CREM | c.677C>T | p.S226L | 5 (1) |
| PCDH15 | c.3127C>T | p.P1043S | 1 (0.2) |
| NPFFR1 | c.8-2A>G | NA | 6 (1.2) |
| MYO7A | c.5866G>A | p.V1956I | 3 (0.6) |
| PWP1 | c.1402G>A | p.E468K | 7 (1.4) |
| PAQR5 | c.20C>G | p.P7R | 3 (0.6) |
| DNAH3 | c.10382C>G | p.P3461R | 1 (0.2) |
| PLIN4 | c.3260_3263dup | p.F1089Pfs * 32 | 3 (0.6) |
| CYP2A6 | c.289G>A | p.E97K | 1 (0.2) |
| ALDH16A1 | c.1376A>T | p.D459V | 5 (1) |
| MYH9 | c.4396C>T | p.R1466W | 2 (0.4) |
| Gene | Coding Change | Protein Change | F1601-PT0138 | F1506-PT0136 | F845-PT0196 | F1085-PT0134 1 | F1085-PT0135 1 | F1617-PT0090 | F694-PT0128 | F1288-PT0158 | F1543-PT0137 | F439-PT0184 | F1650-PT0142 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EZH2 | c.1786G>A | p.Ala596Thr | x | ||||||||||
| ANKRD55 | c.1126T>C | p.Ser376Pro | x | ||||||||||
| MOV10 | c.2501G>A | p.Arg834Gln | x | ||||||||||
| LRRK2 | c.356T>C | p.Leu119Pro | x |
| Sample ID | TP53 | BRCA1 | CSMD3 | NF1 | CDK12 | FAT3 | GABRA6 | BRCA2 | RB1 |
|---|---|---|---|---|---|---|---|---|---|
| PT0001 | |||||||||
| PT0002 | |||||||||
| PT0003 | |||||||||
| PT0004 | |||||||||
| PT0006 | |||||||||
| PT0005 | |||||||||
| PT0007 | |||||||||
| TCGA-04-1336 | |||||||||
| TCGA-24-1603 | |||||||||
| TCGA-25-2393 | |||||||||
| TCGA-29-2431 | |||||||||
| TCGA-61-1903 | |||||||||
| TCGA-61-2009 | |||||||||
| Total (%) | 11 (85%) | 0 | 0 | 0 | 1 (8%) | 3 (23%) | 0 | 3 (23%) | 0 |
| Cancer Type (TCGA Acronym) | Total No. Cases | No. of FANCI c.1813C>T Carriers | Carrier Frequency of FANCI c.1813C>T (%) |
|---|---|---|---|
| Adrenocortical carcinoma (ACC) | 92 | 3 | 3.3 |
| Kidney chromophobe (KICH) | 66 | 2 | 3 |
| Lung squamous cell carcinoma (LUSC) | 499 | 14 | 2.8 |
| Skin cutaneous melanoma (SKCM) | 470 | 13 | 2.8 |
| Kidney renal clear cell carcinoma (KIRC) | 387 | 10 | 2.6 |
| Colon adenocarcinoma (COAD) | 419 | 10 | 2.4 |
| Cholangiocarcinoma (CHOL) | 45 | 1 | 2.2 |
| Esophageal carcinoma (ESCA) | 184 | 4 | 2.2 |
| Brain lower-grade glioma (LGG) | 515 | 11 | 2.1 |
| Liver hepatocellular carcinoma (LIHC) | 375 | 8 | 2.1 |
| Head and neck squamous cell carcinoma (HNSC) | 526 | 9 | 1.7 |
| Uterine corpus endometrial carcinoma (UCEC) | 543 | 9 | 1.7 |
| Breast invasive carcinoma (BRCA) 1 | 1076 | 17 | 1.6 |
| Cervical squamous cell carcinoma (CESC) | 305 | 5 | 1.6 |
| Sarcoma (SARC) | 255 | 4 | 1.6 |
| Stomach adenocarcinoma (STAD) | 443 | 7 | 1.6 |
| Ovarian serous cystadenocarcinoma (OV) | 412 | 6 | 1.5 |
| Testicular germ cell tumours (TGCT) | 134 | 2 | 1.5 |
| Lung adenocarcinoma (LUAD) | 518 | 7 | 1.4 |
| Rectum adenocarcinoma (READ) | 145 | 2 | 1.4 |
| Uveal melanoma (UVM) | 80 | 1 | 1.3 |
| Bladder urothelial carcinoma (BLCA) | 412 | 5 | 1.2 |
| Mesothelioma (MESO) | 82 | 1 | 1.2 |
| Thyroid carcinoma (THCA) | 499 | 6 | 1.2 |
| Pancreatic adenocarcinoma (PAAD) | 185 | 2 | 1.1 |
| Pheochromocytoma and paraganglioma (PCPG) | 179 | 2 | 1.1 |
| Glioblastoma multiforme (GBM) | 393 | 4 | 1 |
| Prostate adenocarcinoma (PRAD) | 498 | 5 | 1 |
| Acute myeloid leukemia (LAML) | 142 | 1 | 0.7 |
| Diffuse large B-cell carcinoma (DLBC) | 41 | 0 | 0 |
| Kidney renal papillary cell carcinoma (KIRP) | 289 | 0 | 0 |
| Thymoma (THYM) | 123 | 0 | 0 |
| Uterine carcinosarcoma (UCS) | 57 | 0 | 0 |
| Total | 10,389 | 171 | 1.6 |
| gnomAD non-cancer overall 2 | 134,164 | 1787 | 1.3 |
| Cancer type (TCGA Acronym) | Total No. of Cases | No. of Tumours Harbouring FANCI Variants | Frequency of Tumours Harbouring FANCI Variants (%) |
|---|---|---|---|
| Uterine corpus endometrial carcinoma (UCEC) | 517 | 43 | 8.32 |
| Skin cutaneous melanoma (SKCM) | 438 | 20 | 4.57 |
| Bladder urothelial carcinoma (BLCA) | 410 | 13 | 3.17 |
| Colon adenocarcinoma (COAD)/Rectum adenocarcinoma (READ) | 534 | 14 | 2.62 |
| Stomach adenocarcinoma (STAD) | 436 | 9 | 2.06 |
| Cervical squamous cell carcinoma (CESC) | 291 | 6 | 2.06 |
| Uterine carcinosarcoma (UCS) | 57 | 1 | 1.75 |
| Lung squamous cell carcinoma (LUSC) | 484 | 8 | 1.65 |
| Lung adenocarcinoma (LUAD) | 566 | 9 | 1.59 |
| Head and neck squamous cell carcinoma (HNSC) | 515 | 8 | 1.55 |
| Acute myeloid leukemia (LAML) | 200 | 3 | 1.50 |
| Mesothelioma (MESO) | 86 | 1 | 1.16 |
| Adrenocortical carcinoma (ACC) | 91 | 1 | 1.10 |
| Esophageal carcinoma (ESCA) | 182 | 2 | 1.10 |
| Glioblastoma multiforme (GBM) | 391 | 4 | 1.02 |
| Breast invasive carcinoma (BRCA) | 1066 | 10 | 0.94 |
| Thymoma (THYM) | 123 | 1 | 0.81 |
| Sarcoma (SARC) | 255 | 2 | 0.78 |
| Ovarian serous cystadenocarcinoma (OV) | 523 | 4 | 0.76 |
| Kidney renal clear cell carcinoma (KIRC) | 402 | 3 | 0.75 |
| Testicular germ cell tumours (TGCT) | 149 | 1 | 0.67 |
| Pheochromocytoma and paraganglioma (PCPG) | 178 | 1 | 0.56 |
| Liver hepatocellular carcinoma (LIHC) | 366 | 2 | 0.55 |
| Prostate adenocarcinoma (PRAD) | 494 | 2 | 0.40 |
| Brain lower-grade glioma (LGG) | 514 | 2 | 0.39 |
| Kidney renal papillary cell carcinoma (KIRP) | 276 | 1 | 0.36 |
| Thyroid carcinoma (THCA) | 489 | 1 | 0.20 |
| Cholangiocarcinoma (CHOL) | 36 | 0 | 0 |
| Diffuse large B-cell carcinoma (DLBC) | 41 | 0 | 0 |
| Kidney chromophobe (KICH) | 65 | 0 | 0 |
| Pancreatic adenocarcinoma (PAAD) | 179 | 0 | 0 |
| Uveal melanoma (UVM) | 80 | 0 | 0 |
| Total | 10,434 | 172 | 1.65 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fierheller, C.T.; Alenezi, W.M.; Serruya, C.; Revil, T.; Amuzu, S.; Bedard, K.; Subramanian, D.N.; Fewings, E.; Bruce, J.P.; Prokopec, S.; et al. Molecular Genetic Characteristics of FANCI, a Proposed New Ovarian Cancer Predisposing Gene. Genes 2023, 14, 277. https://doi.org/10.3390/genes14020277
Fierheller CT, Alenezi WM, Serruya C, Revil T, Amuzu S, Bedard K, Subramanian DN, Fewings E, Bruce JP, Prokopec S, et al. Molecular Genetic Characteristics of FANCI, a Proposed New Ovarian Cancer Predisposing Gene. Genes. 2023; 14(2):277. https://doi.org/10.3390/genes14020277
Chicago/Turabian StyleFierheller, Caitlin T., Wejdan M. Alenezi, Corinne Serruya, Timothée Revil, Setor Amuzu, Karine Bedard, Deepak N. Subramanian, Eleanor Fewings, Jeffrey P. Bruce, Stephenie Prokopec, and et al. 2023. "Molecular Genetic Characteristics of FANCI, a Proposed New Ovarian Cancer Predisposing Gene" Genes 14, no. 2: 277. https://doi.org/10.3390/genes14020277