
The Complete Chloroplast Genome Sequence of Laportea bulbifera (Sieb. et Zucc.) Wedd. and Comparative Analysis with Its Congeneric Species
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chloroplast DNA Extraction and Sequencing
2.2. Genome Assembly and Annotation
2.3. Repeat Sequences, Codon Usage and RNA Editing Sites Analysis in L. bulbifera Chloroplast Genome
2.4. Genome Comparison
2.5. Phylogenetic Analysis and the Nucleotide Substitution Rate
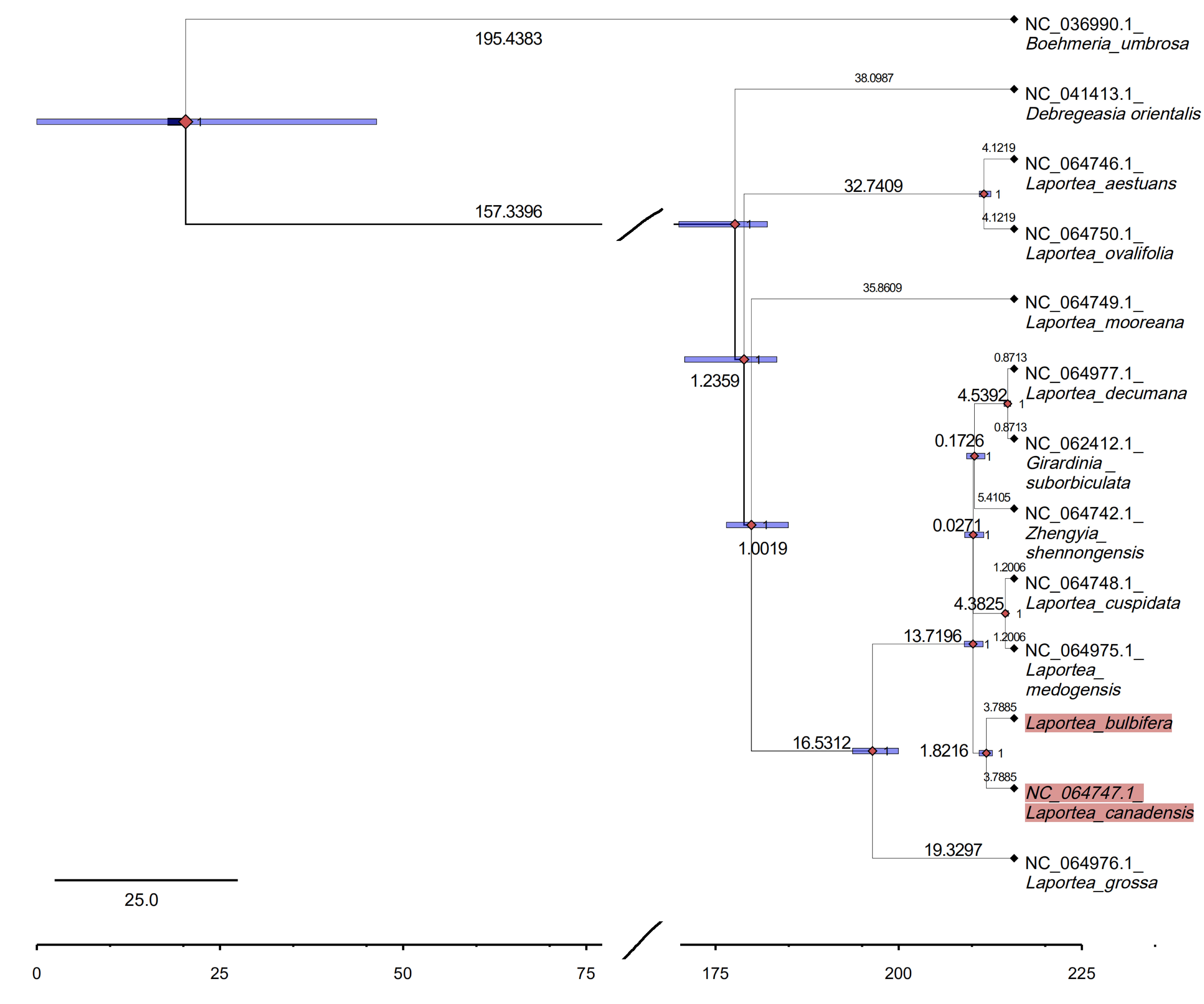
2.6. Molecular Clock Analyses
3. Results
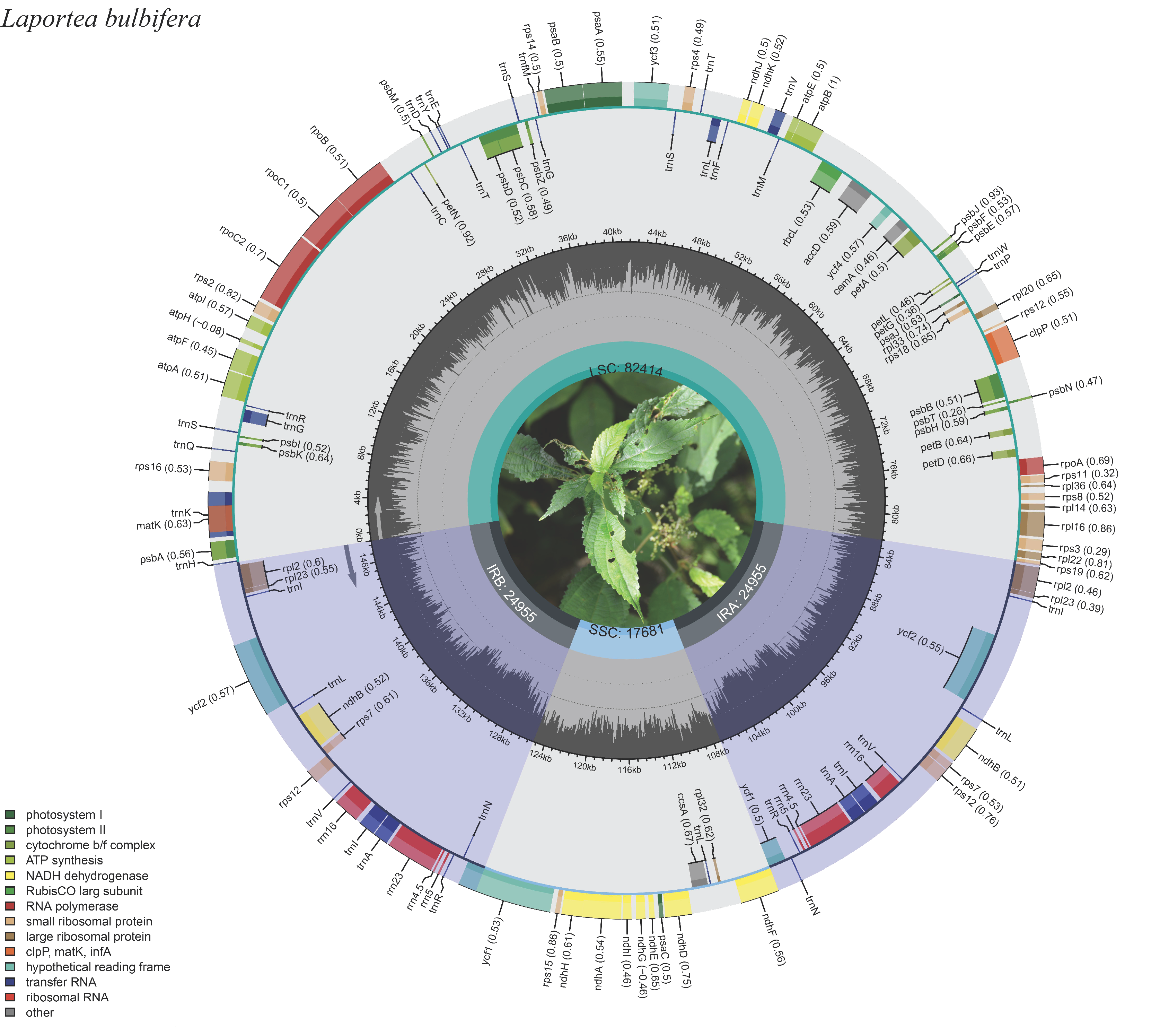
3.1. Plastome Genomes Structure and Features
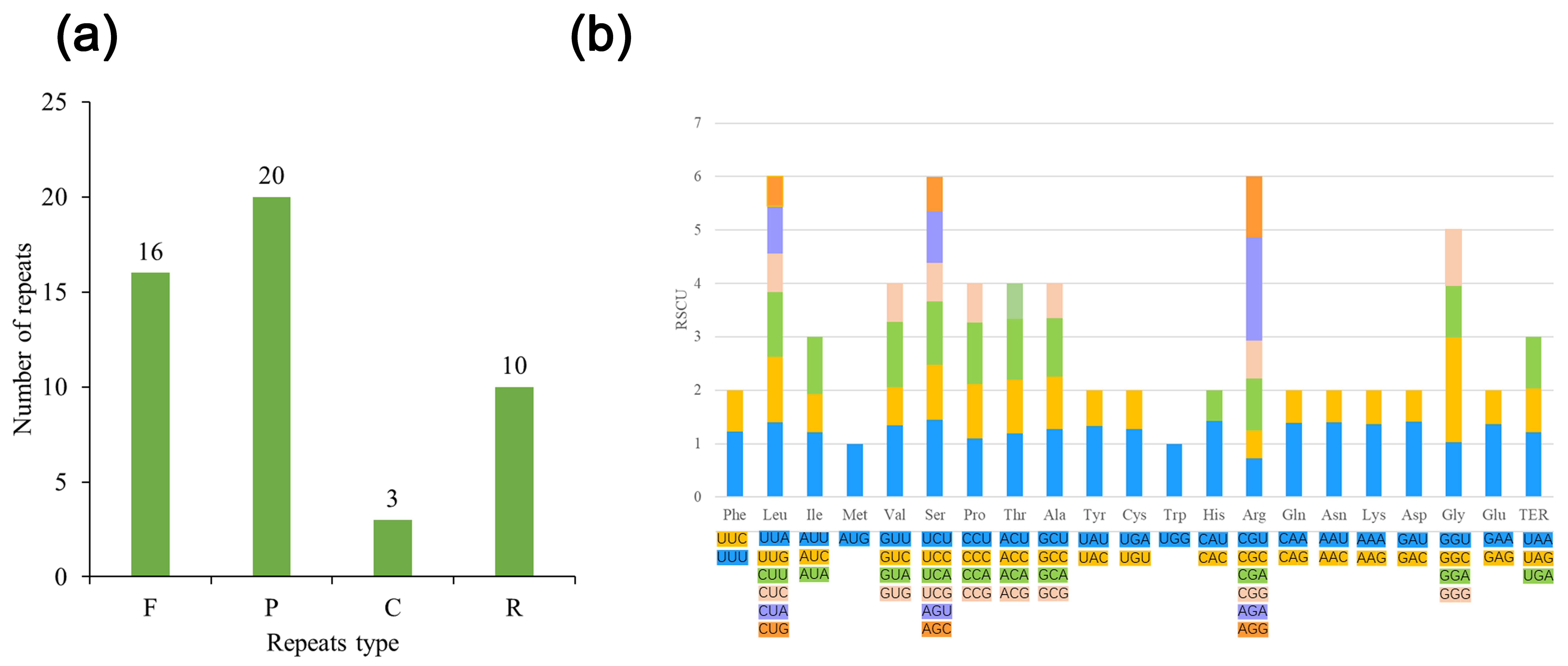
3.2. Analysis of Repeat Sequences and Codon Usage
3.3. Comparative Genomic Analysis and Divergence Hotspot Regions
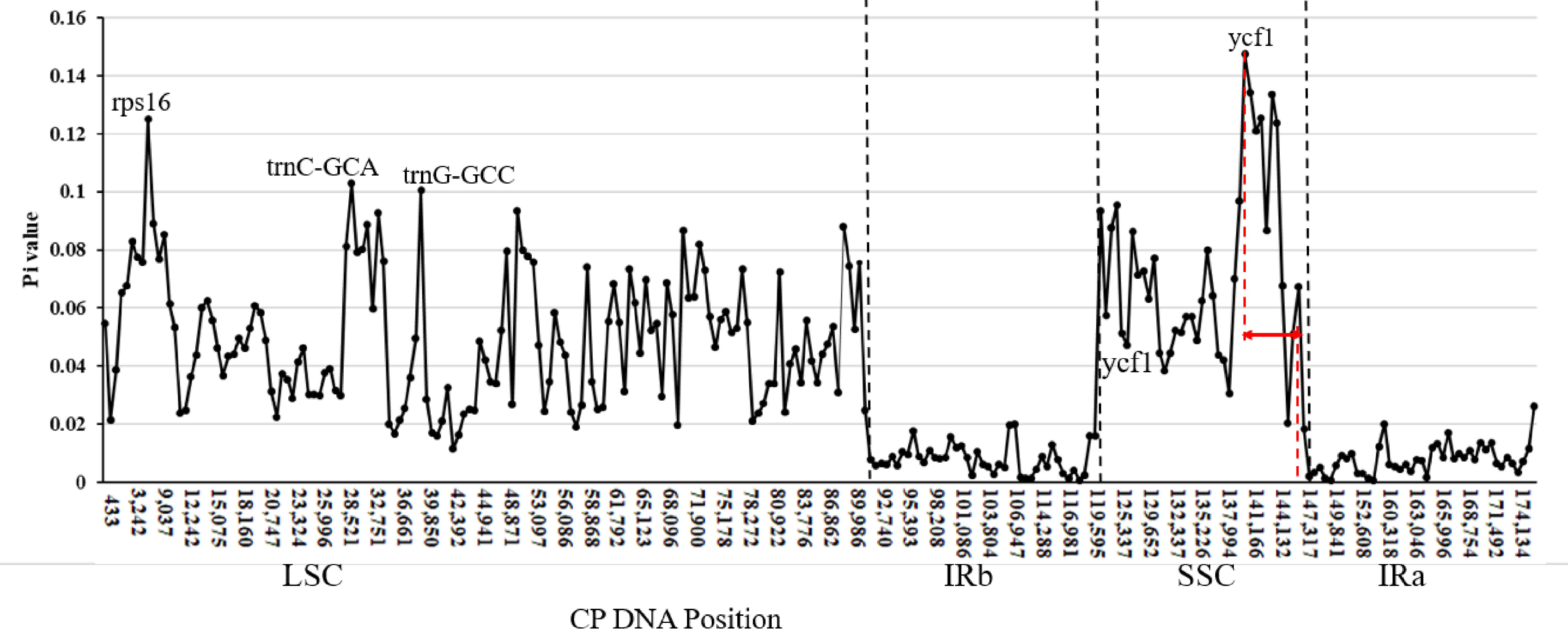
3.3.1. Sliding Window Analysis
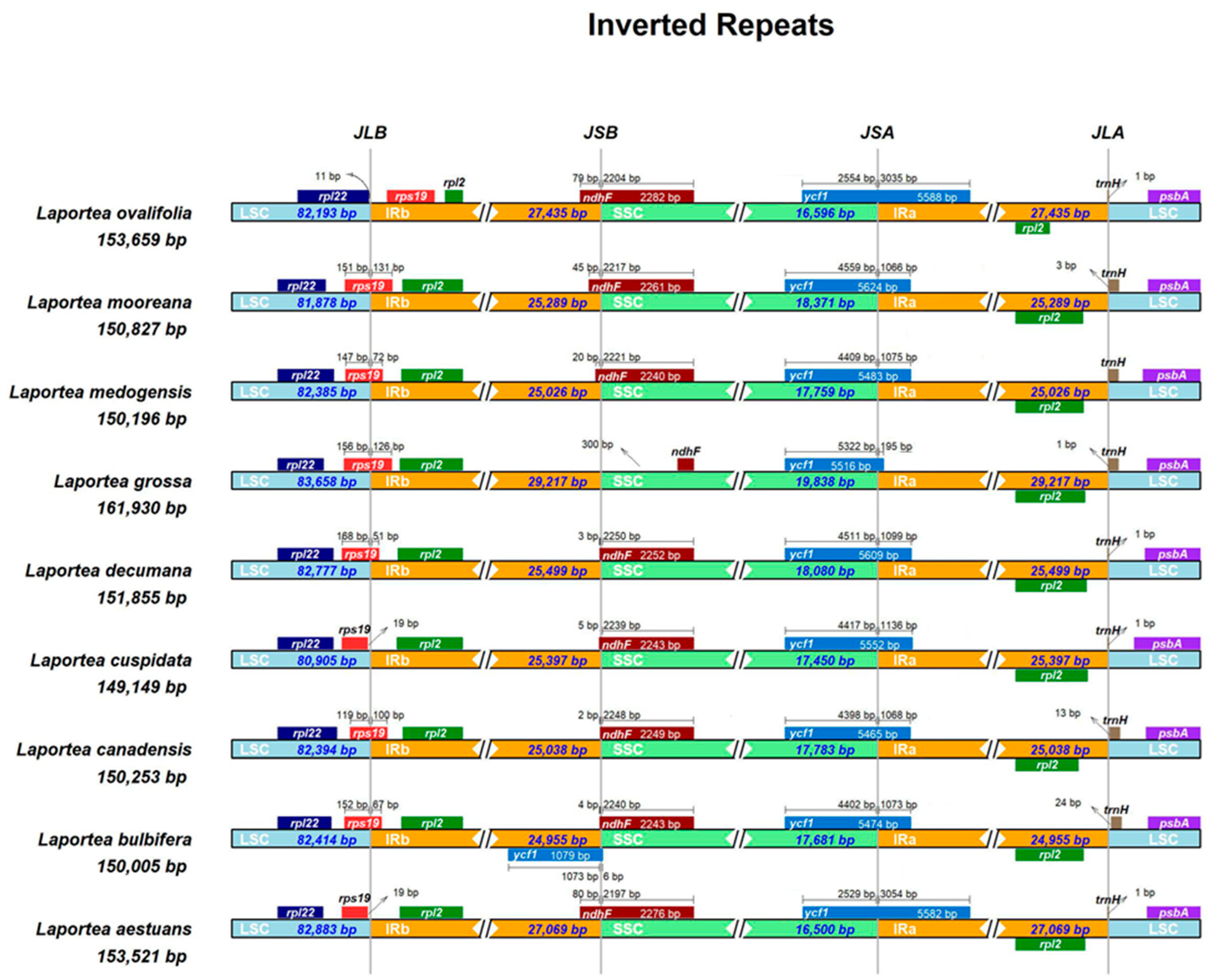
3.3.2. Boundaries of IR
3.3.3. Genome Comparison
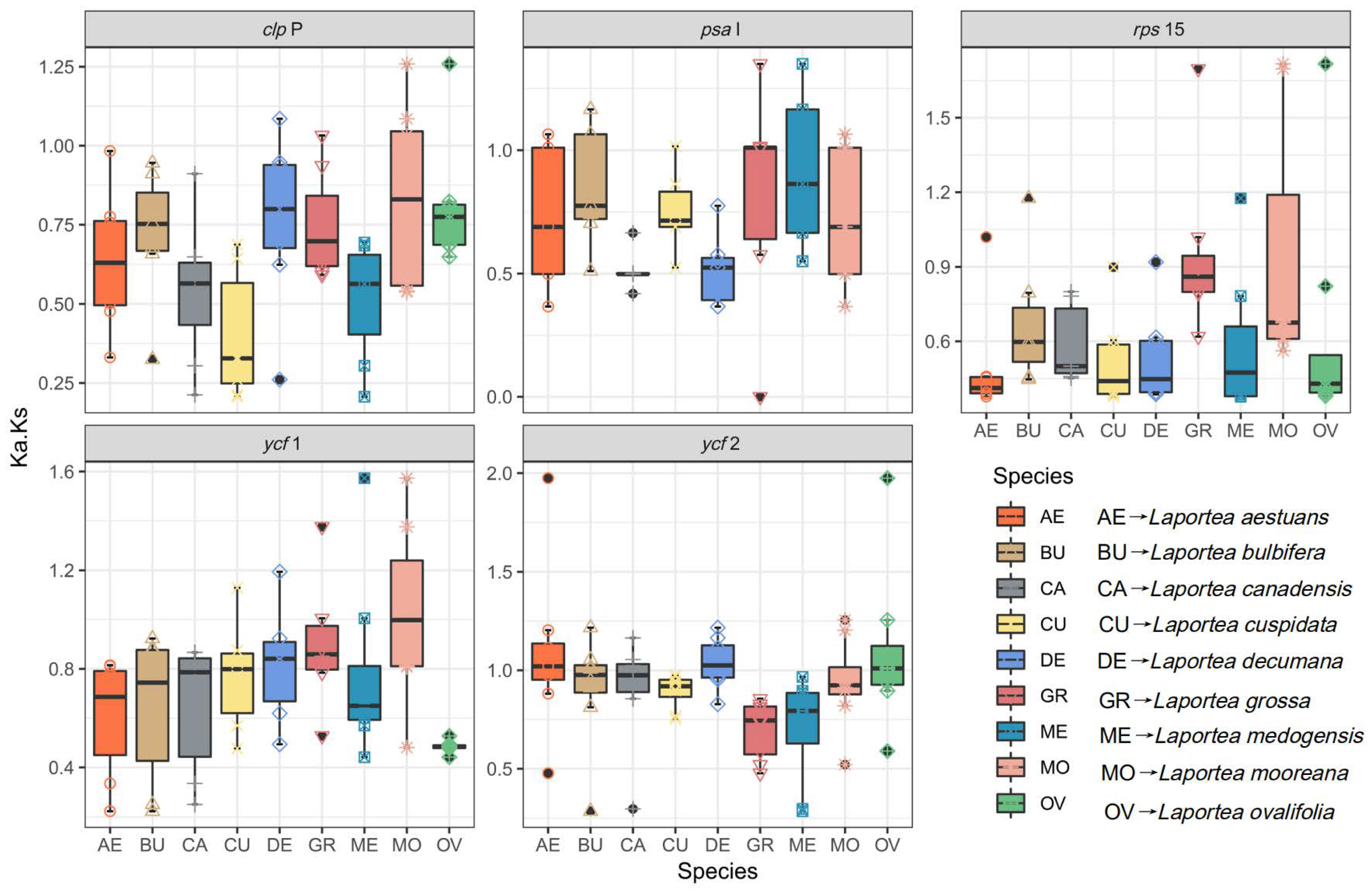
3.4. Gene Selective Pressure Analysis
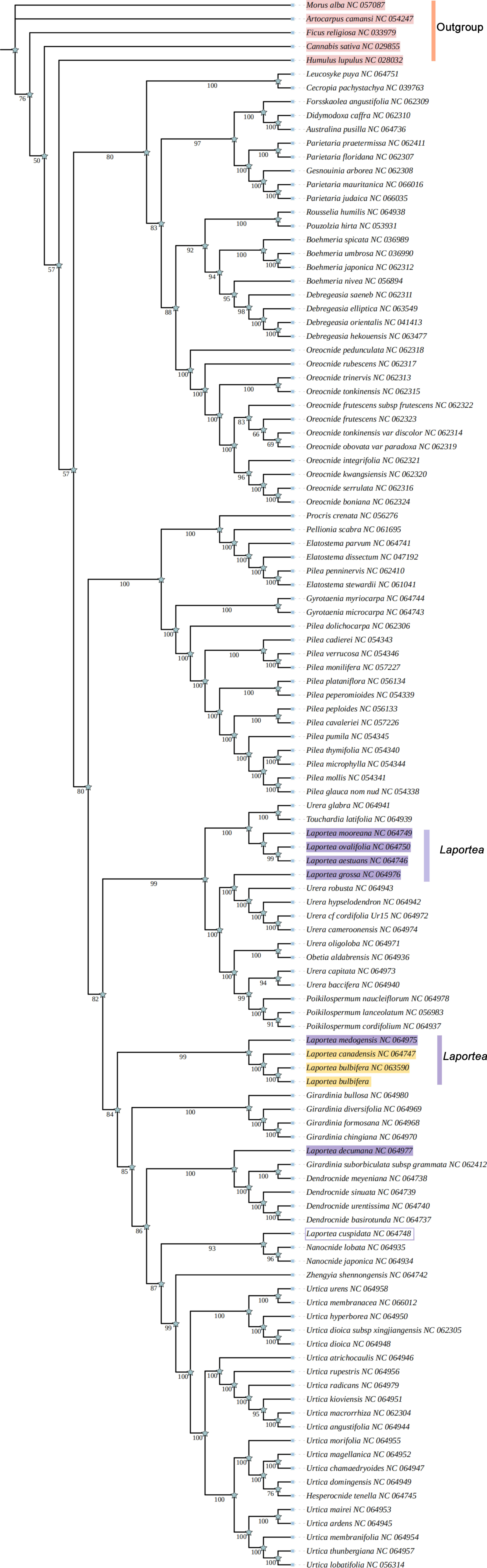
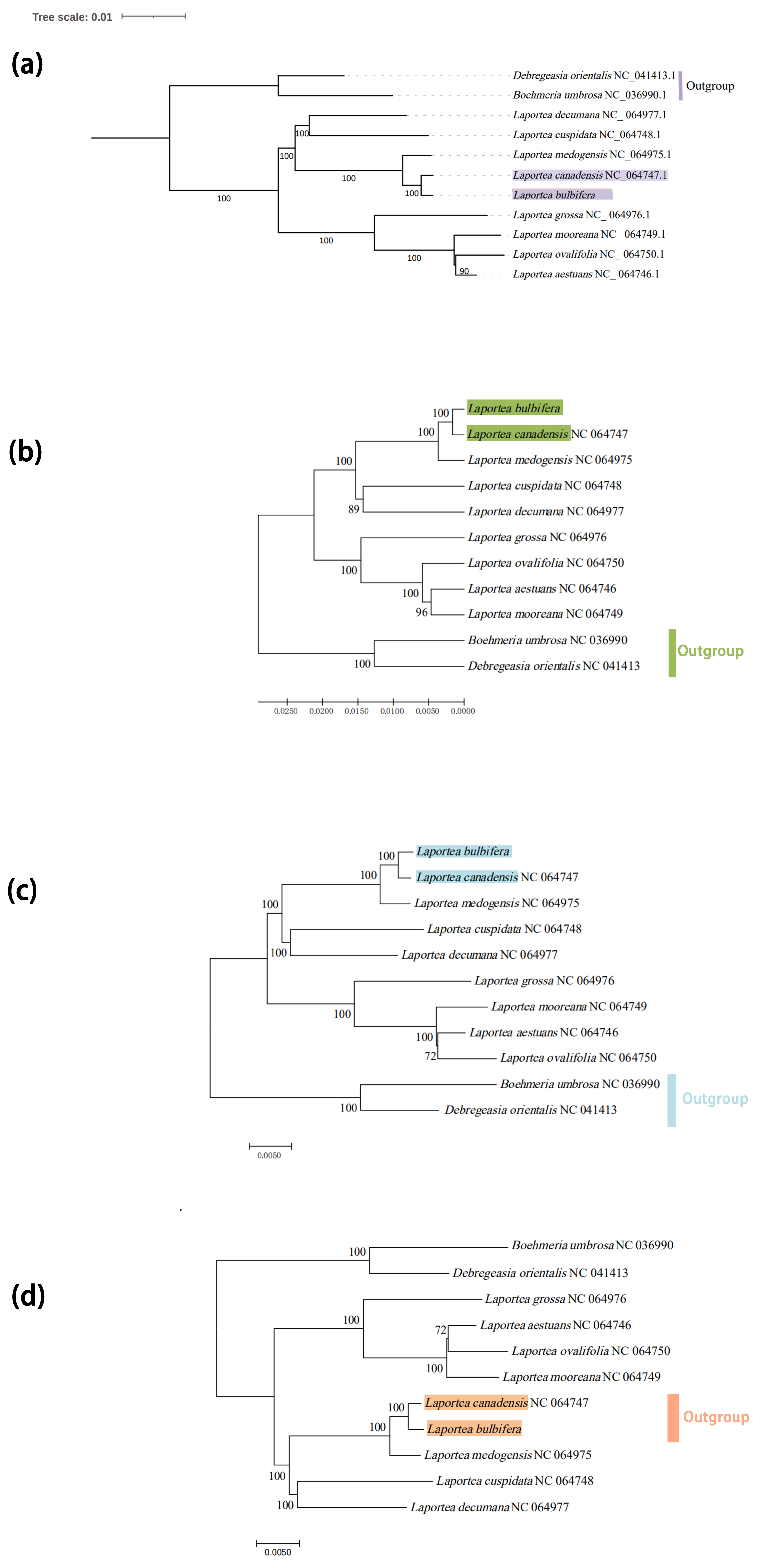
3.5. Phylogenetic Analysis and Divergence Time Estimation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raven, J.A.; Allen, J.F. Genomics and chloroplast evolution: What did cyanobacteria do for plants? Genome Biol. 2003, 4, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.F.; Wicke, S.; Sass, C. Dense infraspecific sampling reveals rapid and independent trajectories of plastome degradation in a heterotrophic orchid complex. New Phytol. 2018, 218, 1192–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, W.-L. Discocnide (Urticaceae). Gard. Bull. Singap. 1969, 24, 355–359. [Google Scholar]
- Ferguson, D.K. Flora of China Volume 9: Pittosporaceae through Connaraceae by Zhengyi Wu; Peter H. Raven. Systematic Botany 2004, 29, 464–465. [Google Scholar] [CrossRef]
- Flint, R.L.; Norton, S.A.; Strausborger, S. Laportea canadensis, the Canada Wood Nettle: Similar to Stinging Nettle in Appearance, Geographic Distribution, and Pharmacologic Effects on the Skin. Dermatitis 2022. [Google Scholar] [CrossRef]
- Wu, Z.-Y.; Monro, A.K.; Milne, R.I.; Wang, H.; Yi, T.-S.; Liu, J.; Li, D.-Z. Molecular phylogeny of the nettle family (Urticaceae) inferred from multiple loci of three genomes and extensive generic sampling. Mol. Phylogenet. Evol. 2013, 69, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Boufford, D.E. Urticaceae nettle family. J. Ariz. -Nev. Acad. Sci. 1992, 26, 42–49. [Google Scholar]
- Kim, C.; Deng, T.; Chase, M.; Zhang, D.-G.; Nie, Z.-L.; Sun, H. Generic phylogeny and character evolution in Urticeae (Urticaceae) inferred from nuclear and plastid DNA regions. Taxon 2015, 64, 65–78. [Google Scholar] [CrossRef]
- Macdonald, A. Theoretical problems of interpreting floral organogenesis of Laportea canadensis. Can. J. Bot. 2011, 52, 639–644. [Google Scholar] [CrossRef]
- Wang, J.; Lu, J.; Lan, Y.; Zhou, H.; Li, W.; Xiang, M. Total coumarins from Urtica dentata Hand prevent murine autoimmune diabetes via suppression of the TLR4-signaling pathways. J. Ethnopharmacol. 2013, 146, 379–392. [Google Scholar] [CrossRef]
- Chen, Y.; Zou, S.; Xu, W.; Sun, Q.; Yun, L. Spectrum–effect relationship of antioxidant and anti-inflammatory activities of Laportea bulbifera based on multivariate statistical analysis. Biomedical Chromatography 2020, 34, e4734–e4746. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhao, Y.; Li, B.; Feng, W.; Qi, J.; Feng, B. Phytochemical, Chemotaxonomic and Bioinformatics Study on Laportea bulbifera (Urticaceae). Chem. Biodivers. 2022, 19, e202200070. [Google Scholar] [CrossRef] [PubMed]
- Chaniad, P.; Tewtrakul, S.; Sudsai, T.; Langyanai, S.; Kaewdana, K. Anti-inflammatory, wound healing and antioxidant potential of compounds from Dioscorea bulbifera L. bulbils. PLoS ONE 2020, 15, e0243632. [Google Scholar] [CrossRef]
- Christensen, C.B.; Soelberg, J.; Jäger, A.K. Antacid activity of Laportea aestuans (L.) Chew. J. Ethnopharmacol. 2015, 171, 1–3. [Google Scholar] [CrossRef]
- Ogoma, C.A.; Liu, J.; Stull, G.W.; Wambulwa, M.C.; Oyebanji, O.; Milne, R.I.; Monro, A.K.; Zhao, Y.; Li, D.-Z.; Wu, Z.-Y. Deep Insights Into the Plastome Evolution and Phylogenetic Relationships of the Tribe Urticeae (Family Urticaceae). Front. Plant Sci. 2022, 13, 870949–870966. [Google Scholar] [CrossRef]
- Zhu, Z.; Ma, L.; Zhu, H.; Yang, X.; Hao, X. Studies on the chemical constituents of Laportea bulbifera. J. Chin. Med. Mater. 2011, 34, 223–225. [Google Scholar]
- Qiu Dewen, D.J. Chinese Materia Medica Miao Medicine Volume; Guizhou Science and Technology Publishing House: Guiyang, China, 2005. [Google Scholar]
- Guizhou Medical Products Administration. Quality Standards of Traditional Chinese Medicine and Ethnic Medicine in Guizhou Province; Beijing Science and Technology Press: Beijing, China, 2020. [Google Scholar]
- Arseneau, J.R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Peden, J. CodonW, version 1.4.2; University of Nottingham: Nottingham, UK, 2005. [Google Scholar]
- Peden, J. Analysis of Codon Usage. Ph.D. Thesis, Department of Genetics, University of Nottingham, Nottingham, UK, 1999. [Google Scholar]
- Sharp, P.M.; Li, W.-H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [Green Version]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef]
- Rzhetsky, A.; Nei, M. A Simple Method for Estimating and Testing Minimum-Evolution Trees. Mol. Biol. Evol. 1991, 9. [Google Scholar] [CrossRef]
- Som, A. Theoretical foundation to estimate the relative efficiencies of the Jukes-Cantor+gamma model and the Jukes-Cantor model in obtaining the correct phylogenetic tree. Gene 2006, 385, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A.; Arif, I.A.; Bahkali, A.H.; Al Farhan, A.H.; Al Homaidan, A.A. Bayesian, maximum parsimony and UPGMA models for inferring the phylogenies of antelopes using mitochondrial markers. Evol. Bioinform. Online 2008, 4, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 2013, 30, 1229–1235. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Takhtajan, A. Ulmaceae-Betulaceae. Foss. Flower. Plants USSR 1982, 2, 216. [Google Scholar]
- Deng, T.; Kim, C.; Zhang, D.-G.; Zhang, J.-W.; Li, Z.-M.; Nie, Z.-L.; Sun, H. Zhengyia shennongensis: A new bulbiliferous genus and species of the nettle family (Urticaceae) from central China exhibiting parallel evolution of the bulbil trait. Taxon 2013, 62, 89–99. [Google Scholar] [CrossRef]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Fang, Y.; Wang, X.; Deng, X.; Zhang, X.; Hu, S.; Yu, J. The complete chloroplast and mitochondrial genome sequences of Boea hygrometrica: Insights into the evolution of plant organellar genomes. PLoS ONE 2012, 7, e30531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Miao, Y.; Sun, X.; Jiang, Y.; Zeng, T.; Zheng, Y.; Huang, L. The complete chloroplast genome sequence of Gentiana triflora and comparative analysis with its congeneric species. J. Appl. Bot. Food Qual. 2022, 95, 51–59. [Google Scholar]
- Zhang, X.; Zhou, T.; Kanwal, N.; Zhao, Y.; Bai, G.; Zhao, G. Completion of Eight Gynostemma BL. (Cucurbitaceae) Chloroplast Genomes: Characterization, Comparative Analysis, and Phylogenetic Relationships. Front. Plant Sci. 2017, 8, 1583. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Zhou, T.; Han, K.; Yang, Y.; Zhao, J.; Liu, Z.L. The Complete Chloroplast Genome Sequences of Six Rehmannia Species. Genes 2017, 8, 103–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, T.; Wang, H.L.; Li, Z.J.; Ni, J.W.; Su, S.; Xu, X.Q. Comparative and Phylogenetic Analyses of the Complete Chloroplast Genomes of Six Almond Species (Prunus spp. L.). Sci. Rep. 2020, 10, 10137. [Google Scholar] [CrossRef]
- Goulding, S.E.; Wolfe, K.; Olmstead, R.; Morden, C. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. MGG 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Duchene, D.; Bromham, L. Rates of molecular evolution and diversification in plants: Chloroplast substitution rates correlate with species-richness in the Proteaceae. BMC Evol. Biol. 2013, 13, 65–76. [Google Scholar] [CrossRef]
- Miao, Y.; Chen, H.; Xu, W.; Yang, Q.; Liu, C.; Huang, L. Structural mutations of small single copy (SSC) region in the plastid genomes of five Cistanche species and inter-species identification. BMC Plant Biol. 2022, 22, 412–425. [Google Scholar] [CrossRef]
- Hjelmen, C.E.; Johnston, J.S. The mode and tempo of genome size evolution in the subgenus Sophophora. PLoS ONE 2017, 12, e0173505. [Google Scholar] [CrossRef]
- Banerjee, R.; Chaudhari, N.M.; Lahiri, A.; Gautam, A.; Bhowmik, D.; Dutta, C.; Chattopadhyay, S.; Huson, D.H.; Paul, S. Interplay of Various Evolutionary Modes in Genome Diversification and Adaptive Evolution of the Family Sulfolobaceae. Front. Microbiol. 2021, 12, 639995. [Google Scholar] [CrossRef]
- Wilson, D.J. GenomegaMap: Within-Species Genome-Wide dN/dS Estimation from over 10,000 Genomes. Mol. Biol. Evol. 2020, 37, 2450–2460. [Google Scholar] [CrossRef] [Green Version]
- Conn, B.J.; Hadiah, J.T. Nomenclature of tribes within the Urticaceae. Kew Bull. 2009, 64, 349–352. [Google Scholar] [CrossRef]
- Sytsma, K.J.; Morawetz, J.; Pires, J.C.; Nepokroeff, M.; Conti, E.; Zjhra, M.; Hall, J.C.; Chase, M.W. Urticalean rosids: Circumscription, rosid ancestry, and phylogenetics based on rbcL, trnL-F, and ndhF sequences. Am. J. Bot. 2002, 89, 1531–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadiah, J.T.; Conn, B.J.; Quinn, C.J. Infra-familial phylogeny of Urticaceae, using chloroplast sequence data. Aust. Syst. Bot. 2008, 21, 375–385. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Tang, J.; Zeng, S.; Han, F.; Yuan, J.; Yu, J. Comparative plastid genomics of four Pilea (Urticaceae) species: Insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 2021, 21, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Fritsch, P.W.; Moore, M.J.; Feng, T.; Meng, A.; Yang, J.; Deng, T.; Zhao, C.; Yao, X.; Sun, H. Plastid phylogenomics resolves infrafamilial relationships of the Styracaceae and sheds light on the backbone relationships of the Ericales. Mol. Phylogenet. Evol. 2018, 121, 198–211. [Google Scholar] [CrossRef]
- Xue, S.; Shi, T.; Luo, W.; Ni, X.; Iqbal, S.; Ni, Z.; Huang, X.; Yao, D.; Shen, Z.; Gao, Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic. Res. 2019, 6, 89–111. [Google Scholar] [CrossRef] [Green Version]
- Qi, W.H.; Jiang, X.M.; Yan, C.C.; Zhang, W.Q.; Xiao, G.S.; Yue, B.S.; Zhou, C.Q. Distribution patterns and variation analysis of simple sequence repeats in different genomic regions of bovid genomes. Sci. Rep. 2018, 8, 14407. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Jiang, J.; Chen, S.; Qi, X.; Peng, H.; Li, P.; Song, A.; Guan, Z.; Fang, W.; Liao, Y.; et al. Next-generation sequencing of the Chrysanthemum nankingense (Asteraceae) transcriptome permits large-scale unigene assembly and SSR marker discovery. PLoS ONE 2013, 8, e62293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Rong, C.; Qin, L.; Mo, C.; Fan, L.; Yan, J.; Zhang, M. Complete Chloroplast Genome Sequence of Malus hupehensis: Genome Structure, Comparative Analysis, and Phylogenetic Relationships. Molecules 2018, 23, 2917–2934. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.; Li, J.; Zhang, H.; Cai, B.; Gao, Z.; Qiao, Y.; Mi, L. The complete chloroplast genome sequence of strawberry (Fragaria × ananassa Duch.) and comparison with related species of Rosaceae. Peer. J. 2017, 5, e3919. [Google Scholar] [CrossRef] [Green Version]
- Hishamuddin, M.S.; Lee, S.Y.; Ng, W.L.; Ramlee, S.I.; Lamasudin, D.U.; Mohamed, R. Comparison of eight complete chloroplast genomes of the endangered Aquilaria tree species (Thymelaeaceae) and their phylogenetic relationships. Sci. Rep. 2020, 10, 13034. [Google Scholar] [CrossRef]
- Huang, L.S.; Sun, Y.Q.; Jin, Y.; Gao, Q.; Hu, X.G.; Gao, F.L.; Yang, X.L.; Zhu, J.J.; El-Kassaby, Y.A.; Mao, J.F. Development of high transferability cpSSR markers for individual identification and genetic investigation in Cupressaceae species. Ecol. Evol. 2018, 8, 4967–4977. [Google Scholar] [CrossRef] [Green Version]
- Alzahrani, D.; Albokhari, E.; Yaradua, S.; Abba, A. Complete chloroplast genome sequences of Dipterygium glaucum and Cleome chrysantha and other Cleomaceae Species, comparative analysis and phylogenetic relationships. Saudi J. Biol. Sci. 2021, 28, 2476–2490. [Google Scholar] [CrossRef]
- Bondar, E.I.; Troukhan, M.E.; Krutovsky, K.V.; Tatarinova, T.V. Genome-Wide Prediction of Transcription Start Sites in Conifers. Int. J. Mol. Sci. 2022, 23, 1735–1751. [Google Scholar] [CrossRef]
- Wang, R.-N.; Milne, R.I.; Du, X.-Y.; Liu, J.; Wu, Z.-Y. Characteristics and mutational hotspots of plastomes in Debregeasia (Urticaceae). Front. Genet. 2020, 11, 729–740. [Google Scholar] [CrossRef]
- Li, Y.; Dong, Y.; Liu, Y.; Yu, X.; Yang, M.; Huang, Y. Comparative analyses of Euonymus chloroplast genomes: Genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 2021, 11, 593984–593999. [Google Scholar] [CrossRef]
- Menges, E.S. Biomass Allocation and Geometry of the Clonal Forest Herb Laportea canadensis: Adaptive Responses to the Environment or Allometric Constraints? Am. J. Bot. 1987, 74, 551–563. [Google Scholar] [CrossRef]
- Dong, W.; Liu, J.; Yu, J.; Wang, L.; Zhou, S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS ONE 2012, 7, e35071. [Google Scholar] [CrossRef]
- Bhellum, B.L.; Singh, B. A new species of Laportea Gaudich.(Urticaceae) from Himalaya, India. Bangladesh J. Plant Taxon. 2016, 23, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Sun, Q.; Xu, W.; Chen, C.; Wang, B.; Wang, Y. The complete chloroplast genome of Laportea bulbifera (Sieb. et Zucc.) Wedd. and its phylogenetic analysis. Mitochondrial DNA Part B 2022, 7, 658–660. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | L. bulbifera | |
|---|---|---|
| Length (bp) | Total | 150,005 |
| LSC | 82,414 | |
| SSC | 17,681 | |
| IR | 24,955 | |
| GC content (%) | Total | 36.81 |
| LSC | 34.44 | |
| SSC | 30.64 | |
| IR | 42.91 | |
| Gene numbers | Total | 126 |
| Protein-coding gene | 81 | |
| tRNA gene | 37 | |
| rRNA gene | 8 |
| Category of Genes | Group of Genes | Name of Genes |
|---|---|---|
| rRNA | rRNA gene | rrn16s (×2), rrn23s (×2), rrn4.5s (×2), rrn5s (×2) |
| tRNA | tRNA gene | trnR-UCU, trnG-UCC, trnH-GUG, trnK-UUU, trnQ-UUG, trnC-GCA, trnD-GUC, trnY-GUA, trnE-UUC, trnL-UAG, trnT-GGU, trnS-UGA, trnG-GCC, trnfM-CAU, trnT-UGU, trnL-UAA, trnF-GAA, trnV-UAC, trnM-CAU, trnW-CCA, trnP-UGG, trnI-CAU (×2), trnL-CAA (×2), trnV-GAC (×2), trnI-GAU (×2), trnA-UGC (×2), trnR-ACG (×2), trnN-GUU (×2), trnS-GCU (×2) |
| Self-replication | Large subunit of ribosome | rpl14, rpl16, rpl2(×2), rpl20, rpl22, rpl23(×2), rpl32, rpl33, rpl36 |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Small subunit of ribosome | rps11, rps12(×2), rps14, rps15, rps16, rps18, rps19, rps2, rps3, rps4, rps7(×2), rps8 | |
| Photosynthesis | Subunits of ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI |
| Subunits of photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbI, psbJ, psbK, psbM, psbN, psbT, psbZ, ycf3 | |
| Subunits of NADH-dehydrogenase | ndhA, ndhB (×2), ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | |
| Subunits of cytochrome b/f complex | petA, petB, petD, petG, petL, petN | |
| Subunits of photosystem I | psaA, psaB, psaC, psaJ | |
| Subunit of rubisco | rbcL | |
| Other genes | Subunit of Acetyl-CoA-carboxylase | accD |
| c-type cytochrom synthesis gene | ccsA | |
| Envelop membrane protein | cemA | |
| Protease | clpP | |
| Maturase | matK | |
| Genes of unknown | Conserved open reading frames | ycf1 (×2), ycf2 (×2), ycf4 |
| Gene | Strand | Length (bp) | ||||||
|---|---|---|---|---|---|---|---|---|
| Start | End | ExonI | IntronI | ExonII | IntronII | ExonIII | ||
| trnK-UUU | — | 1613 | 4232 | 37 | 2546 | 37 | ||
| rps16 | — | 4891 | 6016 | 42 | 865 | 219 | ||
| trnG-UCC | + | 8582 | 9344 | 23 | 692 | 48 | ||
| atpF | — | 11,364 | 12,614 | 145 | 708 | 398 | ||
| rpoC1 | — | 20,130 | 22,961 | 432 | 753 | 1647 | ||
| ycf3 | — | 41,692 | 43,696 | 124 | 776 | 232 | 722 | 151 |
| trnL-UAA | + | 46,505 | 47,065 | 35 | 476 | 50 | ||
| trnV-UAC | — | 49,963 | 50,587 | 37 | 551 | 37 | ||
| clpP | — | 68,438 | 70,389 | 71 | 746 | 294 | 615 | 226 |
| rpl16 | — | 79,433 | 80,856 | 9 | 1016 | 399 | ||
| rpl2 | — | 82,603 | 84,102 | 391 | 675 | 434 | ||
| ndhB | — | 92,571 | 94,788 | 775 | 685 | 758 | ||
| trnI-GAU | + | 99,678 | 100,648 | 37 | 897 | 37 | ||
| trnA-UGC | + | 100,707 | 101,573 | 38 | 794 | 35 | ||
| ycf1 | + | 106,297 | 107,376 | 762 | 30 | 288 | ||
| ndhA | — | 116,555 | 118,805 | 553 | 1153 | 545 | ||
| ycf1 | — | 120,649 | 126,123 | 762 | 30 | 4683 | ||
| trnA-UGC | — | 130,847 | 131,713 | 38 | 794 | 35 | ||
| trnI-GAU | — | 131,772 | 132,742 | 37 | 897 | 37 | ||
| ndhB | + | 137,632 | 139,849 | 775 | 685 | 758 | ||
| rpl2 | + | 148,318 | 149,817 | 391 | 675 | 434 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Miao, Y.; Zhang, X.; Zhang, G.; Sun, X.; Zhang, M.; Feng, Z.; Huang, L. The Complete Chloroplast Genome Sequence of Laportea bulbifera (Sieb. et Zucc.) Wedd. and Comparative Analysis with Its Congeneric Species. Genes 2022, 13, 2230. https://doi.org/10.3390/genes13122230
Zhang H, Miao Y, Zhang X, Zhang G, Sun X, Zhang M, Feng Z, Huang L. The Complete Chloroplast Genome Sequence of Laportea bulbifera (Sieb. et Zucc.) Wedd. and Comparative Analysis with Its Congeneric Species. Genes. 2022; 13(12):2230. https://doi.org/10.3390/genes13122230
Chicago/Turabian StyleZhang, Huihui, Yujing Miao, Xinke Zhang, Guoshuai Zhang, Xiao Sun, Min Zhang, Zhan Feng, and Linfang Huang. 2022. "The Complete Chloroplast Genome Sequence of Laportea bulbifera (Sieb. et Zucc.) Wedd. and Comparative Analysis with Its Congeneric Species" Genes 13, no. 12: 2230. https://doi.org/10.3390/genes13122230