
Identification of Potential Key Genes in Prostate Cancer with Gene Expression, Pivotal Pathways and Regulatory Networks Analysis Using Integrated Bioinformatics Methods

 , ,
, ,
Abstract
:1. Introduction
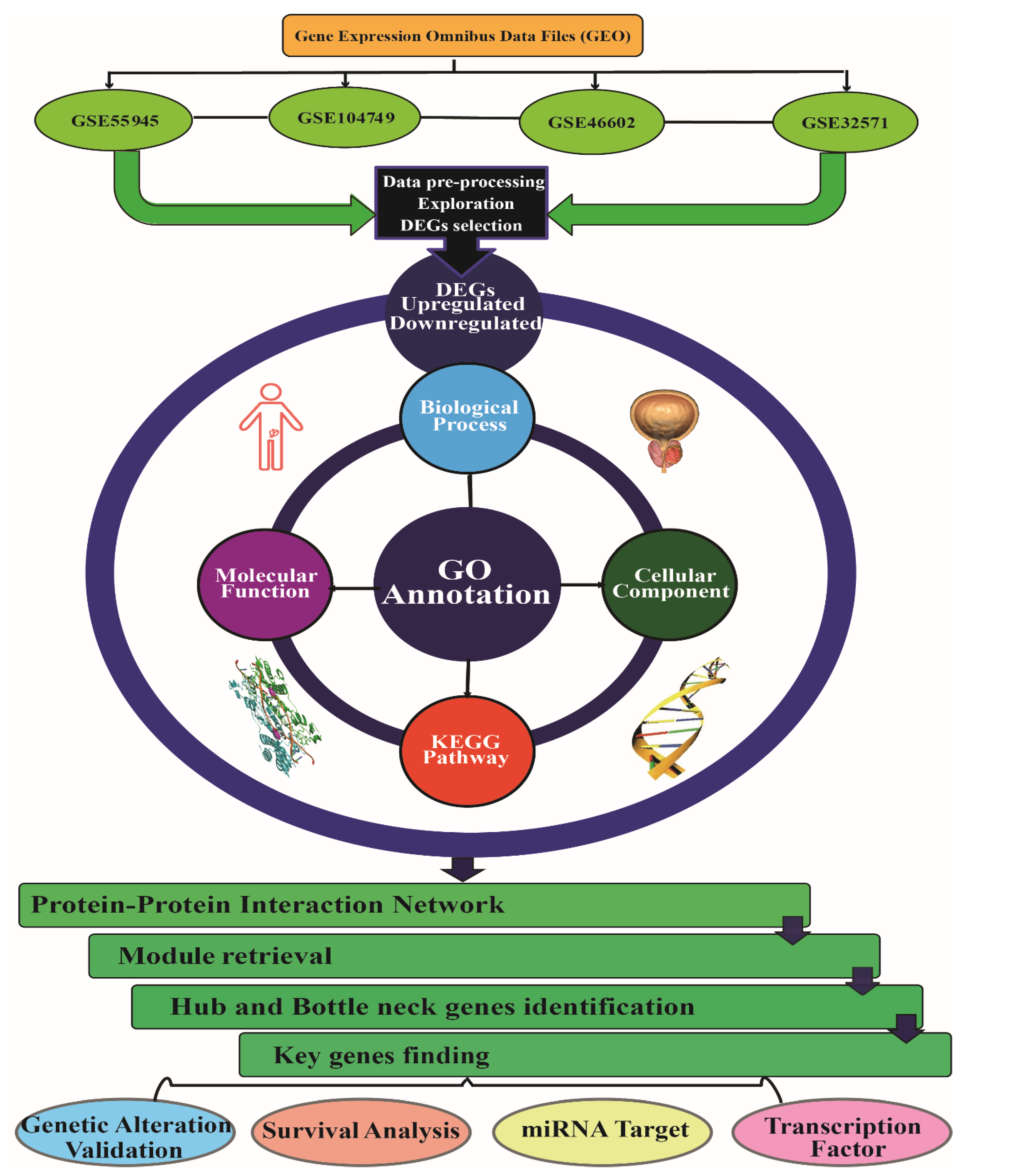
2. Materials and Methods
2.1. Microarray Data Extraction
2.2. Differentially Expressed Genes Identification
2.3. Gene Ontology and Pathway Enrichment Analysis
2.4. Protein-Protein Network Screening (PPI), Key Genes Identification and Module Network Construction
2.5. Genetic Alteration and Validation of Key Genes Expression Paradigm
2.6. Survival Analysis of Key Genes
2.7. miRNA and Transcription Factor Associated Network with Key Genes
3. Result
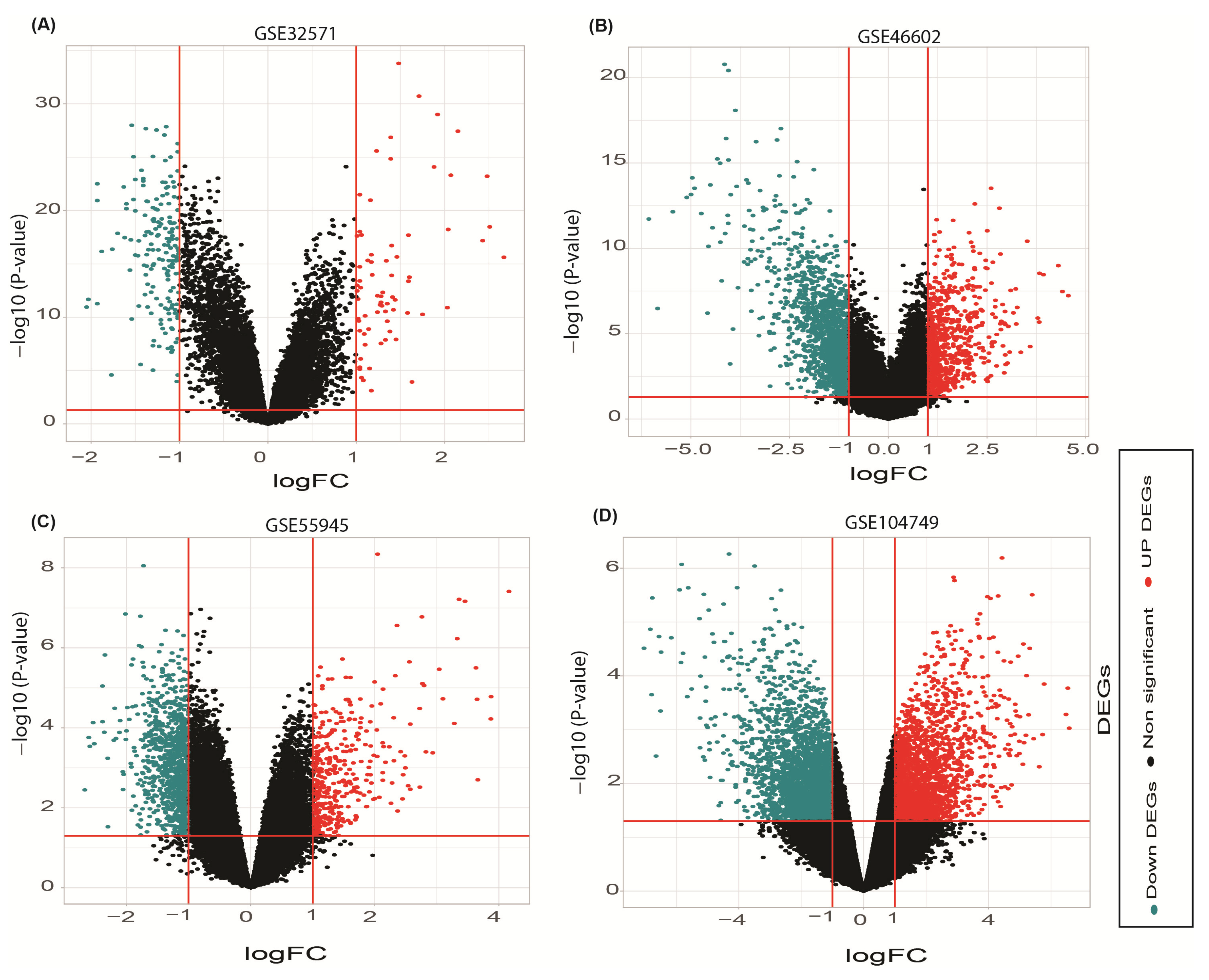
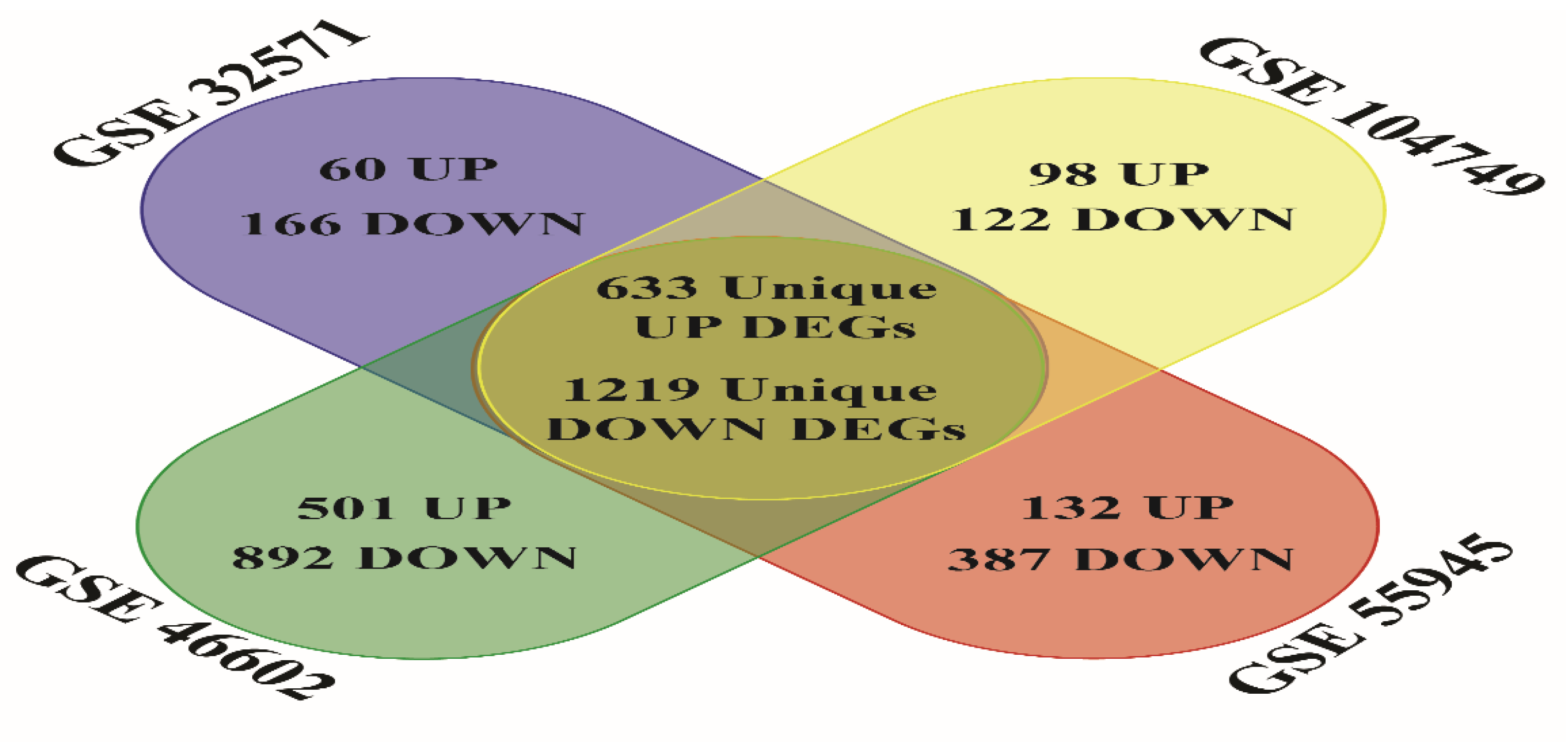
3.1. Deferentially Expressed Genes Discovery
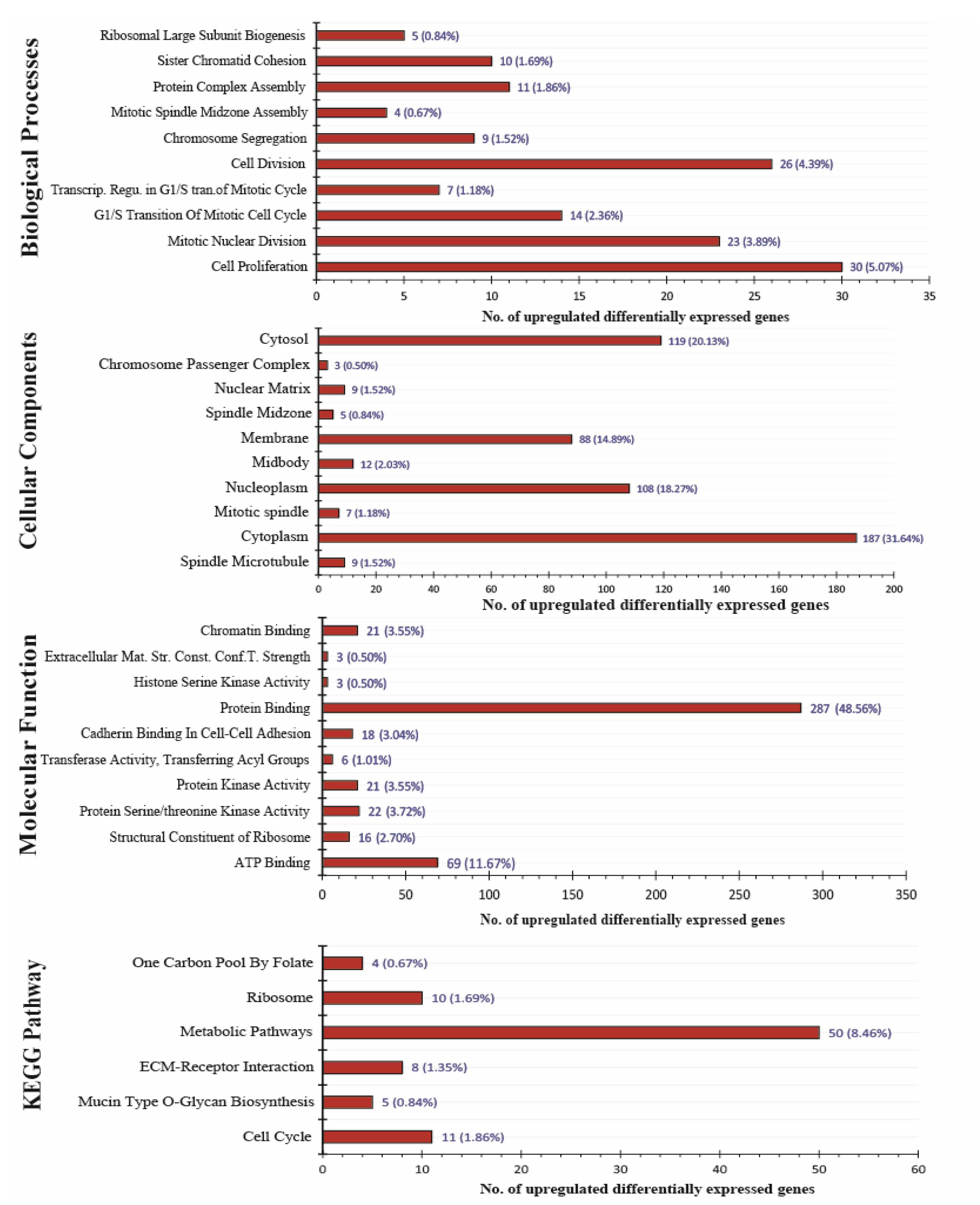
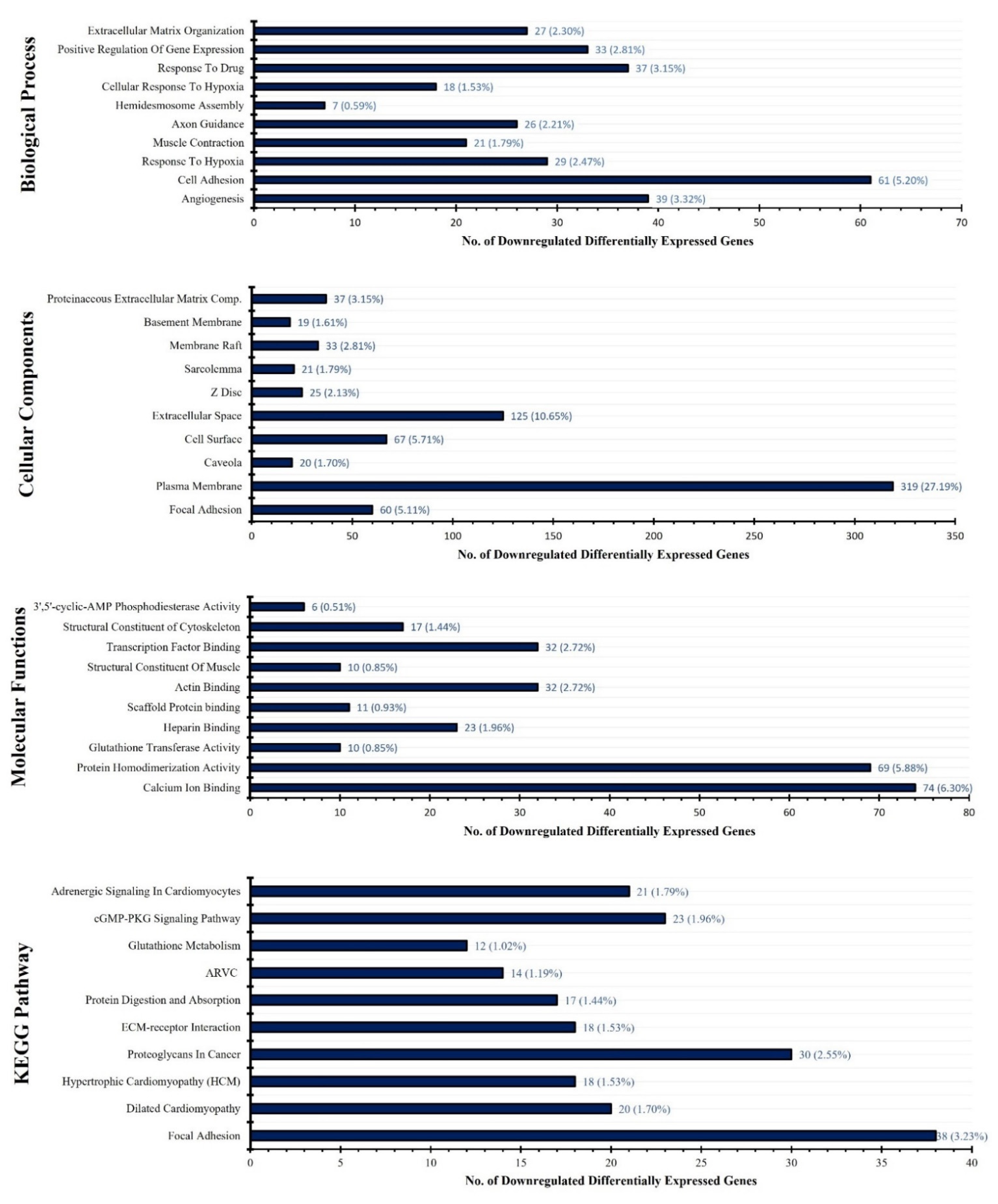
3.2. DEGs Functional Annotation and KEGG Pathway Analysis
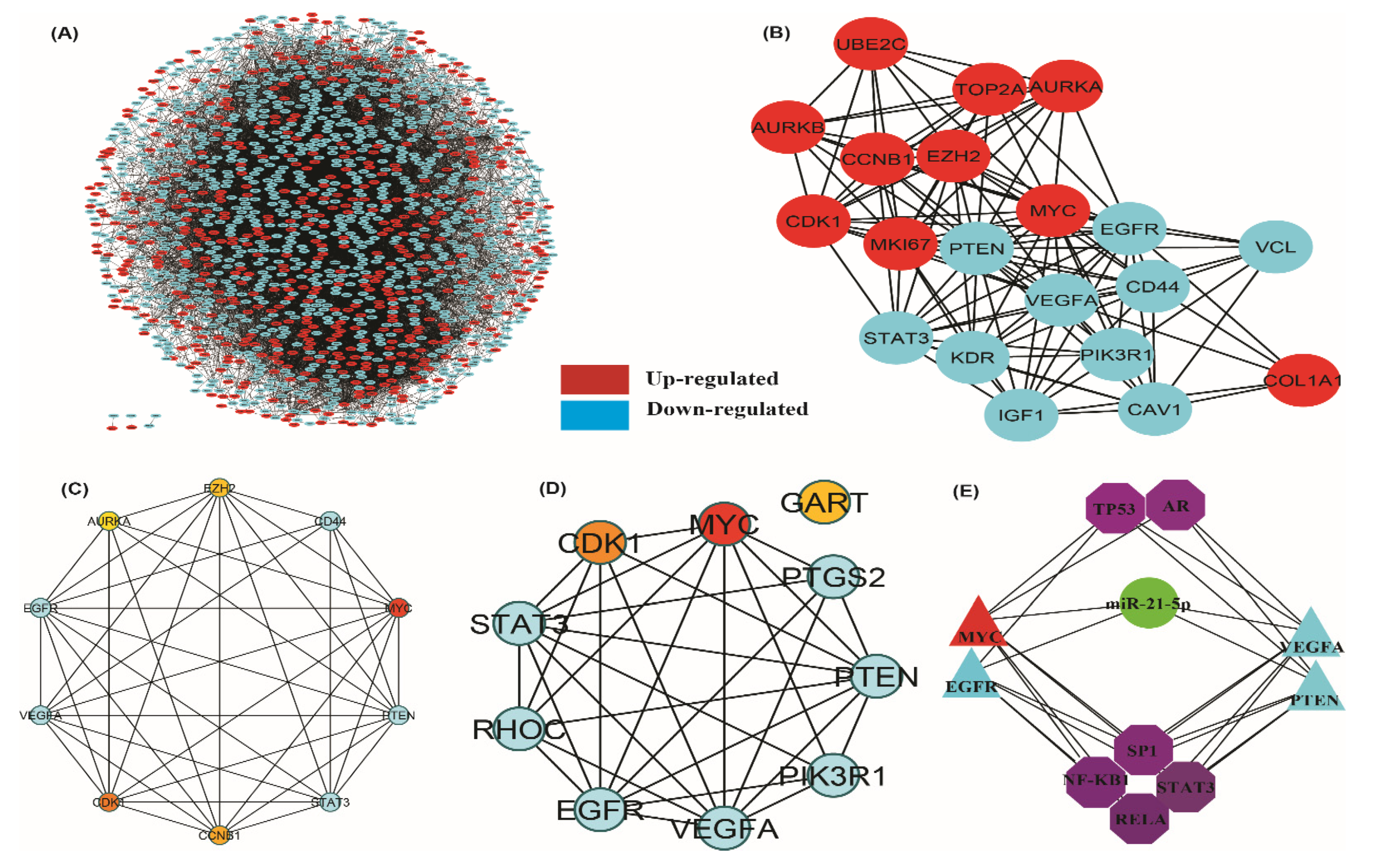
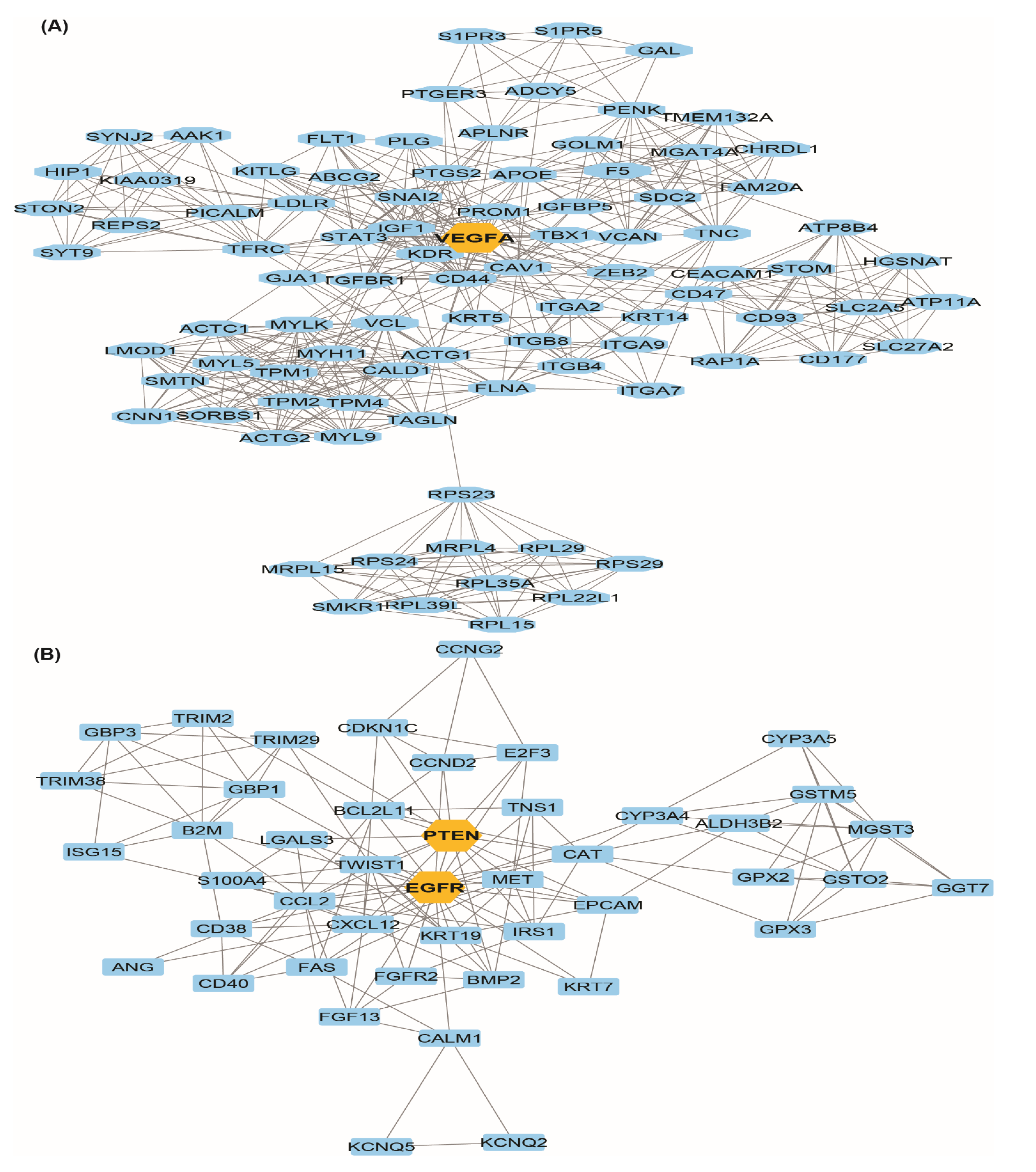
3.3. Protein-Protein (PPI) Network and Module Analysis
3.4. Hub-Bottle Neck Genes and Key Genes Identification
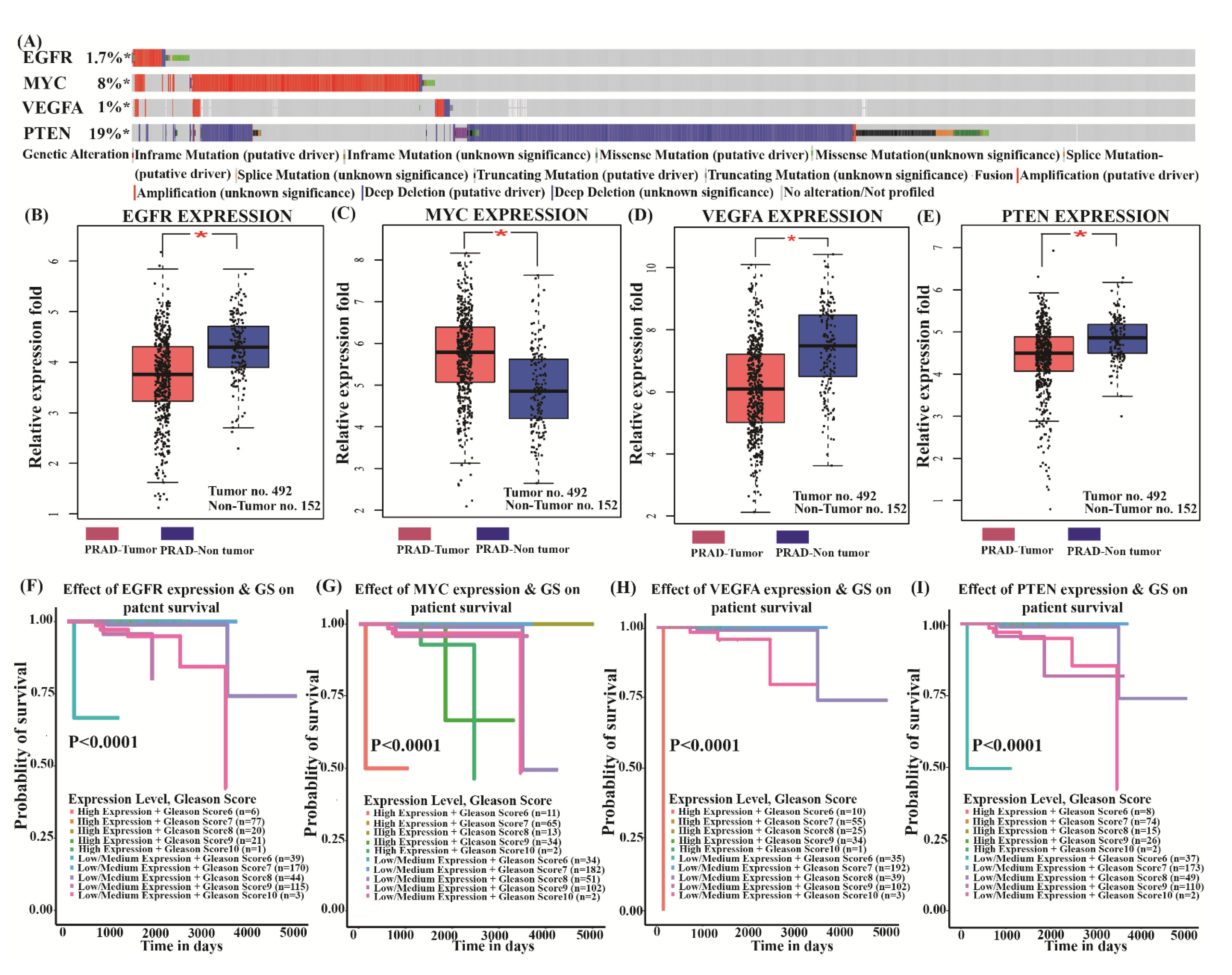
3.5. Genetic Alteration and Validation of Four Key Genes Expression Paradigm
3.6. Survival Analysis of Key Genes
3.7. miRNA and Transcription Factor Associated Network with Key Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stuopelyte, K.; Daniunaite, K.; Bakavicius, A.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. The utility of urine-circulating miRNAs for detection of prostate cancer. Br. J. Cancer 2016, 115, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Saxena, S.; Kumar, A. Epidemiology of prostate cancer in India. Meta Gene 2014, 2, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Center, M.M.; Jemal, A.; Lortet-Tieulent, J.; Ward, E.; Ferlay, J.; Brawley, O.; Bray, F. International variation in prostate cancer incidence and mortality rates. Eur. Urol. 2012, 61, 1079–1092. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, M.; Wang, M.C.; Papsidero, L.D.; Killian, C.S.; Shimano, T.; Valenzuela, L.; Nishiura, T.; Murphy, G.P.; Chu, T.M. Quantitation of prostate-specific antigen in serum by a sensitive enzyme immunoassay. Cancer Res. 1980, 40, 4658–4662. [Google Scholar]
- Herget, K.A.; Patel, D.P.; Hanson, H.A.; Sweeney, C.; Lowrance, W.T. Recent decline in prostate cancer incidence in the United States, by age, stage, and Gleason score. Cancer Med. 2016, 5, 136–141. [Google Scholar] [CrossRef]
- Deng, J.; Tang, J.; Wang, G.; Zhu, Y.S. Long non-coding RNA as potential biomarker for prostate cancer: Is it making a difference? Int. J. Environ. Res. Public Health 2017, 14, 270. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liu, Y.; Yuan, B.; Yin, L.; Peng, Y.; Yu, X.; Zhou, W.; Gong, Z.; Liu, J.; He, L.; et al. FOXM1 promotes the progression of prostate cancer by regulating PSA gene transcription. Oncotarget 2017, 8, 17027. [Google Scholar] [CrossRef] [Green Version]
- Albertsen, P. Predicting survival for men with clinically localized prostate cancer: What do we need in contemporary practice? Cancer 2008, 112, 1–3. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Gleave, M.; Hurtado-Col, A.; Nelson, C. Mechanisms of the development of androgen independence in prostate cancer. World J. Urol. 2005, 23, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yu, N.; Guo, T.; Hou, Y.; Zeng, Z.; Yang, X.; Hu, P.; Tang, X.; Wang, J.; Liu, M. Tissue biomarkers for prognosis of prostate cancer: A systematic review and meta-analysis. Cancer Epidemiol. Prev. Biomark. 2014, 23, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.J.; Chen, H.; Chen, L.Z.; Ru, G.M.; Guo, J.J.; Ding, Q.N. The potential of microRNAs as human prostate cancer biomarkers: A meta-analysis of related studies. J. Cell. Biochem. 2018, 119, 2763–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filella, X.; Fernández-Galan, E.; Fernández Bonifacio, R.; Foj, L. Emerging biomarkers in the diagnosis of prostate cancer. Pharm. Pers. Med. 2018, 11, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Arredouani, M.S.; Lu, B.; Bhasin, M.; Eljanne, M.; Yue, W.; Mosquera, J.M.; Bubley, G.J.; Li, V.; Rubin, M.A.; Libermann, T.A.; et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin. Cancer Res. 2009, 15, 5794–5802. [Google Scholar] [CrossRef] [Green Version]
- Shan, M.; Xia, Q.; Yan, D.; Zhu, Y.; Zhang, X.; Zhang, G.; Guo, J.; Hou, J.; Chen, W.; Zhu, T.; et al. Molecular analyses of prostate tumors for diagnosis of malignancy on fine-needle aspiration biopsies. Oncotarget 2017, 8, 104761–104771. [Google Scholar] [CrossRef]
- Mortensen, M.M.; Høyer, S.; Lynnerup, A.S.; Ørntoft, T.F.; Sørensen, K.D.; Borre, M.; Dyrskjøt, L. Expression profiling of prostate cancer tissue delineates genes associated with recurrence after prostatectomy. Sci. Rep. 2015, 5, 16018. [Google Scholar] [CrossRef]
- Kuner, R.; Fälth, M.; Pressinotti, N.C.; Brase, J.C.; Puig, S.B.; Metzger, J.; Gade, S.; Schäfer, G.; Bartsch, G.; Steiner, E.; et al. The maternal embryonic leucine zipper kinase (MELK) is upregulated in high-grade prostate cancer. J. Mol. Med. 2013, 91, 237–248. [Google Scholar] [CrossRef]
- Ostano, P.; Mello-Grand, M.; Sesia, D.; Gregnanin, I.; Peraldo-Neia, C.; Guana, F.; Jachetti, E.; Farsetti, A.; Chiorino, G. Gene Expression Signature Predictive of Neuroendocrine Transformation in Prostate Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Qu, C.B.; Zhang, Y.; Zhang, W.F.; Wang, D.D.; Gao, C.C.; Ma, L.; Chen, J.S.; Liu, K.L.; Zheng, B.; et al. Dysregulation of p53-RBM25-mediated circAMOTL1L biogenesis contributes to prostate cancer progression through the circAMOTL1L-miR-193a-5p-Pcdha pathway. Oncogene 2019, 38, 2516–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreuz, M.; Otto, D.J.; Fuessel, S.; Blumert, C.; Bertram, C.; Bartsch, S.; Loeffler, D.; Puppel, S.H.; Rade, M.; Buschmann, T.; et al. ProstaTrend—A Multivariable Prognostic RNA Expression Score for Aggressive Prostate Cancer. Eur. Urol. 2020, 78, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Saito, R.; Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Lotia, S.; Pico, A.R.; Bader, G.D.; Ideker, T. A travel guide to Cytoscape plugins. Nat. Methods 2012, 9, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [Green Version]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Cho, J.W.; Lee, S.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Cui, Q.; Wang, J.; Zhou, Y. TransmiR v2.0: An updated transcription factor-microRNA regulation database. Nucleic Acids Res. 2019, 47, D253–D258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, L.; Zhou, G.; Soufan, O.; Xia, J. miRNet 2.0: Network-based visual analytics for miRNA functional analysis and systems biology. Nucleic Acids Res. 2020, 48, W244–W251. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Zelefsky, M.J.; Sjoberg, D.D.; Nelson, J.B.; Egevad, L.; Magi-Galluzzi, C.; Vickers, A.J.; Parwani, A.V.; Reuter, V.E.; Fine, S.W.; et al. A contemporary prostate cancer grading system: A validated alternative to the Gleason score. Eur. Urol. 2016, 69, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Kojima, S.; Goto, Y.; Naya, Y. The roles of microRNAs in the progression of castration-resistant prostate cancer. J. Hum. Genet. 2017, 62, 25–31. [Google Scholar] [CrossRef]
- Lee, K.C.; Bradley, D.A.; Hussain, M.; Meyer, C.R.; Chenevert, T.L.; Jacobson, J.A.; Johnson, T.D.; Galban, C.J.; Rehemtulla, A.; Pienta, K.J.; et al. A feasibility study evaluating the functional diffusion map as a predictive imaging biomarker for detection of treatment response in a patient with metastatic prostate cancer to the bone. Neoplasia 2007, 9, 1003–1011. [Google Scholar] [CrossRef] [Green Version]
- Perabo, F.G.; Von Löw, E.C.; Ellinger, J.; Von Rücker, A.; Müller, S.C.; Bastian, P.J. Soy isoflavone genistein in prevention and treatment of prostate cancer. Prostate Cancer Prostatic Dis. 2008, 11, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Guérin, O.; Fischel, J.L.; Ferrero, J.M.; Bozec, A.; Milano, G. EGFR targeting in hormone-refractory prostate cancer: Current appraisal and prospects for treatment. Pharmaceuticals 2010, 3, 2238–2247. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, G.; Tortora, G.; D’Armiento, F.P.; De Rosa, G.; Staibano, S.; Autorino, R.; D’Armiento, M.; De Laurentiis, M.; De Placido, S.; Catalano, G.; et al. Expression of epidermal growth factor receptor correlates with disease relapse and progression to androgen-independence in human prostate cancer. Clin. Cancer Res. 2002, 8, 3438–3444. [Google Scholar] [PubMed]
- Roepstorff, K.; Grøvdal, L.; Grandal, M.; Lerdrup, M.; van Deurs, B. Endocytic downregulation of ErbB receptors: Mechanisms and relevance in cancer. Histochem. Cell Biol. 2008, 129, 563–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashmi, S.K.; Irfan, M.; Asif, H.; Nisar, L.; Naeem, M.; Khan, E.Y.; Baloch, S.; Faridi, N. Prognostic utility of epidermal growth factor receptor (EGFR) expression in prostatic acinar adenocarcinoma. Appl. Cancer Res. 2019, 39, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Mandel, A.; Larsson, P.; Sarwar, M.; Semenas, J.; Syed Khaja, A.S.; Persson, J.L. The interplay between AR, EGF receptor and MMP-9 signaling pathways in invasive prostate cancer. Mol. Med. 2018, 24, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Baek, K.H.; Hong, M.E.; Jung, Y.Y.; Lee, C.H.; Lee, T.J.; Park, E.S.; Kim, M.K.; Yoo, J.H.; Lee, S.W. Correlation of AR, EGFR, and HER2 expression levels in prostate cancer: Immunohistochemical analysis and chromogenic in situ hybridization. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2012, 44, 50. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, M.; Zhu, S.; Wu, F.; Wu, H.; Walia, V.; Kumar, S.; Elble, R.; Watabe, K.; Mo, Y.Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc. Natl. Acad. Sci. USA 2009, 106, 3207–3212. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Cho, W.C.; Chow, A.S.; Au, J.S. MiR-145 inhibits cell proliferation of human lung adenocarcinoma by targeting EGFR and NUDT1. RNA Biol. 2011, 8, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Liu, C.; Zhang, D.; Liu, B.; Chen, X.; Rycaj, K.; Jeter, C.; Calhoun-Davis, T.; Li, Y.; Yang, T.; et al. miR-199a-3p targets stemness-related and mitogenic signaling pathways to suppress the expansion and tumorigenic capabilities of prostate cancer stem cells. Oncotarget 2016, 7, 56628. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Huang, Y.; Dong, S.; Qiao, C.; Yang, G.; Zhang, S.; Wang, C.; Xu, Y.; Zheng, F.; Yan, M. MiR-199a-3p/5p participated in TGF-β and EGF induced EMT by targeting DUSP5/MAP3K11 in pterygium. J. Transl. Med. 2020, 18, 1–9. [Google Scholar] [CrossRef]
- Nesbit, C.E.; Grove, L.E.; Yin, X.; Prochownik, E.V. Differential apoptotic behaviors of c-myc, N-myc, and L-myc oncoproteins. Cell Growth Differ. Publ. Am. Assoc. Cancer Res. 1998, 9, 731–742. [Google Scholar]
- Barrett, J.; Birrer, M.J.; Kato, G.J.; Dosaka-Akita, H.; Dang, C.V. Activation domains of L-Myc and c-Myc determine their transforming potencies in rat embryo cells. Mol. Cell. Biol. 1992, 12, 3130–3137. [Google Scholar] [PubMed] [Green Version]
- Tansey, W.P. Mammalian MYC proteins and cancer. New J. Sci. 2014, 2014, 757534. [Google Scholar] [CrossRef] [Green Version]
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; De Marzo, A.M. MYC and prostate cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Gorrini, C.; Donzelli, M.; Torriglia, A.; Supino, R.; Brison, O.; Bernardi, R.; Negri, C.; Denegri, M.; Counis, M.F.; Ranzani, G.N.; et al. Effect of apoptogenic stimuli on colon carcinoma cell lines with a different c-myc expression level. Int. J. Mol. Med. 2003, 11, 737–742. [Google Scholar] [CrossRef]
- Nadiminty, N.; Tummala, R.; Lou, W.; Zhu, Y.; Zhang, J.; Chen, X.; White, R.W.D.; Kung, H.J.; Evans, C.P.; Gao, A.C.; et al. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of Myc expression in prostate cancer cells. J. Biol. Chem. 2012, 287, 1527–1537. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7, e29722. [Google Scholar] [CrossRef] [Green Version]
- Fulciniti, M.; Amodio, N.; Bandi, R.L.; Cagnetta, A.; Samur, M.K.; Acharya, C.; Prabhala, R.; D’Aquila, P.; Bellizzi, D.; Passarino, G.; et al. miR-23b/SP1/c-myc forms a feed-forward loop supporting multiple myeloma cell growth. Blood Cancer J. 2016, 6, e380. [Google Scholar] [CrossRef]
- Bender, R.J.; Mac Gabhann, F. Dysregulation of the vascular endothelial growth factor and semaphorin ligand-receptor families in prostate cancer metastasis. BMC Syst. Biol. 2015, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Ding, Z. Identification of key genes in prostate cancer gene expression profile by bioinformatics. Andrologia 2019, 51, e13169. [Google Scholar] [CrossRef]
- Song, Z.Y.; Wang, F.; Cui, S.X.; Qu, X.J. Knockdown of CXCR4 inhibits CXCL12-induced angiogenesis in HUVECs through downregulation of the MAPK/ERK and PI3K/AKT and the Wnt/β-catenin pathways. Cancer Investig. 2018, 36, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Jiang, J.B.; Li, Y.; Wang, Y.L.; Dai, Y. MicroRNA-299-3p suppresses proliferation and invasion by targeting VEGFA in human colon carcinoma. Biomed. Pharmacother. 2017, 93, 1047–1054. [Google Scholar] [CrossRef] [PubMed]
- Östling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Mäkelä, R.; Hagman, Z.; Edsjö, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffari, M.; Ghaderian, S.M.; Omrani, M.D.; Afsharpad, M.; Shankaie, K.; Samadaian, N. The Association of miR-let 7b and miR-548 with PTEN in Prostate Cancer. Urol. J. 2019, 16, 267–273. [Google Scholar]
- Morais, C.E.; Gurgel, D.C.; Teixeira, A.C.; Mattos, T.V.A.; Silva, A.V.A.D.; Tavora, F. Prevalence of ERG expression and PTEN loss in a Brazilian prostate cancer cohort. Braz. J. Med. Biol. Res. 2019, 52, e8483. [Google Scholar] [CrossRef]
- Nodouzi, V.; Nowroozi, M.; Hashemi, M.; Javadi, G.; Mahdian, R. Concurrent down-regulation of PTEN and NKX3. 1 expression in Iranian patients with prostate cancer. Int. Braz. J. Urol. 2015, 41, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Song, M.S.; Salmena, L.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 2012, 13, 283–296. [Google Scholar] [CrossRef]
- Mangangcha, I.R.; Malik, M.Z.; Kucuk, O.; Ali, S.; Singh, R.B. Kinless hubs are potential target genes in prostate cancer network. Genomics 2020, 112, 6. [Google Scholar] [CrossRef]
- Mangangcha, I.R.; Malik, M.; Küçük, Ö.; Ali, S.; Singh, R.K. Identification of key regulators in Prostate cancer from gene expression datasets of patients. Sci. Rep. 2019, 9, 16420. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.K.; Hu, Y.C.; Lee, D.K.; Chang, C. Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells. Mol. Endocrinol. 2004, 18, 2409–2423. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample (Accession No.) | Prostate Cancer | Benign Prostate Hyperplasia | Organism | Sample Type | Platform | Reference | Included/ Excluded |
|---|---|---|---|---|---|---|---|
| GSE55945 | 13 | 8 | Homo sapiens | Radical prostatectomy tissue | Affymetrix GPL570 | [17] | Included |
| GSE104749 | 4 | 4 | Homo sapiens | Fine-Needle Aspiration tissue | Affymetrix GPL570 | [18] | Included |
| GSE46602 | 36 | 14 | Homo sapiens | Laser micro dissected tissue | Affymetrix GPL570 | [19] | Included |
| GSE32571 | 59 | 39 | Homo sapiens | Fresh frozen tissue | Illumina GPL6947 | [20] | Included |
| GSE142288 | 48 | Nil | Homo sapiens | Tissue | Agilent GPL13264 | [21] | Excluded |
| GSE155792 | 1 | Nil | Homo sapiens | Tissue | Agilent GPL28148 | NA | Excluded |
| GSE113153 | 10 | Nil | Homo sapiens | Tissue | GPL21825 | [22] | Excluded |
| GSE134160 | 164 | Nil | Homo sapiens | Fresh frozen tissue | Agilent GPL26898 | [23] | Excluded |
| Status | Gene Symbol | Degree | Status | Gene Symbol | Degree |
|---|---|---|---|---|---|
| Upregulated | MYC | 166 | Downregulated | EGFR | 190 |
| CDK1 | 111 | VEGFA | 178 | ||
| CCNB1 | 97 | STAT3 | 117 | ||
| EZH2 | 95 | CD44 | 113 | ||
| AURKA | 89 | PTEN | 110 | ||
| UBE2C | 82 | VCL | 86 | ||
| AURKB | 80 | IGF1 | 84 | ||
| COL1A1 | 76 | CAV1 | 82 | ||
| MKI67 | 76 | KDR | 79 | ||
| TOP2A | 75 | PIK3R1 | 72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.M.; Mohsen, M.T.; Malik, M.Z.; Bagabir, S.A.; Alkhanani, M.F.; Haque, S.; Serajuddin, M.; Bharadwaj, M. Identification of Potential Key Genes in Prostate Cancer with Gene Expression, Pivotal Pathways and Regulatory Networks Analysis Using Integrated Bioinformatics Methods. Genes 2022, 13, 655. https://doi.org/10.3390/genes13040655
Khan MM, Mohsen MT, Malik MZ, Bagabir SA, Alkhanani MF, Haque S, Serajuddin M, Bharadwaj M. Identification of Potential Key Genes in Prostate Cancer with Gene Expression, Pivotal Pathways and Regulatory Networks Analysis Using Integrated Bioinformatics Methods. Genes. 2022; 13(4):655. https://doi.org/10.3390/genes13040655
Chicago/Turabian StyleKhan, Mohd Mabood, Mohammad Taleb Mohsen, Md. Zubbair Malik, Sali Abubaker Bagabir, Mustfa F. Alkhanani, Shafiul Haque, Mohammad Serajuddin, and Mausumi Bharadwaj. 2022. "Identification of Potential Key Genes in Prostate Cancer with Gene Expression, Pivotal Pathways and Regulatory Networks Analysis Using Integrated Bioinformatics Methods" Genes 13, no. 4: 655. https://doi.org/10.3390/genes13040655