
DNA Methylation Level Changes in Transgenic Chinese Cabbage (Brassica rapa ssp. pekinensis) Plants and Their Effects on Corresponding Gene Expression Patterns


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Bisulfite Sequencing
2.2. Sequence DATA Processing and Analysis
2.3. Selection of DMR Candidates
2.4. Expression Analysis of Genes Associated with DMR Candidates
2.5. MSP for DMR Candidates
3. Results
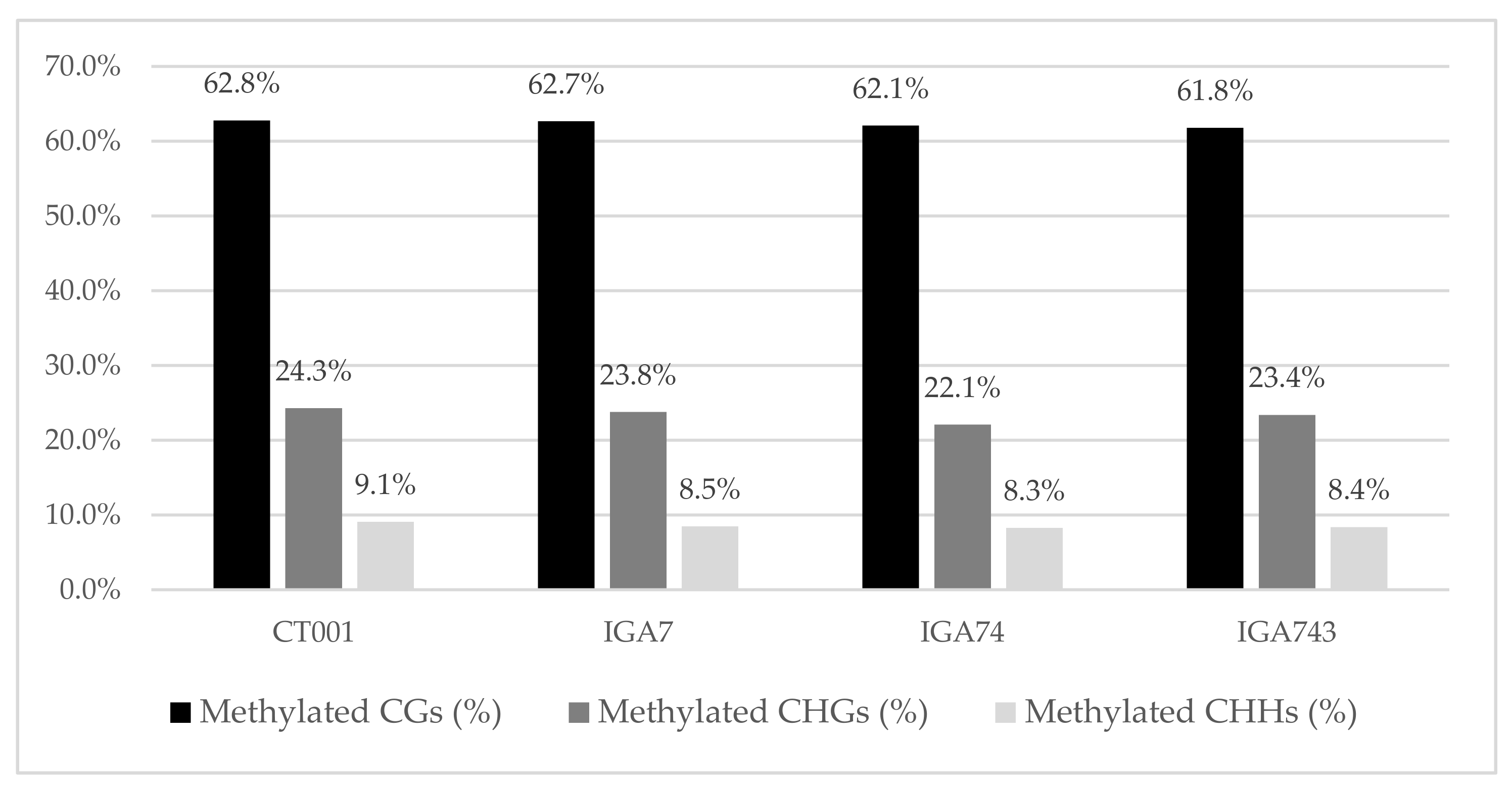
3.1. Bisulfite Sequencing and Mapping
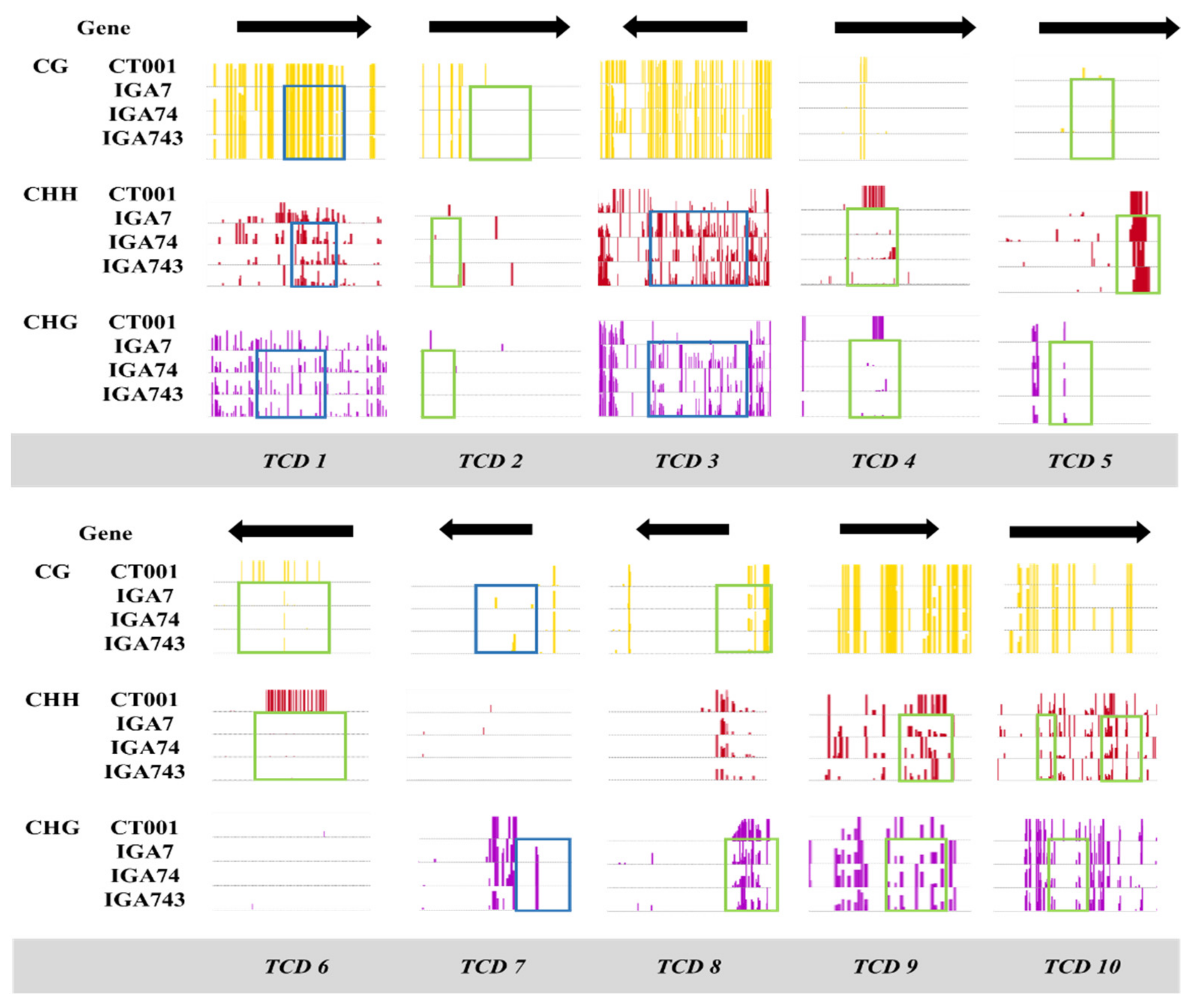
3.2. Confirmation of Selected TCDs
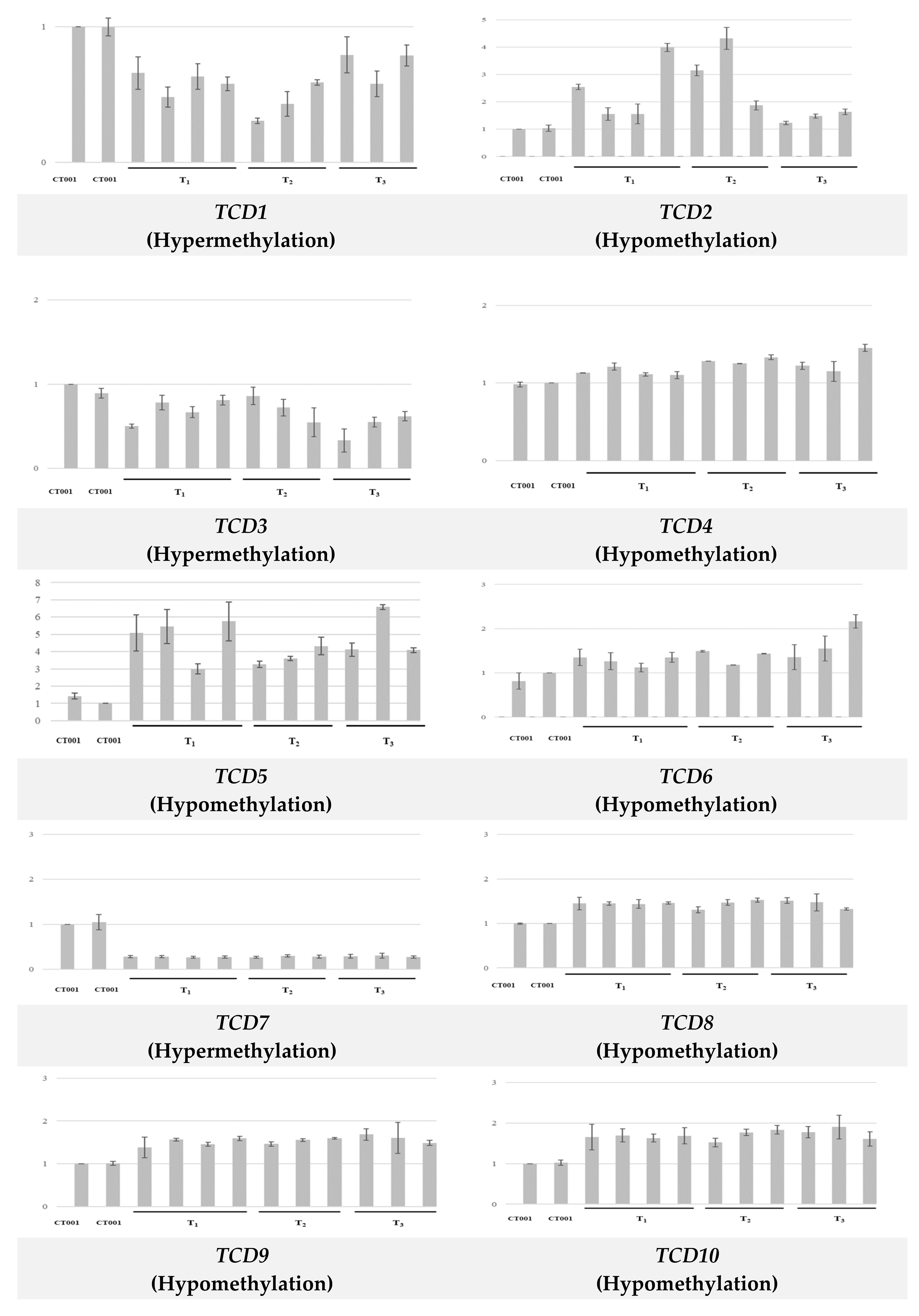
3.3. Correlation between Methylation Status and Expression of TCD Genes
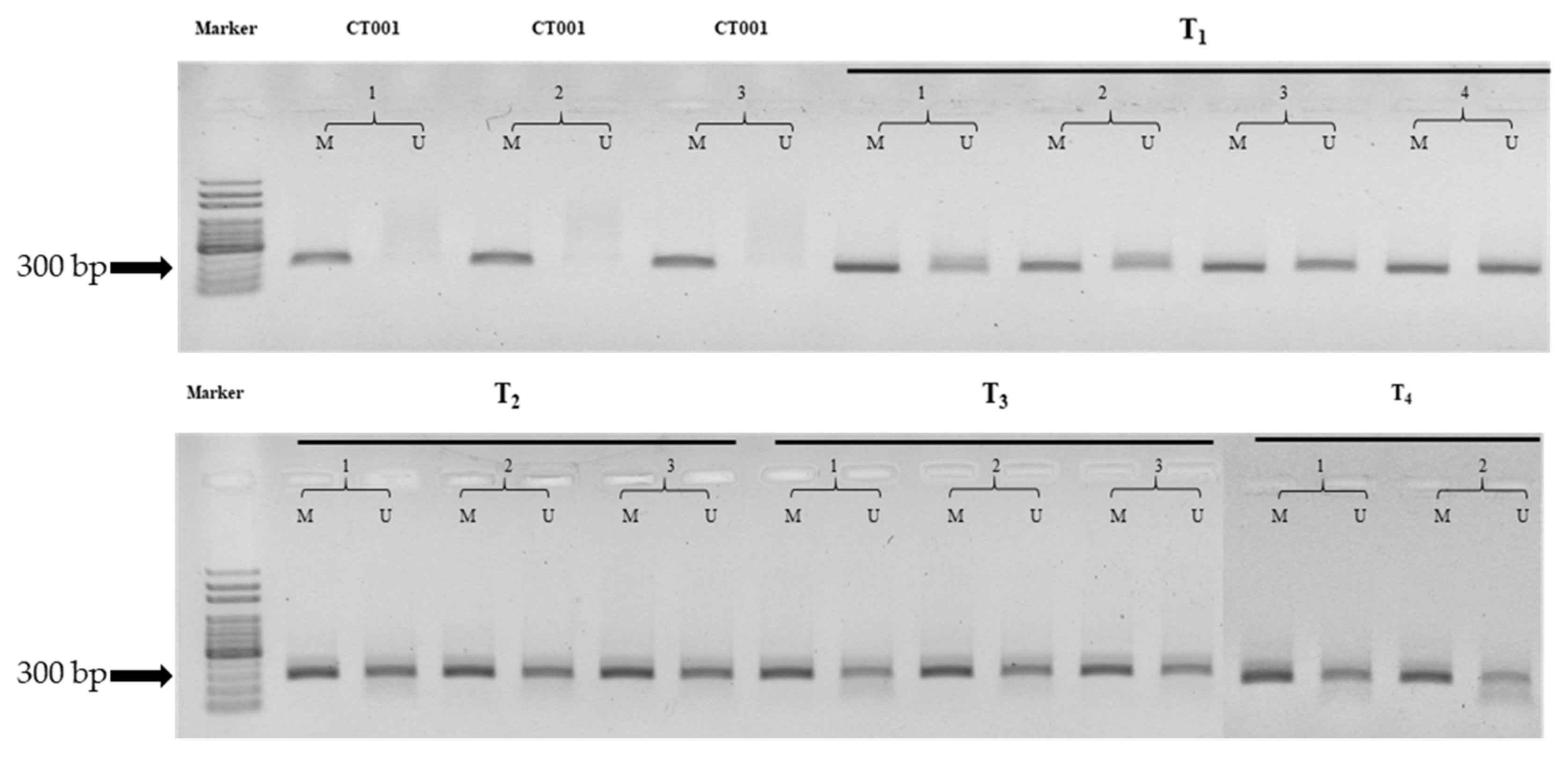
3.4. Determination of DNA Methylation Patterns by MSP
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Husaini, A.M.; Abdin, M.Z.; Parray, G.A.; Sanghera, G.S.; Murtaza, I.; Alam, T.; Srivastava, D.K.; Farooqi, H.; Khan, H.N. Vehicles and ways for efficient nuclear transformation in plants. GM Crop. 2010, 1, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Larkin, P.J.; Scowcroft, W.R. Somaclonal variation—A novel source of variability from cell cultures for plant improvement. Theor. Appl. Genet. 1981, 60, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, Y.P.S. Somaclonal variation—Origin, induction, cryopreservation and implication in plant breeding. In Somaclonal Variation in Crop. Improvement I; Springer: Berlin/Heidelberg, Germany, 1990; pp. 3–48. [Google Scholar]
- Miguel, C.; Marum, L. An epigenetic view of plant cells cultured in vitro: Somaclonal variation and beyond. J. Exp. Bot. 2011, 62, 3713–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaeppler, S.; Phillips, R.L. Tissue culture-induced DNA methylation variation in maize. Proc. Natl. Acad. Sci. USA 1993, 90, 8773–8776. [Google Scholar] [CrossRef] [Green Version]
- Kaeppler, S.M.; Kaeppler, H.F.; Rhee, Y. Epigenetic aspects of somaclonal variation in plants. Plant Mol. Biol. 2000, 43, 179–188. [Google Scholar] [CrossRef]
- Thorpe, T.A. History of plant tissue culture. Mol. Biotechnol. 2007, 37, 169–180. [Google Scholar] [CrossRef]
- Rhee, Y.; Sekhon, R.S.; Chopra, S.; Kaeppler, S. Tissue Culture-Induced Novel Epialleles of a Myb Transcription Factor Encoded by pericarp color1 in Maize. Genetics 2010, 186, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Neelakandan, A.; Wang, K. Recent progress in the understanding of tissue culture-induced genome level changes in plants and potential applications. Plant Cell Rep. 2011, 31, 597–620. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014, 26, 577–590. [Google Scholar] [CrossRef] [Green Version]
- To, T.K.; Saze, H.; Kakutani, T. DNA Methylation within Transcribed Regions. Plant Physiol. 2015, 168, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; A Jeddeloh, J.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 Cytosine Methyltransferase Mutants. Genetics 2003, 163, 1109–1122. [Google Scholar] [CrossRef]
- Shemer, O.; Landau, U.; Candela, H.; Zemach, A.; Williams, L.E. Competency for shoot regeneration from Arabidopsis root explants is regulated by DNA methylation. Plant Sci. 2015, 238, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell. Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubis, S.E.; Castilho, A.; Vershinin, A.V.; Heslop-Harrison, J.S. Retroelements, transposons and methylation status in the genome of oil palm (Elaeis guineensis) and the relationship to somaclonal variation. Plant Mol. Biol. 2003, 52, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [Green Version]
- Smulders, M.J.M.; De Klerk, G.J. Epigenetics in plant tissue culture. Plant Growth Regul. 2011, 63, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Coronel, C.J.; González, A.I.; Ruiz, M.L.; Polanco, C. Analysis of somaclonal variation in transgenic and regenerated plants of Arabidopsis thaliana using methylation related metAFLP and TMD markers. Plant Cell Rep. 2017, 37, 137–152. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schuebeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Masser, D.R.; Berg, A.S.; Freeman, W.M. Focused, high accuracy 5-methylcytosine quantitation with base resolution by benchtop next-generation sequencing. Epigenetics Chromatin 2013, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Park, J.H.; Park, Y.D. Construction of pseudomolecule sequences of Brassica rapa ssp. pekinensis inbred line CT001 and analysis of spontaneous mutations derived via sexual propagation. PLoS ONE 2019, 14, e0222283. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, S.J.; Kim, B.R.; Woo, E.T.; Lee, J.S.; Han, E.H.; Lee, Y.H.; Park, Y.D. Isolation of Myrosinase and Glutathione S-transferase Genes and Transformation of These Genes to Develop Phenylethylisothiocyanate Enriching Chinese Cabbage. Kor. J. Hort. Sci. Technol. 2011, 29, 623–632. [Google Scholar]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, J.S.; Shin, Y.H.; Park, Y.D. Identification and Validation of Genetic Variations in Transgenic Chinese Cabbage Plants (Brassica rapa ssp. pekinensis) by Next-Generation Sequencing. Genes 2021, 12, 621. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Wu, H. Differential methylation analysis for BS-seq data under general experimental design. Bioinformatics 2016, 32, 1446–1453. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Ecker, J.R. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009, 19, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Rubbi, L.; Jacobsen, S.E.; Pellegrini, M. Determining DNA Methylation Profiles Using Sequencing. High-Throughput Next Gener. Seq. 2011, 733, 223–238. [Google Scholar] [CrossRef]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.L.; Meyers, B.C.; Jacobsen, S.E. Plants regenerated from tissue culture contain stable epigenome changes in rice. elife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Stelpflug, S.C.; Eichten, S.R.; Hermanson, P.J.; Springer, N.M.; Kaeppler, S.M. Consistent and Heritable Alterations of DNA Methylation Are Induced by Tissue Culture in Maize. Genetics 2014, 198, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Lauria, M.; Echegoyen-Nava, R.A.; Rodríguez-Ríos, D.; Zaina, S.; Lund, G. Inter-individual variation in DNA methylation is largely restricted to tissue-specific differentially methylated regions in maize. BMC Plant Biol. 2017, 17, 52. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Li, N.; Zhang, Z.; Meng, X.; Dong, Q.; Xu, C.; Gong, L.; Liu, B. CG hypomethylation leads to complex changes in DNA methylation and transpositional burst of diverse transposable elements in callus cultures of rice. Plant J. 2019, 101, 188–203. [Google Scholar] [CrossRef]
- Fujimoto, R.; Sasaki, T.; Inoue, H.; Nishio, T. Hypomethylation and transcriptional reactivation of retrotransposon-like sequences in ddm1 transgenic plants of Brassica rapa. Plant Mol. Biol. 2008, 66, 463–473. [Google Scholar] [CrossRef]
- Karim, R.; Nuruzzaman, M.; Khalid, N.; Harikrishna, J.A. Importance of DNA and histone methylation in in vitro plant propagation for crop improvement: A review. Ann. Appl. 2016, 169, 1–16. [Google Scholar] [CrossRef]
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116. [Google Scholar] [CrossRef] [Green Version]
- Takuno, S.; Ran, J.H.; Gaut, B.S. Evolutionary patterns of genic DNA methylation vary across land plants. Nat. Plants 2016, 2, 15222. [Google Scholar] [CrossRef]
- Velásquez, A.C.; Nomura, K.; Cooper, M.D.; Herrin, B.R.; He, S.Y. Leucine-rich-repeat-containing variable lymphocyte receptors as modules to target plant-expressed proteins. Plant Methods 2017, 13, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosher, S.; Kemmerling, B. PSKR1 and PSY1R-mediated regulation of plant defense responses. Plant Signal. Behav. 2013, 8, e24119. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, K.; Kannangara, R.; Jørgensen, K.; Fuglsang, A.T. Analysis of peptide PSY1 responding transcripts in the two Arabidopsis plant lines: Wild type and psy1r receptor mutant. BMC Genom. 2014, 15, 441. [Google Scholar] [CrossRef] [Green Version]
- Brocard, I.M.; Lynch, T.J.; Finkelstein, R.R. Regulation and Role of the Arabidopsis Abscisic Acid-Insensitive 5 Gene in Abscisic Acid, Sugar, and Stress Response. Plant Physiol. 2002, 129, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, M.E.; Lynch, T.; Peeters, J.; Snowden, C.; Finkelstein, R. A small plant-specific protein family of ABI five binding proteins (AFPs) regulates stress response in germinating Arabidopsis seeds and seedlings. Plant Mol. Biol. 2008, 67, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Tsai, M.C.; Wu, S.S.; Chang, I.F. Regulation of ABI5 expression by ABF3 during salt stress responses in Arabidopsis thaliana. Bot. Stud. 2019, 60, 16. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Nakamura, M.; Calarco, J.P.; Susaki, D.; Lim, P.Q.; Kinoshita, T.; Higashiyama, T.; Martienssen, R.A.; Berger, F. RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 2013, 140, 2953–2960. [Google Scholar] [CrossRef] [Green Version]
- Arıkan, B.; Özden, S.; Turgut-Kara, N. DNA methylation related gene expression and morphophysiological response to abiotic stresses in Arabidopsis thaliana. Environ. Exp. Bot. 2018, 149, 17–26. [Google Scholar] [CrossRef]
- Zhong, X.; Hale, C.J.; Nguyen, M.; Ausin, I.; Groth, M.; Hetzel, J.; Vashisht, A.A.; Henderson, I.; Wohlschlegel, J.A.; Jacobsen, S.E. DOMAINS REARRANGED METHYLTRANSFERASE3 controls DNA methylation and regulates RNA polymerase V transcript abundance in Arabidopsis. Proc. Natl. Acad. Sci. USA 2015, 112, 911–916. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Kwak, J.M.; Uozumi, N.; Schroeder, J.I. AtKUP1: An Arabidopsis Gene Encoding High-Affinity Potassium Transport Activity. Plant Cell. 1998, 10, 51–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, M.D.; Urbez, C.; Perez-Amador, M.A.; Carbonell, J. Characterization of constricted fruit (ctf) Mutant Uncovers a Role for AtMYB117/LOF1 in Ovule and Fruit Development in Arabidopsis thaliana. PLoS ONE 2011, 6, e18760. [Google Scholar] [CrossRef] [Green Version]
- Bolaños-Villegas, P.; Yang, X.; Wang, H.J.; Juan, C.T.; Chuang, M.H.; Makaroff, C.A.; Jauh, G.Y. Arabidopsis CHROMOSOME TRANSMISSION FIDELITY 7 (AtCTF7/ECO1) is required for DNA repair, mitosis and meiosis. Plant J. 2013, 75, 927–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, S.; Chen, G.; Pan, D.; Chen, J.; She, W. Molecular Analysis of 14-3-3 Genes in Citrus sinensis and Their Responses to Different Stresses. Int. J. Mol. Sci. 2021, 22, 568. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster Enrichment Score | Category z | Description | Count | p Value |
|---|---|---|---|---|
| 1.45 | INTERPRO | Leucine-rich repeat, typical subtype | 4 | 3.1 × 10−3 |
| SMART | LRR TYP | 4 | 5.3 × 10−1 | |
| INTERPRO | Leucine-rich repeat | 4 | 2.3 × 10−2 | |
| INTERPRO | Leucine-rich repeat-containing N-terminal, type 2 | 4 | 5.0 × 10−2 | |
| UP_KEYWORDS | Receptor | 4 | 6.2 × 10−2 | |
| UP_KEYWORDS | Leucine-rich repeat | 4 | 2.3 × 10−1 | |
| GOTERM_MF_DIRECT | Kinase activity | 4 | 2.6 × 10−1 | |
| 0.43 | GOTERM_CC_DIRECT | Integral component of membrane | 11 | 3.0 × 10−1 |
| UP_SEQ_FEATURE | Transmembrane region | 5 | 3.2 × 10−1 | |
| UP_KEYWORDS | Transport | 5 | 3.7 × 10−1 | |
| UP_KEYWORDS | Transmembrane helix | 11 | 4.0 × 10−1 | |
| UP_KEYWORDS | Transmembrane | 11 | 4.1 × 10−1 | |
| UP_KEYWORDS | Membrane | 12 | 4.8 × 10−1 | |
| 0.42 | UP_KEYWORDS | Nucleotide-binding | 8 | 6.2 × 10−2 |
| UP_KEYWORDS | ATP-binding | 6 | 2.0 × 10−1 | |
| UP_KEYWORDS | Serine/threonine-protein kinase | 3 | 3.3 × 10−1 | |
| GOTERM_MF_DIRECT | ATP binding | 6 | 3.8 × 10−1 | |
| INTERPRO | Serine/threonine-protein kinase, active site | 3 | 4.0 × 10−1 | |
| GOTERM_MF_DIRECT | Protein serine/threonine kinase activity | 3 | 4.0 × 10−1 | |
| SMART | S TKc | 3 | 5.2 × 10−1 | |
| INTERPRO | Protein kinase, catalytic domain | 3 | 5.3 × 10−1 | |
| INTERPRO | Protein kinase-like domain | 3 | 5.5 × 10−1 | |
| INTERPRO | P-loop containing nucleoside triphosphate hydrolase | 3 | 5.7 × 10−1 | |
| UP_KEYWORDS | Transferase | 6 | 5.9 × 10−1 | |
| UP_KEYWORDS | Kinase | 3 | 6.2 × 10−1 |
| Name | Context | Methyl in Transformants Z | Gene ID | Brassica ID | TAIR ID | Description |
|---|---|---|---|---|---|---|
| TCD 1 | CG CHG CHH | Hyper | CT001_A07241320 | - | - | - |
| TCD 2 | CG CHG CHH | Hypo | CT001_A07421310 | - | - | - |
| TCD 3 | CHG CHH | Hyper | CT001_A08284340 | Bra034540 | - | - |
| TCD 4 | CHG CHH | Hypo | CT001_A05184770 | Bra027247 | AT3G15390 | Putative nuclear RNA export factor SDE5 isoform X1 |
| TCD 5 | CHG CHH | Hypo | CT001_A07260420 | Bra004227 | AT1G67480 | F-box/kelch-repeat protein |
| TCD 6 | CG CHH | Hypo | CT001_A07264360 | Bra016068 | AT1G72300 | Tyrosine-sulfated glycopeptide receptor 1 |
| TCD 7 | CG CHG | Hyper | CT001_A07243410 | Bra012325 | AT1G78300 | G-box binding factor GF14 omega encoding a 14-3-3 protein |
| TCD 8 | CG CHG | Hypo | CT001_A09347510 | Bra031188 | AT3G17310 | Probable inactive DNA (cytosine-5)-methyltransferase DRM3 |
| TCD 9 | CHH CHG | Hypo | CT001_A01011320 | Bra013553 | AT2G30070 | High affinity potassium transporter |
| TCD 10 | CHG CHH | Hypo | CT001_A03126570 | Bra024010 | AT4G31400 | Protein chromosome transmission fidelity 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.-S.; Shin, Y.-H.; Park, Y.-D. DNA Methylation Level Changes in Transgenic Chinese Cabbage (Brassica rapa ssp. pekinensis) Plants and Their Effects on Corresponding Gene Expression Patterns. Genes 2021, 12, 1563. https://doi.org/10.3390/genes12101563
Park J-S, Shin Y-H, Park Y-D. DNA Methylation Level Changes in Transgenic Chinese Cabbage (Brassica rapa ssp. pekinensis) Plants and Their Effects on Corresponding Gene Expression Patterns. Genes. 2021; 12(10):1563. https://doi.org/10.3390/genes12101563
Chicago/Turabian StylePark, Jee-Soo, Yun-Hee Shin, and Young-Doo Park. 2021. "DNA Methylation Level Changes in Transgenic Chinese Cabbage (Brassica rapa ssp. pekinensis) Plants and Their Effects on Corresponding Gene Expression Patterns" Genes 12, no. 10: 1563. https://doi.org/10.3390/genes12101563