Clustering Analysis Supports the Detection of Biological Processes Related to Autism Spectrum Disorder

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Database
2.2. Analysis of the Dataset
2.3. Hierarchical Agglomerative Clustering (HAC)
2.4. Enrichment Analysis
2.5. Statistical Analysis
3. Results
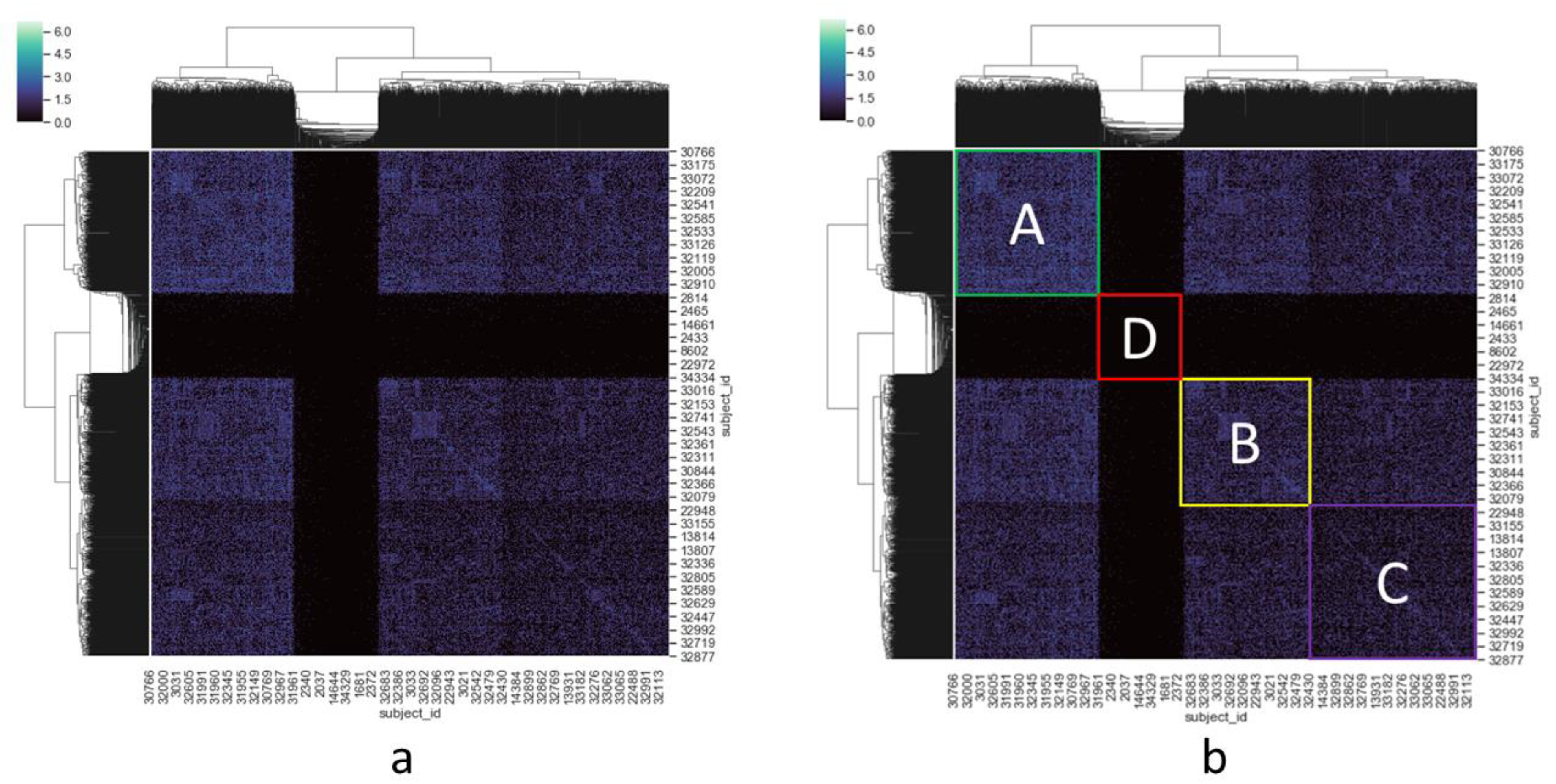
3.1. Cluster Heatmap
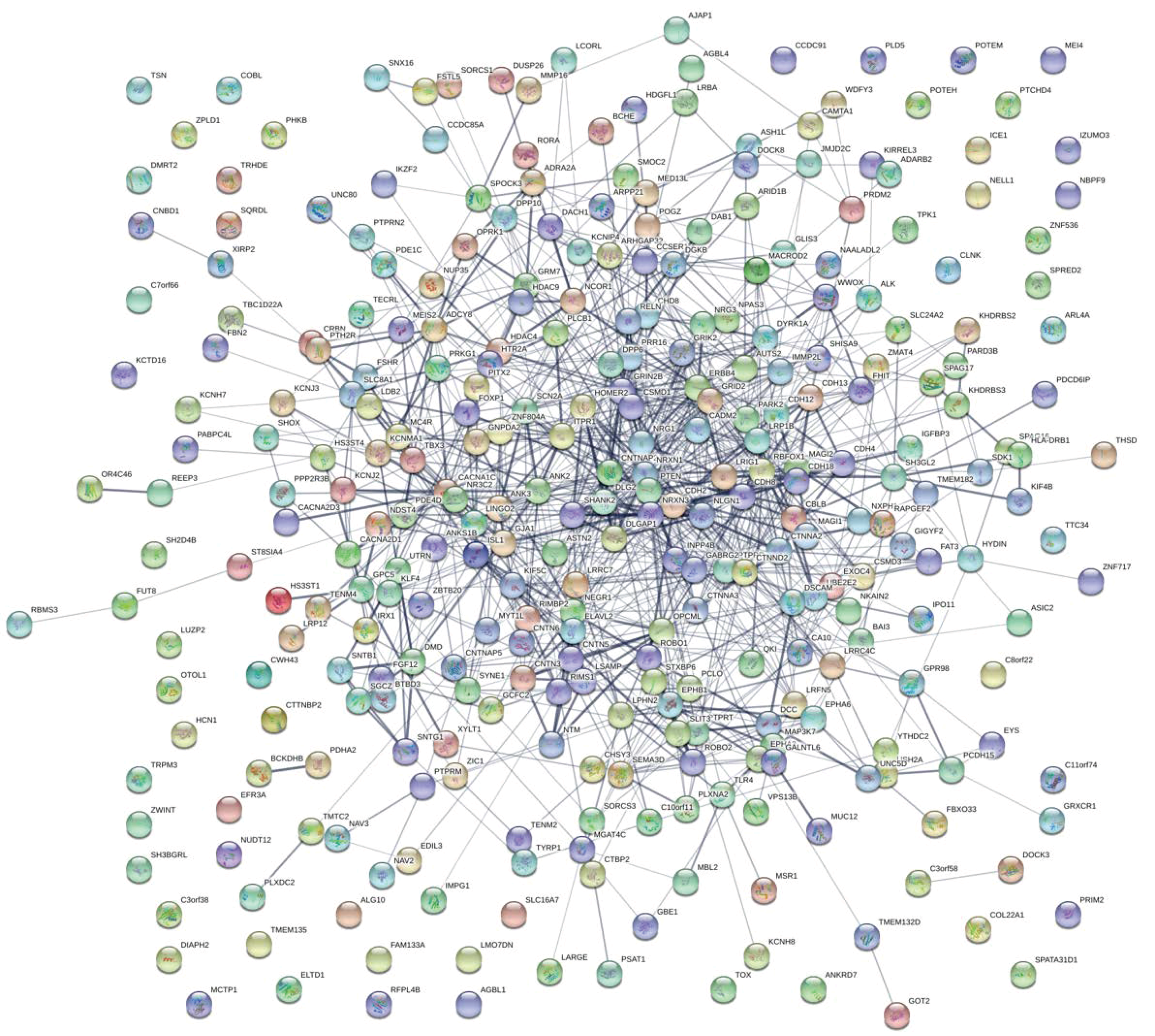
3.2. Enrichment Analysis
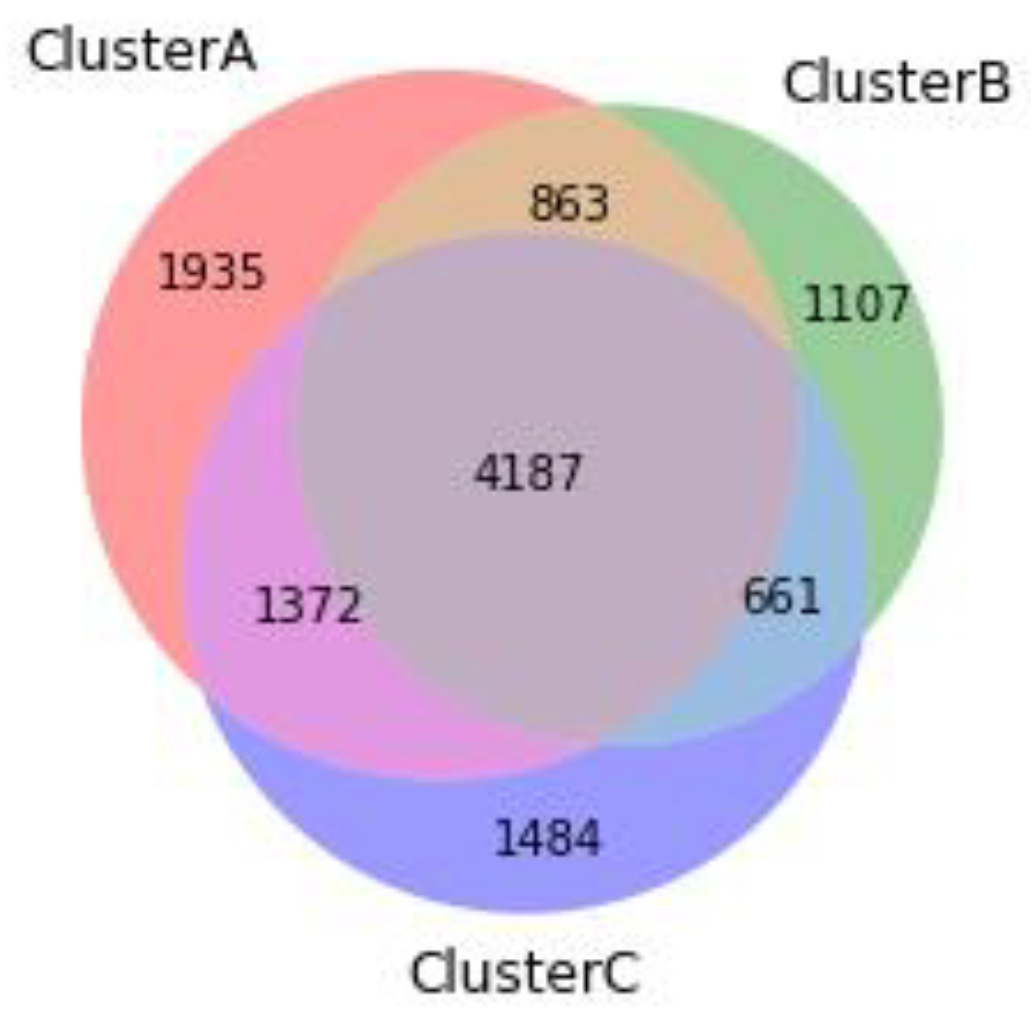
3.3. Cluster Comparisons
3.4. Enrichment Analysis on ExAC Filtered Variants
3.5. Enrichment Analysis on CADD Filtered Variants
4. Discussion
4.1. Enrichment Analysis
4.2. Cluster Comparisons
4.3. Translation into Clinical Research
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorder (DSM-5), 5th ed.; American Psychiatric Publishing: Arlington, VA, USA, 2013; Section II; pp. 50–59. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; He, X.; Willsey, J.A.; Ercan-Sencicek, G.A.; Samocha, K.E.; Cicek, E.A.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, M.; Halayem, S.; Mrad, R.; Bourgou, S.; Charfi, F.; Belhadj, A. Epigenetics’ implication in autism spectrum disorders: A review. Encephale 2017, 43, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Gialloreti, L.E.; Mazzone, L.; Benvenuto, A.; Fasano, A.; García-Alcón, A.; Kraneveld, A.; Moavero, R.; Raz, R.; Riccio, M.P.; Siracusano, M.; et al. Risk and Protective Environmental Factors Associated with Autism Spectrum Disorder: Evidence-Based Principles and Recommendations. J. Clin. Med. 2019, 8, 217. [Google Scholar] [CrossRef] [Green Version]
- Rylaarsdam, L.E.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- De Rubeis, S.; Menachem, F.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Yuen, R.K.C.; Thiruvahindrapuram, B.; Merico, D.; Walker, S.P.; Tammimies, K.; Hoang, N.; Chrysler, C.; Nalpathamkalam, T.; Pellecchia, G.; Liu, Y.; et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat. Med. 2015, 21, 185–191. [Google Scholar] [CrossRef]
- Lombardo, M.V.; Lai, M.-C.; Baron-Cohen, S. Big data approaches to decomposing heterogeneity across the autism spectrum. Mol. Psychiatry 2019, 24, 1435–1450. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, B.S.; Arking, D.E.; Campbell, D.B.; Mefford, H.C.; Morrow, E.M.; Weiss, L.A.; Menashe, I.; Wadkins, T.; Banerjee-Basu, S.; Packer, A. SFARI Gene 2.0: A community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 2013, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; De Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.L.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H.; State, M.W. Gene hunting in autism spectrum disorder: On the path to precision medicine. Lancet Neurol. 2015, 14, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Asif, M.; Martiniano, H.F.M.C.; Marques, A.R.; Santos, J.X.; Vilela, J.; Rasga, C.; Oliveira, G.; Couto, F.M.; Vicente, A.M. Identification of biological mechanisms underlying a multidimensional ASD phenotype using machine learning. Transl. Psychiatry 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of Genes and Cellular Pathways Dysregulated in Autism Spectrum Disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosca, E.; Bersanelli, M.; Gnocchi, M.; Moscatelli, M.; Castellani, G.; Milanesi, L.; Mezzelani, A. Network Diffusion-Based Prioritization of Autism Risk Genes Identifies Significantly Connected Gene Modules. Front. Genet. 2017, 8, 129. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, L.; Guo, H.; Shi, L.; Zhang, K.; Tang, M.; Hu, S.; Dong, S.; Liu, C.; Wang, T.; et al. Targeted sequencing and functional analysis reveal brain-size-related genes and their networks in autism spectrum disorders. Mol. Psychiatry 2017, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Duda, M.; Zhang, H.; Li, H.-D.; Wall, D.P.; Burmeister, M.; Guan, Y. Brain-specific functional relationship networks inform autism spectrum disorder gene prediction. Transl. Psychiatry 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Brueggeman, L.; Koomar, T.; Michaelson, J.J. Forecasting risk gene discovery in autism with machine learning and genome-scale data. Sci. Rep. 2020, 10, 4569. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Hu, T. PANDA: Prioritization of autism-genes using network-based deep-learning approach. Genet Epidemiol. 2020, 44, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Hu, V.W.; Steinberg, M.E. Novel clustering of items from the Autism Diagnostic Interview-Revised to define phenotypes within autism spectrum disorders. Autism Res. 2009, 2, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.N.; Kollu, R.; Banerjee-Basu, S. AutDB: A gene reference resource for autism research. Nucleic Acids Res. 2009, 37, D832–D836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Li, J.; Wu, Q.; Yang, X.; Huang, A.Y.; Zhang, J.; Ye, A.Y.; Dou, Y.; Yan, L.; Zhou, W.-Z.; et al. AutismKB 2.0: A knowledge for the genetic evidence of autism spectrum disorder. Database (Oxford) 2018. [Google Scholar] [CrossRef] [PubMed]
- Yuen, R.K.C.; Merico, D.; Bookman, M.; Howe, J.L.; Thiruvahindrapuram, B.; Patel, R.V.; Whitney, J.; Deflaux, N.; Bingham, J.; Wang, Z.; et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017, 20, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Belmadani, M.; Jacobson, M.; Holmes, N.; Phan, M.; Nguyen, T.; Pavlidis, P.; Rogic, S. VariCarta: A Comprehensive Database of Harmonized Genomic Variants Found in Autism Spectrum Disorder Sequencing Studies. Autism Res. 2019, 12, 1728–1736. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.-A. Patient Similarity: Emerging Concepts in Systems and Precision Medicine. Front. Physiol. 2016, 7, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, J.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Casanova, M.F.; Casanova, E.L.; Frye, R.E.; Baeza-Velasco, C.; Lasalle, J.M.; Hagerman, R.J.; Scherer, S.W.; Natowicz, M.R. Editorial: Secondary vs. Idiopathic Autism. Front. Psychiatry 2020, 11, 297. [Google Scholar] [CrossRef]
- Seaborn. Available online: https://seaborn.pydata.org/index.html (accessed on 18 May 2020).
- Johnson, S.C. Hierarchical clustering schemes. Psychometrika 1967, 32, 241–254. [Google Scholar] [CrossRef]
- Wilkinson, L.; Friendly, M. The History of the Cluster Heat Map. Am. Stat. 2009, 63, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Wallace, M.; Akrivas, G.; Stamou, G. Automatic thematic categorization of documents using a fuzzy taxonomy and fuzzy hierarchical clustering. In Proceedings of the 12th IEEE International Conference on Fuzzy Systems (FUZZ 2003), St Louis, MO, USA, 25–28 May 2003. [Google Scholar]
- Rohlf, F.J. Adaptive hierarchical clustering schemes. Syst. Biol. 1970, 19, 58–82. [Google Scholar] [CrossRef]
- Seo, J.; Shneiderman, B. Interactively Exploring Hierarchical Clustering Results. Craft Inf. Vis. 2003, 35, 334–340. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Ge, Y.; Kohane, Y. Comorbidity Clusters in Autism Spectrum Disorders: An Electronic Health Record Time-Series Analysis. Pediatrics 2014, 133, e53–e63. [Google Scholar] [CrossRef] [Green Version]
- Hu, V.W.; Sarachana, T.; Kim, K.S.; Nguyen, A.; Kulkarni, S.; Steinberg, M.E.; Luu, T.; Lai, Y.; Lee, N.H. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: Evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009, 2, 78–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obafemi-Ajayi, T.; Lam, D.; Takahashi, T.N.; Kanne, S.; Wunsch, D. Sorting the phenotypic heterogeneity of autism spectrum disorders: A hierarchical clustering model. In Proceedings of the 2015 IEEE Conference on Computational Intelligence in Bioinformatics and Computational Biology (CIBCB), Niagara Falls, ON, Canada, 12–15 August 2015; Institute of Electrical and Electronics Engineers (IEEE): Piscataway, NJ, USA, 2015; pp. 1–7. [Google Scholar]
- Mullner, D. Modern hierarchical, agglomerative clustering algorithms. arXiv 2011, arXiv:1109.2378. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Ontology. Available online: http://geneontology.org (accessed on 18 May 2020).
- Thomas, P.D.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Harrison, P.F.; Pattison, A.D.; Powell, D.R.; Beilharz, T.H. Topconfects: A package for confident effect sizes in differential expression analysis provides a more biologically useful ranked gene list. Genome Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Liu, Y.-Y.; Wang, X.; Tan, G.-H.; Li, H.-P.; Hulbert, S.W.; Li, C.-Y.; Yong-Hui, J.; Xiong, Z.-Q.; Xu, X.; et al. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Schafer, S.T.; Paquola, A.C.M.; Stern, S.; Gosselin, D.; Ku, M.; Pena, M.; Kuret, T.J.M.; Liyanage, M.; Mansour, A.A.F.; Jaeger, B.N.; et al. Pathological priming causes developmental gene network heterochronicity in autism patient-derived neurons. Nat. Neurosci. 2019, 22, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Ciarrusta, J.; Dimitrova, R.; Batalle, D.; O’Muircheartaigh, J.; Cordero-Grande, L.; Price, A.; Hughes, E.; Kangas, J.; Perry, E.; Javed, A.; et al. Emerging functional connectivity differences in newborn infants vulnerable to autism spectrum disorders. Transl. Psychiatry 2020, 10, 1–10. [Google Scholar] [CrossRef]
- García-Cabezas, M.Á.; Barbas, H.; Zikopoulos, B. Parallel Development of Chromatin Patterns, Neuron Morphology, and Connections: Potential for Disruption in Autism. Front. Neuroanat. 2018, 12, 70. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Hashimoto, R.; Nakazawa, T.; Tsurusaki, Y.; Yasuda, Y.; Nagayasu, K.; Matsumura, K.; Kawashima, H.; Yamamori, H.; Fujimoto, M.; Ohi, K.; et al. Whole-exome sequencing and neurite outgrowth analysis in autism spectrum disorder. J. Hum. Genet. 2016, 61, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zikopoulos, B.; Barbas, H. Changes in Prefrontal Axons May Disrupt the Network in Autism. J. Neurosci. 2010, 30, 14595–14609. [Google Scholar] [CrossRef] [Green Version]
- Bakos, J.; Bacova, Z.; Grant, S.G.; Castejon, A.M.; Ostatnikova, D. Are Molecules Involved in Neuritogenesis and Axon Guidance Related to Autism Pathogenesis? NeuroMolecular Med. 2015, 17, 297–304. [Google Scholar] [CrossRef]
- Nishiyama, J. Plasticity of dendritic spines: Molecular function and dysfunction in neurodevelopmental disorders. Psychiatry Clin. Neurosci. 2019, 73, 541–550. [Google Scholar] [CrossRef]
- Gabrielsen, T.P.; Anderson, J.S.; Stephenson, K.G.; Beck, J.; King, J.B.; Kellems, R.; Top, D.N.; Russell, N.C.C.; Anderberg, E.; Lundwall, R.A.; et al. Functional MRI connectivity of children with autism and low verbal and cognitive performance. Mol. Autism 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rolls, E.T.; Zhou, Y.; Cheng, W.; Gilson, M.; Deco, G.; Feng, J. Effective connectivity in autism. Autism Res. 2020, 13, 32–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehdizadehfar, V.; Ghassemi, F.; Fallah, A. Brain Connectivity Reflected in Electroencephalogram Coherence in Individuals with Autism: A Meta-analysis. Basic Clin. Neurosci. J. 2019, 10, 409–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickinson, A.; Varcin, K.J.; Sahin, M.; Nelson, C.A.; Jeste, S.S. Early patterns of functional brain development associated with autism spectrum disorder in tuberous sclerosis complex. Autism Res. 2019, 12, 1758–1773. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, J.; Yanlin, C.; Chen, Y.; Liu, L.; Gu, H.; Luo, X.; Hou, F.; Zhang, J.; Song, R. Neurexin gene family variants as risk factors for autism spectrum disorder. Autism Res. 2018, 11, 37–43. [Google Scholar] [CrossRef]
- Baig, D.N.; Yanagawa, T.; Tabuchi, K. Distortion of the normal function of synaptic cell adhesion molecules by genetic variants as a risk for autism spectrum disorders. Brain Res. Bull. 2017, 129, 82–90. [Google Scholar] [CrossRef]
- McAuliffe, D.; Zhao, Y.; Pillai, A.S.; Ament, K.; Adamek, J.; Caffo, B.S.; Mostofsky, S.H.; Ewen, J.B. Learning of skilled movements via imitation in ASD. Autism Res. 2020, 13, 777–784. [Google Scholar] [CrossRef]
- Su, W.-C.; Culotta, M.; Mueller, J.; Tsuzuki, D.; Pelphrey, K.; Bhat, A. Differences in cortical activation patterns during action observation, action execution, and interpersonal synchrony between children with or without autism spectrum disorder (ASD): An fNIRS pilot study. PLoS ONE 2020, 15, e0240301. [Google Scholar] [CrossRef]
- Sigmon, E.R.; Kelleman, M.; Susi, A.; Nylund, C.M.; Oster, M.E. Congenital Heart Disease and Autism: A Case-Control Study. Pediatrics 2019, 144, e20184114. [Google Scholar] [CrossRef]
- Wang, Y.; Kou, Y.; Meng, D. Network Structure Analysis Identifying Key Genes of Autism and Its Mechanism. Comput. Math. Methods Med. 2020, 2020, 3753080-9. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder—current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Roque, F.S.; Jensen, P.B.; Schmock, H.; Dalgaard, M.; Andreatta, M.; Hansen, T.; Søeby, K.; Bredkjær, S.; Juul, A.; Werge, T.; et al. Using Electronic Patient Records to Discover Disease Correlations and Stratify Patient Cohorts. PLoS Comput. Biol. 2011, 7, e1002141. [Google Scholar] [CrossRef] [Green Version]
- Bolouri, H.; Zhao, L.P.; Holland, E.C. Big data visualization identifies the multidimensional molecular landscape of human gliomas. Proc. Natl. Acad. Sci. USA 2016, 113, 5394–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, B.; Walter, S.R.; Day, R.; Dunn, A.G.; Sivaraman, V.; Shah, N.; Longhurst, A.C.; Coiera, E. Bringing cohort studies to the bedside: Framework for a ‘green button’ to support clinical decision-making. J. Comp. Eff. Res. 2015, 4, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, A.; Stein, G.Y.; Ruppin, E.; Altman, R.B.; Sharan, R. A method for inferring medical diagnoses from patient similarities. BMC Med. 2013, 11, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Wang, F.; Hu, J.; Sorrentino, R. Towards Personalized Medicine: Leveraging Patient Similarity and Drug Similarity Analytics. AMIA Jt. Summits Transl. Sci. Proc. 2014, 2014, 132–136. [Google Scholar]
- Yip, B.H.; Bai, D.; Mahjani, B.; Klei, L.; Pawitan, Y.; Hultman, C.M.; Grice, D.E.; Roeder, K.; Buxbaum, J.D.; Devlin, B.; et al. Heritable Variation, With Little or No Maternal Effect, Accounts for Recurrence Risk to Autism Spectrum Disorder in Sweden. Biol. Psychiatry 2018, 83, 589–597. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Cluster A | Cluster B | Cluster C | ||||||
|---|---|---|---|---|---|---|---|---|
| GO Biological Process | FE | FDR | GO Biological Process | FE | FDR | GO Biological Process | FE | FDR |
| neuron projection guidance (GO:0097485) | 1.75 | 6.88 × 10−6 | dendrite morphogenesis (GO:0048813) | 2.38 | 3.03 × 10−3 | neuron recognition (GO:0008038) | 2.46 | 2.19 × 10−3 |
| axon guidance (GO:0007411) | 1.74 | 9.84 × 10−6 | neuron projection guidance (GO:0097485) | 2.07 | 7.85 × 10−10 | ventricular septum development (GO:0003281) | 2.18 | 2.85 × 10−3 |
| regulation of axonogenesis (GO:0050770) | 1.71 | 9.35 × 10−4 | axon guidance (GO:0007411) | 2.07 | 1.29 × 10−9 | cardiac septum development (GO:0003279) | 2.01 | 5.15 × 10−4 |
| axonogenesis (GO:0007409) | 1.68 | 3.30 × 10−7 | synapse assembly (GO:0007416) | 2.07 | 1.28 × 10−3 | negative regulation of developmental growth (GO:0048640) | 1.89 | 4.10 × 10−3 |
| neuron projection morphogenesis (GO:0048812) | 1.68 | 2.13 × 10−9 | action potential (GO:0001508) | 2.06 | 2.60 × 10−3 | cell–cell junction assembly (GO:0007043) | 1.87 | 3.43 × 10−3 |
| plasma membrane bounded cell projection morphogenesis (GO:0120039) | 1.68 | 1.95 × 10−9 | developmental growth involved in morphogenesis (GO:0060560) | 2.05 | 4.71 × 10−4 | neuron projection morphogenesis (GO:0048812) | 1.86 | 1.45 × 10−13 |
| regulation of JNK cascade (GO:0046328) | 1.68 | 2.23 × 10−3 | regulation of synapse assembly (GO:0051963) | 2.02 | 1.89 × 10−3 | cell morphogenesis involved in neuron differentiation (GO:0048667) | 1.85 | 5.59 × 10−12 |
| cell morphogenesis involved in neuron differentiation (GO:0048667) | 1.68 | 3.49 × 10−8 | cell morphogenesis involved in neuron differentiation (GO:0048667) | 2.00 | 1.31 × 10−14 | neuron projection guidance (GO:0097485) | 1.84 | 5.84 × 10−7 |
| cell projection morphogenesis (GO:0048858) | 1.67 | 2.25 × 10−9 | axonogenesis (GO:0007409) | 2.00 | 2.11 × 10−12 | plasma membrane bounded cell projection morphogenesis (GO:0120039) | 1.84 | 3.30 × 10−13 |
| regulation of cell junction assembly (GO:1901888) | 1.67 | 1.34 × 10−3 | negative regulation of cell morphogenesis involved in differentiation (GO:0010771) | 2.00 | 4.30 × 10−3 | axon guidance (GO:0007411) | 1.84 | 6.70 × 10−7 |
| axon development (GO:0061564) | 1.67 | 1.76 × 10−7 | neuron projection morphogenesis (GO:0048812) | 2.00 | 3.64 × 10−16 | axonogenesis (GO:0007409) | 1.84 | 6.78 × 10−10 |
| cell part morphogenesis (GO:0032990) | 1.66 | 2.93 × 10−9 | plasma membrane bounded cell projection morphogenesis (GO:0120039) | 1.99 | 4.61 × 10−16 | cell projection morphogenesis (GO:0048858) | 1.83 | 4.04 × 10−13 |
| renal system development (GO:0072001) | 1.62 | 1.87 × 10−4 | regulation of cell junction assembly (GO:1901888) | 1.99 | 2.52 × 10−6 | axon development (GO:0061564) | 1.82 | 1.99 × 10−10 |
| telencephalon development (GO:0021537) | 1.62 | 4.40 × 10−4 | cell projection morphogenesis (GO:0048858) | 1.97 | 1.10 × 10−15 | regulation of axonogenesis (GO:0050770) | 1.82 | 1.27 × 10−4 |
| kidney development (GO:0001822) | 1.62 | 3.42 × 10−4 | cell junction assembly (GO:0034329) | 1.95 | 7.79 × 10−8 | cell part morphogenesis (GO:0032990) | 1.80 | 8.91 × 10−13 |
| regulation of small GTPase mediated signal transduction (GO:0051056) | 1.61 | 6.55 × 10−5 | cell-cell junction assembly (GO:0007043) | 1.93 | 3.16 × 10−3 | regulation of small GTPase mediated signal transduction (GO:0051056) | 1.78 | 3.01 × 10−7 |
| regulation of neuron projection development (GO:0010975) | 1.61 | 5.54 × 10−8 | synapse organization (GO:0050808) | 1.93 | 2.32 × 10−8 | cell junction assembly (GO:0034329) | 1.76 | 8.13 × 10−6 |
| regulation of cell morphogenesis involved in differentiation (GO:0010769) | 1.60 | 1.26 × 10−4 | axon development (GO:0061564) | 1.92 | 6.41 × 10−12 | cell–cell junction organization (GO:0045216) | 1.76 | 1.54 × 10−3 |
| positive regulation of neuron differentiation (GO:0045666) | 1.60 | 1.03 × 10−5 | cell part morphogenesis (GO:0032990) | 1.92 | 5.66 × 10−15 | neuron projection development (GO:0031175) | 1.75 | 8.64 × 10−15 |
| cell morphogenesis involved in differentiation (GO:0000904) | 1.60 | 1.97 × 10−8 | cell morphogenesis involved in differentiation (GO:0000904) | 1.90 | 1.35 × 10−15 | regulation of cell junction assembly (GO:1901888) | 1.73 | 5.78 × 10−4 |
| GO Biological Process | FE | FDR |
|---|---|---|
| cell-cell adhesion mediated by cadherin (GO:0044331) | 3.85 | 1.74 × 10−3 |
| outflow tract septum morphogenesis (GO:0003148) | 3.62 | 4.27 × 10−3 |
| synaptic transmission, glutamatergic (GO:0035249) | 3.36 | 8.03 × 10−4 |
| neuron recognition (GO:0008038) | 3.30 | 1.50 × 10−4 |
| glutamate receptor signaling pathway (GO:0007215) | 3.18 | 4.96 × 10−4 |
| dendrite morphogenesis (GO:0048813) | 3.05 | 1.98 × 10−4 |
| receptor localization to synapse (GO:0097120) | 3.05 | 2.83 × 10−3 |
| heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules (GO:0007157) | 2.99 | 2.58 × 10−3 |
| regulation of cell–substrate junction organization (GO:0150116) | 2.87 | 2.72 × 10−4 |
| synapse assembly (GO:0007416) | 2.84 | 2.45 × 10−6 |
| heart valve morphogenesis (GO:0003179) | 2.82 | 2.78 × 10−3 |
| regulation of focal adhesion assembly (GO:0051893) | 2.81 | 6.54 × 10−4 |
| regulation of cell–substrate junction assembly (GO:0090109) | 2.81 | 6.52 × 10−4 |
| retina morphogenesis in camera-type eye (GO:0060042) | 2.79 | 1.71 × 10−3 |
| neuron projection guidance (GO:0097485) | 2.75 | 2.15 × 10−15 |
| axon guidance (GO:0007411) | 2.73 | 5.03 × 10−15 |
| negative regulation of axonogenesis (GO:0050771) | 2.67 | 8.24 × 10−4 |
| adherens junction organization (GO:0034332) | 2.66 | 2.70 × 10−3 |
| regulation of glutamate receptor signaling pathway (GO:1900449) | 2.64 | 1.56 × 10−3 |
| cardiac septum morphogenesis (GO:0060411) | 2.64 | 6.87 × 10−4 |
| negative regulation of cell morphogenesis involved in differentiation (GO:0010771) | 2.59 | 6.52 × 10−5 |
| positive regulation of synapse assembly (GO:0051965) | 2.59 | 2.49 × 10−3 |
| protein localization to synapse (GO:0035418) | 2.56 | 4.75 × 10−3 |
| regulation of neurotransmitter receptor activity (GO:0099601) | 2.52 | 1.13 × 10−3 |
| mechanoreceptor differentiation (GO:0042490) | 2.52 | 4.82 × 10−3 |
| regulation of synaptic transmission, glutamatergic (GO:0051966) | 2.50 | 3.75 × 10−3 |
| cell morphogenesis involved in neuron differentiation (GO:0048667) | 2.48 | 8.42 × 10−20 |
| neuron projection morphogenesis (GO:0048812) | 2.47 | 1.37 × 10−21 |
| axonogenesis (GO:0007409) | 2.47 | 1.72 × 10−16 |
| regulation of synapse assembly (GO:0051963) | 2.46 | 1.41 × 10−4 |
| plasma membrane bounded cell projection morphogenesis (GO:0120039) | 2.45 | 2.14 × 10−21 |
| cell projection morphogenesis (GO:0048858) | 2.43 | 4.51 × 10−21 |
| positive regulation of phosphatidylinositol 3-kinase signaling (GO:0014068) | 2.43 | 1.35 × 10−3 |
| dendrite development (GO:0016358) | 2.42 | 1.01 × 10−4 |
| neural crest cell differentiation (GO:0014033) | 2.42 | 1.85 × 10−3 |
| regulation of sodium ion transport (GO:0002028) | 2.39 | 2.01 × 10−3 |
| axon development (GO:0061564) | 2.38 | 2.47 × 10−16 |
| regulation of potassium ion transmembrane transport (GO:1901379) | 2.38 | 3.48 × 10−3 |
| cell part morphogenesis (GO:0032990) | 2.38 | 1.50 × 10−20 |
| regulation of potassium ion transport (GO:0043266) | 2.37 | 6.70 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emberti Gialloreti, L.; Enea, R.; Di Micco, V.; Di Giovanni, D.; Curatolo, P. Clustering Analysis Supports the Detection of Biological Processes Related to Autism Spectrum Disorder. Genes 2020, 11, 1476. https://doi.org/10.3390/genes11121476
Emberti Gialloreti L, Enea R, Di Micco V, Di Giovanni D, Curatolo P. Clustering Analysis Supports the Detection of Biological Processes Related to Autism Spectrum Disorder. Genes. 2020; 11(12):1476. https://doi.org/10.3390/genes11121476
Chicago/Turabian StyleEmberti Gialloreti, Leonardo, Roberto Enea, Valentina Di Micco, Daniele Di Giovanni, and Paolo Curatolo. 2020. "Clustering Analysis Supports the Detection of Biological Processes Related to Autism Spectrum Disorder" Genes 11, no. 12: 1476. https://doi.org/10.3390/genes11121476