Divergent Mutational Landscapes of Consensus and Minority Genotypes of West Nile Virus Demonstrate Host and Gene-Specific Evolutionary Pressures

Abstract
:1. Introduction
2. Materials and Methods
3. Results
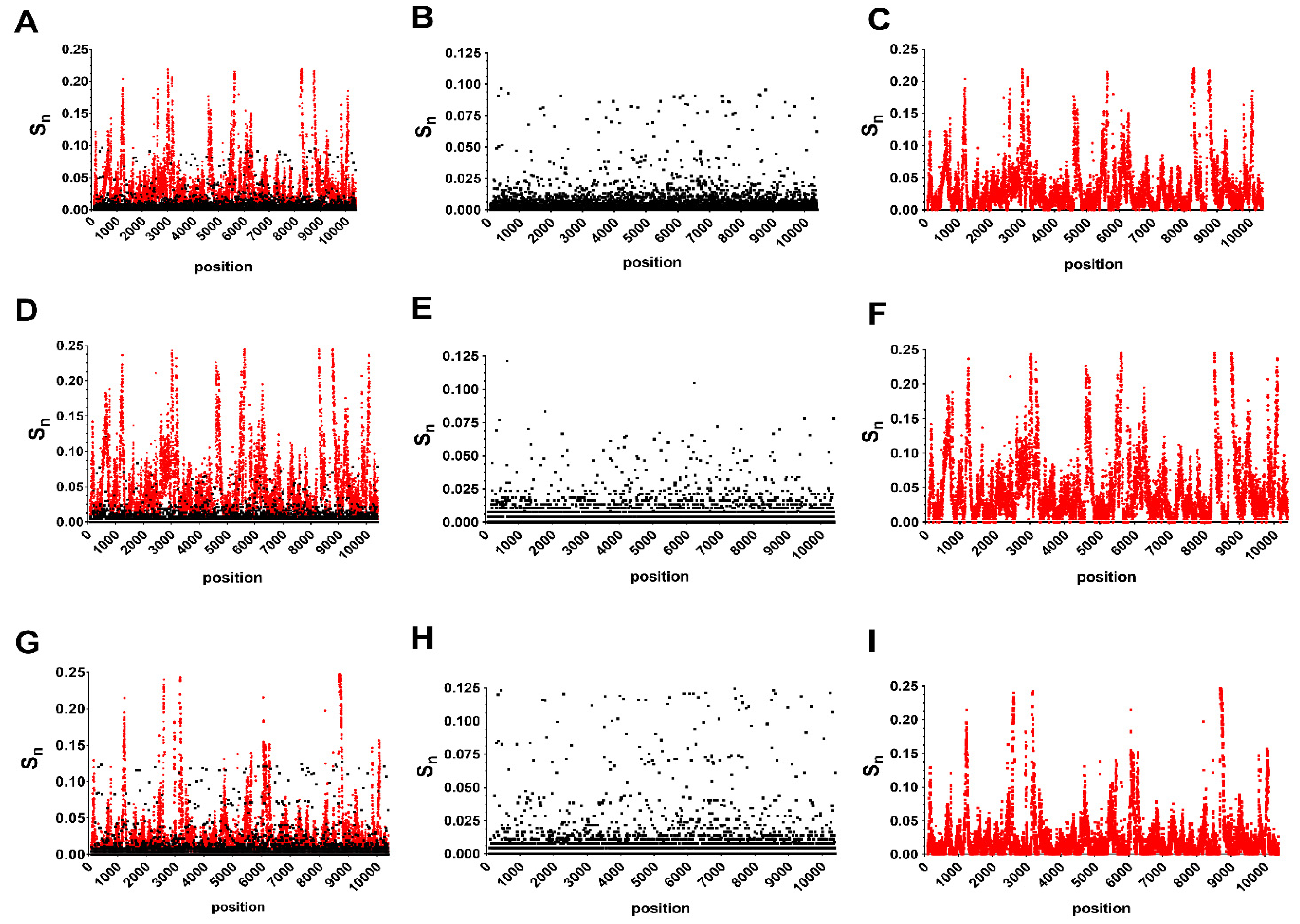
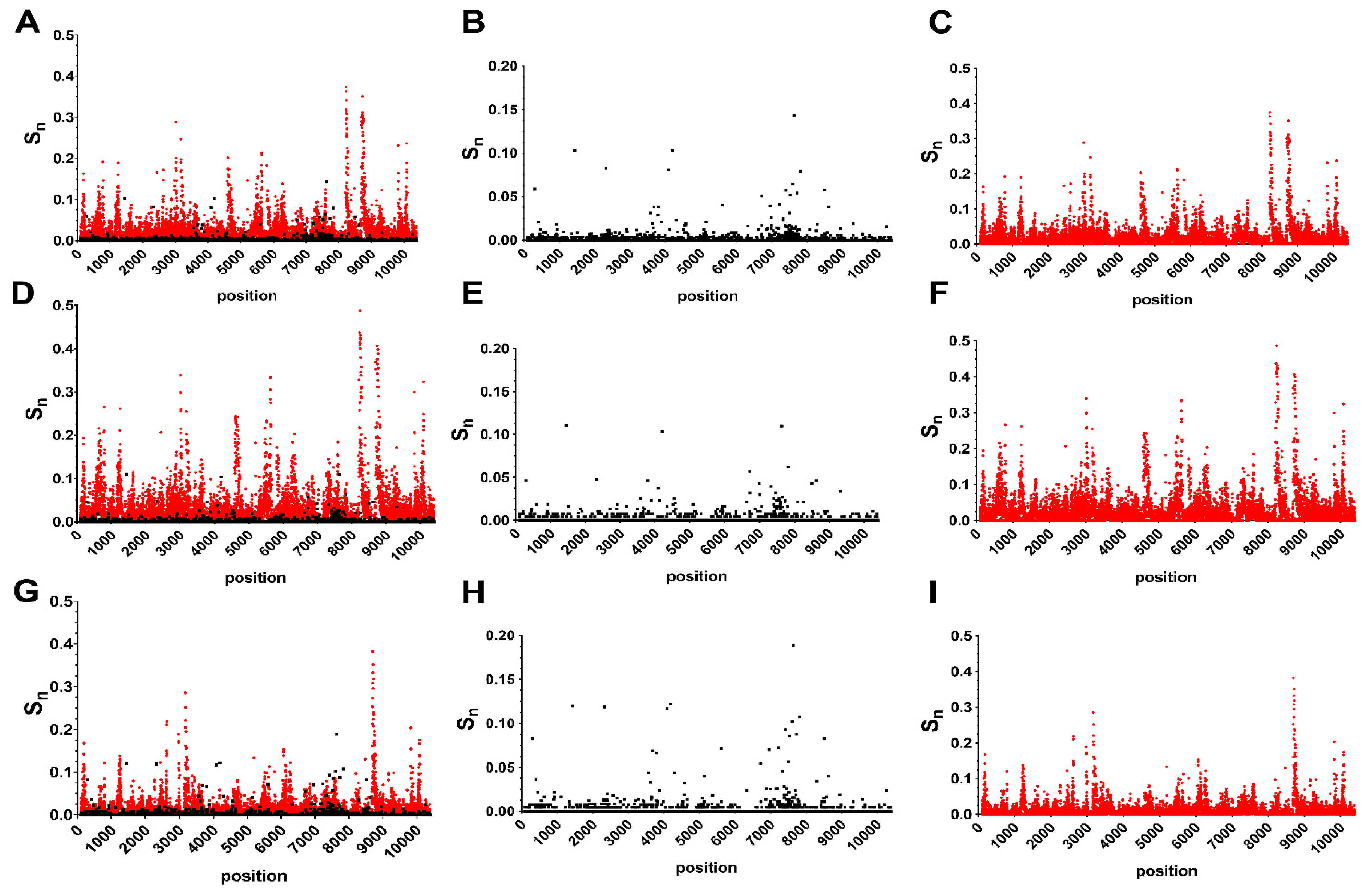
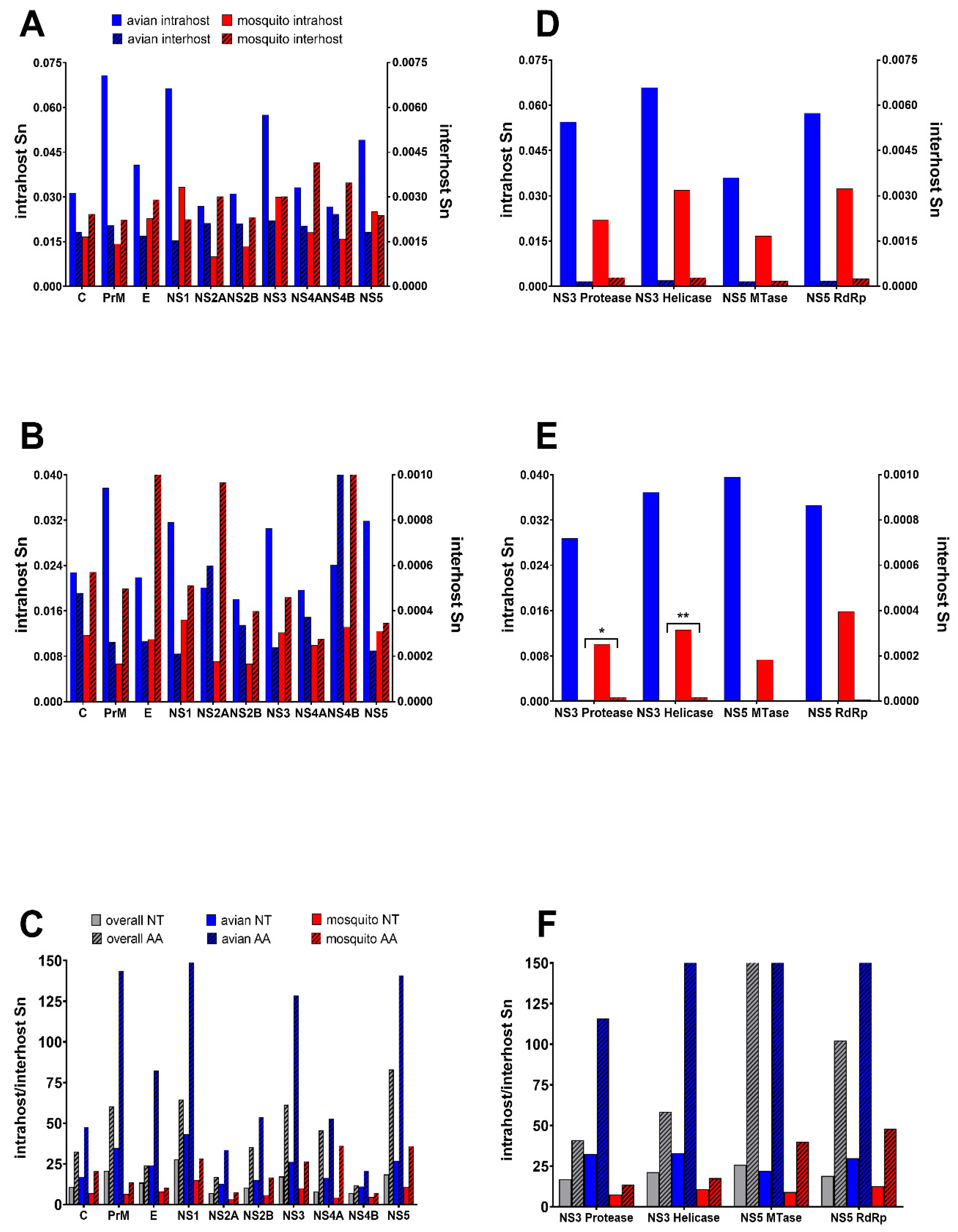
3.1. Host and Gene-Specific Genetic Diversity and Selective Pressures
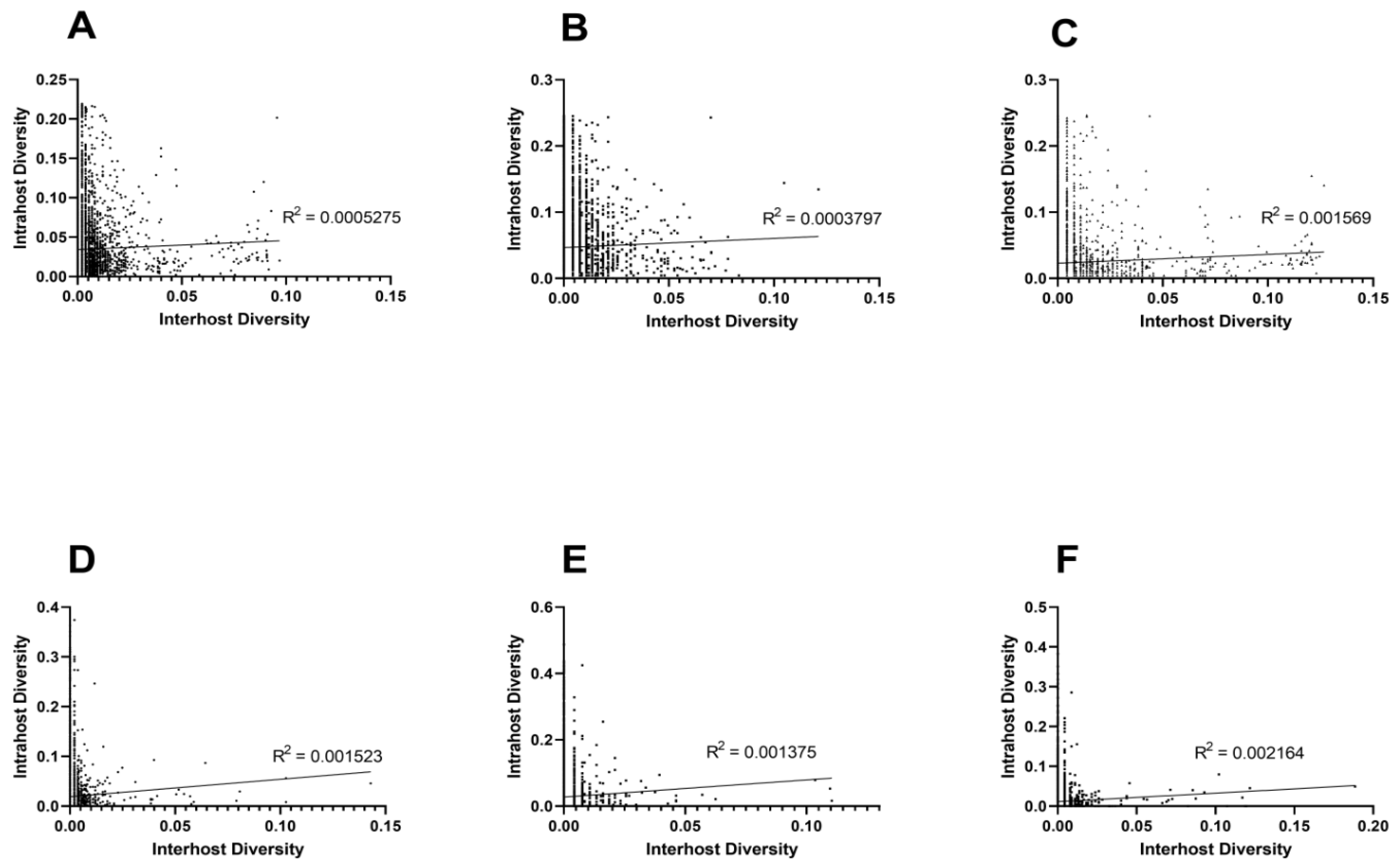
3.2. Unique Intra and Interhost Genetic Signatures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gubler, D.J. Dengue and Dengue Hemorrhagic Fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, E.B.; Gubler, D.J. West Nile Virus: Epidemiology and Clinical Features of an Emerging Epidemic in the United States. Annu. Rev. Med. 2006, 57, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ciota, A.T.; Kramer, L.D. Vector-Virus Interactions and Transmission Dynamics of West Nile Virus. Viruses 2013, 5, 3021–3047. [Google Scholar] [CrossRef] [PubMed]
- Chancey, C.; Grinev, A.; Volkova, E.; Rios, M. The Global Ecology and Epidemiology of West Nile Virus. BioMed Res. Int. 2015, 2015, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klema, V.J.; Padmanabhan, R.; Choi, K.H. Flaviviral Replication Complex: Coordination between RNA Synthesis and 5′-RNA Capping. Viruses 2015, 7, 4640–4656. [Google Scholar] [CrossRef] [PubMed]
- Brinton, M.A. The Molecular Biology of West Nile Virus: A New Invader of the Western Hemisphere. Annu. Rev. Microbiol. 2002, 56, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Kim, B.S.; Chipman, P.R.; Rossmann, M.G.; Kuhn, R.J. Structure of West Nile Virus. Science 2003, 302, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malet, H.; Massé, N.; Selisko, B.; Romette, J.-L.; Alvarez, K.; Guillemot, J.C.; Tolou, H.; Yap, T.L.; Vasudevan, S.G.; Lescar, J.; et al. The flavivirus polymerase as a target for drug discovery. Antivir. Res. 2008, 80, 23–35. [Google Scholar] [CrossRef]
- Li, K.; Phoo, W.W.; Luo, D. Functional interplay among the flavivirus NS3 protease, helicase, and cofactors. Virol. Sin. 2014, 29, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Genet. 2004, 2, 109–122. [Google Scholar] [CrossRef]
- CDC. West Nile Virus: Final Cumulative Maps & Data for 1999–2018: Center for Disease Control. 2019. Available online: https://www.cdc.gov/westnile/statsmaps/cumMapsData.html#two (accessed on 7 September 2020).
- Busch, M.P.; Wright, D.J.; Custer, B.; Tobler, L.H.; Stramer, S.L.; Kleinman, S.H.; Prince, H.E.; Bianco, C.; Foster, G.; Petersen, L.R.; et al. West nile virus infections projected from blood donor screening data, United States, 2003. Emerg. Infect. Dis. 2006, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Peterson, R.K.D.; Macedo, P.A.; Davis, R.S. A Human-Health Risk Assessment for West Nile Virus and Insecticides Used in Mosquito Management. Environ. Heal. Perspect. 2006, 114, 366–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. West Nile Virus & Dead Birds 2015. Available online: http://www.cdc.gov/westnile/faq/deadbirds.html (accessed on 7 September 2020).
- Añez, G.; Grinev, A.; Chancey, C.; Ball, C.; Akolkar, N.; Land, K.J.; Winkelman, V.; Stramer, S.L.; Kramer, L.D.; Rios, M. Evolutionary Dynamics of West Nile Virus in the United States, 1999–2011: Phylogeny, Selection Pressure and Evolutionary Time-Scale Analysis. PLoS Negl. Trop. Dis. 2013, 7, e2245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, L.D.; Styer, L.M.; Ebel, G.D. A Global Perspective on the Epidemiology of West Nile Virus. Annu. Rev. Entomol. 2008, 53, 61–81. [Google Scholar] [CrossRef] [Green Version]
- Lauring, A.S.; Andino, R. Quasispecies Theory and the Behavior of RNA Viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef]
- Eigen, M. Viral Quasispecies. Sci. Am. 1993, 269, 42–49. [Google Scholar] [CrossRef]
- Van Slyke, G.; Ciota, A.T.; Willsey, G.G.; Jaeger, J.; Shi, P.-Y.; Kramer, L.D. Point mutations in the West Nile virus (Flaviviridae; Flavivirus) RNA-dependent RNA polymerase alter viral fitness in a host-dependent manner in vitro and in vivo. Virology 2012, 427, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Dridi, M.; Rosseel, T.; Orton, R.; Johnson, P.C.D.; Lecollinet, S.; Muylkens, B.; Lambrecht, B.; Van Borm, S. Next-generation sequencing shows West Nile virus quasispecies diversification after a single passage in a carrion crow (Corvus corone) in vivo infection model. J. Gen. Virol. 2015, 96, 2999–3009. [Google Scholar] [CrossRef] [Green Version]
- Pfeiffer, J.K.; Kirkegaard, K. Increased Fidelity Reduces Poliovirus Fitness and Virulence under Selective Pressure in Mice. PLoS Pathog. 2005, 1, e11. [Google Scholar] [CrossRef]
- Griesemer, S.B.; Kramer, L.D.; Van Slyke, A.G.; Pata, J.D.; Gohara, D.W.; Cameron, E.C.; Ciota, A.T. Mutagen resistance and mutation restriction of St. Louis encephalitis virus. J. Gen. Virol. 2017, 98, 201–211. [Google Scholar] [CrossRef]
- Van Slyke, G.; Arnold, J.J.; Lugo, A.J.; Griesemer, S.B.; Moustafa, I.M.; Kramer, L.D.; Cameron, C.E.; Ciota, A.T. Sequence-Specific Fidelity Alterations Associated with West Nile Virus Attenuation in Mosquitoes. PLoS Pathog. 2015, 11, e1005009. [Google Scholar] [CrossRef]
- Kautz, T.F.; Guerbois, M.; Khanipov, K.; Patterson, I.E.; Langsjoen, R.M.; Yun, R.; Warmbrod, K.L.; Fofanov, Y.; Weaver, S.C.; Forrester, N.L. Low-fidelity Venezuelan equine encephalitis virus polymerase mutants to improve live-attenuated vaccine safety and efficacy. Virus Evol. 2018, 4, vey004. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Ussel, M.D.; Casado, C.; Yuste, E.; Olivares, I.; López-Galíndez, C. In vitro analysis of human immunodeficiency virus type 1 resistance to nevirapine and fitness determination of resistant variants. J. Gen. Virol. 2002, 83, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Tsetsarkin, K.A.; Weaver, S.C. Sequential Adaptive Mutations Enhance Efficient Vector Switching by Chikungunya Virus and Its Epidemic Emergence. PLoS Pathog. 2011, 7, e1002412. [Google Scholar] [CrossRef] [Green Version]
- Farci, P. The Outcome of Acute Hepatitis C Predicted by the Evolution of the Viral Quasispecies. Science 2000, 288, 339–344. [Google Scholar] [CrossRef]
- Domingo, E.; Martín, V.; Perales, C.; Grande-Pérez, A.; García-Arriaza, J.; Arias, A. Viruses as Quasispecies: Biological Implications. Curr. Top. Microbiol. Immunol. 2006, 299, 51–82. [Google Scholar] [PubMed]
- Ho, C.K.Y.; Raghwani, J.; Koekkoek, S.; Liang, R.H.; Van Der Meer, J.T.M.; Van Der Valk, M.; De Jong, M.; Pybus, O.; Schinkel, J.; Molenkamp, R. Characterization of Hepatitis C Virus (HCV) Envelope Diversification from Acute to Chronic Infection within a Sexually Transmitted HCV Cluster by Using Single-Molecule, Real-Time Sequencing. J. Virol. 2016, 91, e02262-16. [Google Scholar] [CrossRef] [Green Version]
- Brackney, D.E.; Schirtzinger, E.E.; Harrison, T.D.; Ebel, G.D.; Hanley, K.A. Modulation of Flavivirus Population Diversity by RNA Interference. J. Virol. 2015, 89, 4035–4039. [Google Scholar] [CrossRef] [Green Version]
- Metsky, H.C.; Matranga, C.B.; Wohl, S.; Schaffner, S.F.; Freije, C.A.; Winnicki, S.M.; West, K.; Quigley, J.E.; Baniecki, M.L.; Gladden-Young, A.; et al. Zika virus evolution and spread in the Americas. Nat. Cell Biol. 2017, 546, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, P.; Wang, C.; Trivedi, S.B.; Eswarappa, M.; Montoya, M.; Balmaseda, A.; Harris, E. Intrahost Selection Pressures Drive Rapid Dengue Virus Microevolution in Acute Human Infections. Cell Host Microbe 2017, 22, 400–410.e5. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-T.; Chang, B.-L.; Liang, J.-J.; Tsai, H.-J.; Lee, Y.-L.; Lin, R.-J.; Lin, Y.-L. Japanese Encephalitis Virus Nonstructural Protein NS5 Interacts with Mitochondrial Trifunctional Protein and Impairs Fatty Acid β-Oxidation. PLoS Pathog. 2015, 11, e1004750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, L.L.; Vignuzzi, M. Host Alternation of Chikungunya Virus Increases Fitness while Restricting Population Diversity and Adaptability to Novel Selective Pressures. J. Virol. 2010, 85, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nat. Cell Biol. 2006, 439, 344–348. [Google Scholar] [CrossRef]
- Jerzak, G.; Bernard, K.A.; Kramer, L.D.; Ebel, G.D. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 2005, 86, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Jerzak, G.V.S.; Brown, I.; Shi, P.-Y.; Kramer, L.D.; Ebel, G.D. Genetic diversity and purifying selection in West Nile virus populations are maintained during host switching. Virology 2008, 374, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Geoghegan, J.L.; Holmes, E.C. The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 2018, 19, 756–769. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Shi, M.; Holmes, E.C. Using Metagenomics to Characterize an Expanding Virosphere. Cell 2018, 172, 1168–1172. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Glebova, O.; Knyazev, S.; Melnyk, A.; Artyomenko, A.; Khudyakov, Y.; Zelikovsky, A.; Skums, P. Inference of genetic relatedness between viral quasispecies from sequencing data. BMC Genom. 2017, 18, 918. [Google Scholar] [CrossRef] [Green Version]
- Shilts, M.H.; Tan, Y.; Zink, S.D.; Koetzner, A.C.; Maffei, J.G.; Halpin, A.R.; Muller, E.; Novatny, M.; Shilts, M.; Fedorova, N.B.; et al. Evolutionary dynamics and molecular epidemiology of West Nile virus in New York State: 1999–2015. Virus Evol. 2019, 5, vez020. [Google Scholar]
- Ciota, A.T.; Ngo, K.A.; Lovelace, A.O.; Payne, A.F.; Zhou, Y.; Shi, P.-Y.; Kramer, L.D. Role of the mutant spectrum in adaptation and replication of West Nile virus. J. Gen. Virol. 2007, 88, 865–874. [Google Scholar] [CrossRef]
- Eigen, M.; McCaskill, J.S.; Schuster, P. Molecular quasi-species. J. Phys. Chem. 1988, 92, 6881–6891. [Google Scholar] [CrossRef]
- Ciota, A.T.; Ehrbar, D.J.; Van Slyke, G.; Payne, A.F.; Willsey, G.G.; Viscio, R.E.; Kramer, L.D. Quantification of intrahost bottlenecks of West Nile virus in Culex pipiens mosquitoes using an artificial mutant swarm. Infect. Genet. Evol. 2012, 12, 557–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, C.W.; Sibley, S.D.; Kolokotronis, S.-O.; Hamer, G.L.; Newman, C.M.; Anderson, T.; Tavis, K.; Walker, E.D.; Kitron, U.D.; Brawn, J.D.; et al. Selective constraint and adaptive potential of West Nile virus within and among naturally infected avian hosts and mosquito vectors. Virus Evol. 2018, 4, vey013. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Weger-Lucarelli, J.; Murrieta, R.A.; Fauver, J.R.; Garcia-Luna, S.M.; Prasad, A.N.; Iv, W.C.B.; Ebel, G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe 2016, 19, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, E.R.; Fitzpatrick, K.A.; Jerzak, G.V.S.; Shi, P.-Y.; Kramer, L.D.; Ebel, G.D. West Nile Virus Experimental Evolution in vivo and the Trade-off Hypothesis. PLoS Pathog. 2011, 7, e1002335. [Google Scholar] [CrossRef]
- Ebel, G.D.; Fitzpatrick, K.A.; Lim, P.-Y.; Bennett, C.J.; Deardorff, E.R.; Jerzak, G.V.S.; Kramer, L.D.; Zhou, Y.; Shi, P.-Y.; Bernard, K.A. Nonconsensus West Nile Virus Genomes Arising during Mosquito Infection Suppress Pathogenesis and Modulate Virus Fitness In Vivo. J. Virol. 2011, 85, 12605–12613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerzak, G.V.S.; Bernard, K.; Kramer, L.D.; Shi, P.-Y.; Ebel, G.D. The West Nile virus mutant spectrum is host-dependant and a determinant of mortality in mice. Virology 2007, 360, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciota, A.T.; Jia, Y.; Payne, A.F.; Jerzak, G.; Davis, L.J.; Young, D.S.; Ehrbar, D.; Kramer, L.D. Experimental Passage of St. Louis Encephalitis Virus In Vivo in Mosquitoes and Chickens Reveals Evolutionarily Significant Virus Characteristics. PLoS ONE 2009, 4, e7876. [Google Scholar] [CrossRef] [Green Version]
- Ehrbar, D.J.; Ngo, K.A.; Campbell, S.R.; Kramer, L.D.; Ciota, A.T. High levels of local inter- and intra-host genetic variation of West Nile virus and evidence of fine-scale evolutionary pressures. Infect. Genet. Evol. 2017, 51, 219–226. [Google Scholar] [CrossRef]
- Ciota, A.T.; Ehrbar, D.J.; Van Slyke, A.G.; Willsey, G.G.; Kramer, L.D. Cooperative interactions in the West Nile virus mutant swarm. BMC Evol. Biol. 2012, 12, 58. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Brito, A.F.; Swetnam, D.M.; Vogels, C.B.F.; Tokarz, R.E.; Andersen, K.G.; Smith, R.C.; Bedford, T.; Grubaugh, N.D. Twenty years of West Nile virus spread and evolution in the Americas visualized by Nextstrain. PLoS Pathog. 2019, 15, e1008042. [Google Scholar] [CrossRef] [Green Version]
- Arjona, A.; Ledizet, M.; Anthony, K.; Bonafé, N.; Modis, Y.; Town, T.; Fikrig, E. West Nile virus envelope protein inhibits dsRNA-induced innate immune responses. J. Immunol. 2007, 179, 8403–8409. [Google Scholar] [CrossRef] [Green Version]
- Crill, W.D.; Chang, G.-J.J. Localization and Characterization of Flavivirus Envelope Glycoprotein Cross-Reactive Epitopes. J. Virol. 2004, 78, 13975–13986. [Google Scholar] [CrossRef] [Green Version]
- Brault, A.C.; Langevin, S.A.; Ramey, W.N.; Fang, Y.; Beasley, D.W.C.; Barker, C.M.; Sanders, T.A.; Reisen, W.K.; Barrett, A.D.T.; Bowen, R.A. Reduced Avian Virulence and Viremia of West Nile Virus Isolates from Mexico and Texas. Am. J. Trop. Med. Hyg. 2011, 85, 758–767. [Google Scholar] [CrossRef] [Green Version]
- Muller, D.A.; Young, P.R. The flavivirus NS1 protein: Molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antivir. Res. 2013, 98, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Edeling, M.A.; Diamond, M.S.; Fremont, D.H. Structural basis of Flavivirus NS1 assembly and antibody recognition. Proc. Natl. Acad. Sci. USA 2014, 111, 4285–4290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613.e8. [Google Scholar] [CrossRef] [Green Version]
- Mandl, C.W.; Guirakhoo, F.; Holzmann, H.; Heinz, F.X.; Kunz, C. Antigenic structure of the flavivirus envelope protein E at the molecular level, using tick-borne encephalitis virus as a model. J. Virol. 1989, 63, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Cardosa, M.J.; Wang, S.M.; Sum, M.S.H.; Tio, P.H. Antibodies against prM protein distinguish between previous infection with dengue and Japanese encephalitis viruses. BMC Microbiol. 2002, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Sandoval, A.; Ludert, J.E. The Dual Role of the Antibody Response Against the Flavivirus Non-structural Protein 1 (NS1) in Protection and Immuno-Pathogenesis. Front. Immunol. 2019, 10, 1651. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Wang, X.J.; Mokhonov, V.V.; Shi, P.-Y.; Randall, R.; Khromykh, A.A. Inhibition of Interferon Signaling by the New York 99 Strain and Kunjin Subtype of West Nile Virus Involves Blockage of STAT1 and STAT2 Activation by Nonstructural Proteins. J. Virol. 2005, 79, 1934–1942. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.J.; Wang, X.J.; Clark, D.C.; Lobigs, M.; Hall, R.A.; Khromykh, A.A. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 2006, 80, 2396–2404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Xu, Y.-P.; Wang, M.; Miao, M.; Zhou, H.; Xu, J.; Kong, J.; Zheng, D.; Li, R.-T.; Zhang, R.-R.; et al. Flavivirus induces and antagonizes antiviral RNA interference in both mammals and mosquitoes. Sci. Adv. 2020, 6, eaax7989. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Lee, L.T.; Wang, Q.Y.; Xie, X.; Lu, S.; Yau, Y.H.; Yuan, Z.; Shochat, S.G.; Kang, C.; Lescar, J.; et al. Mapping the Interactions between the NS4B and NS3 Proteins of Dengue Virus. J. Virol. 2015, 89, 3471–3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced Autophagy Protects Cells against Death and Enhances Virus Replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [Green Version]
- Blã¡zquez, A.-B.; Martín-Acebes, M.A.; Saiz, J.-C. Amino acid substitutions in the non-structural proteins 4A or 4B modulate the induction of autophagy in West Nile virus infected cells independently of the activation of the unfolded protein response. Front. Microbiol. 2015, 5, 797. [Google Scholar] [CrossRef]
- McMullen, A.R.; May, F.; Li, L.; Guzman, H.; Bueno, R.; Dennett, J.A.; Tesh, R.B.; Barrett, A.D. Evolution of New Genotype of West Nile Virus in North America. Emerg. Infect. Dis. 2011, 17, 785–793. [Google Scholar] [CrossRef]
- Powdrill, M.H.; Tchesnokov, E.P.; Kozak, R.A.; Russell, R.S.; Martin, R.; Svarovskaia, E.S.; Mo, H.; Kouyos, R.D.; Götte, M. Contribution of a mutational bias in hepatitis C virus replication to the genetic barrier in the development of drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 20509–20513. [Google Scholar] [CrossRef] [Green Version]
- Warmbrod, K.L.; Patterson, I.E.; Kautz, T.F.; Stanton, A.; Rockx-Brouwer, D.; Kalveram, B.K.; Khanipov, K.; Thangamani, S.; Fofanov, Y.; Forrester, N.L. Viral RNA-dependent RNA polymerase mutants display an altered mutation spectrum resulting in attenuation in both mosquito and vertebrate hosts. PLoS Pathog. 2019, 15, e1007610. [Google Scholar] [CrossRef]
- Caldwell, H.S.; Ngo, K.; Pata, J.D.; Kramer, L.D.; Ciota, A.T. West Nile Virus fidelity modulates the capacity for host cycling and adaptation. J. Gen. Virol. 2020, 101, 410–419. [Google Scholar] [CrossRef]
- Vogel, F.; Kopun, M. Higher frequencies of transitions among point mutations. J. Mol. Evol. 1977, 9, 159–180. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Miyazawa, S.; Yasunaga, T. Two types of amino acid substitutions in protein evolution. J. Mol. Evol. 1979, 12, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Evidence for the Selective Basis of Transition-to-Transversion Substitution Bias in Two RNA Viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef] [Green Version]
- Moratorio, G.; Iriarte, A.; Moreno, P.; Musto, H.; Cristina, J. A detailed comparative analysis on the overall codon usage patterns in West Nile virus. Infect. Genet. Evol. 2013, 14, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, P.; Charlebois, P.; Tellez, Y.; Nunez, A.; Ryan, E.M.; Malboeuf, C.M.; Levin, J.Z.; Lennon, N.J.; Balmaseda, A.; Harris, E.; et al. Genome-Wide Patterns of Intrahuman Dengue Virus Diversity Reveal Associations with Viral Phylogenetic Clade and Interhost Diversity. J. Virol. 2012, 86, 8546–8558. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interhost | |||||
| AA 1 | NT 2 | pN 3 | Mean Sn NT | Mean Sn AA | |
| All | 4702 | 25,503 | 0.1844 | 0.0023 | 0.0005 |
| Avian | 1896 | 10,739 | 0.1766 | 0.0019 | 0.0004 |
| Mosquito | 2796 | 14,671 | 0.1906 | 0.0028 | 0.0007 |
| Intrahost | |||||
| AA 1 | NT 2 | pN 3 | Mean Sn NT | Mean Sn AA | |
| All | 151,265 | 380,537 | 0.3975 | 0.0346 | 0.0192 |
| Avian | 113,728 | 283,958 | 0.4005 | 0.0470 * | 0.0277 * |
| Mosquito | 36,449 | 91,853 | 0.3968 | 0.0233 * | 0.0114 * |
| Intrahost Substitution Ratio | ||||||||||||
| # A - U | # A - C | # A - G | # U - A | # U - G | # U - C | # G - A | # G - C | # G - U | # C - U | # C - G | # C - A | |
| All | 0.0966 | 0.0866 | 0.0954 | 0.0674 | 0.0597 | 0.0777 | 0.0948 | 0.0910 | 0.0934 | 0.0932 | 0.0717 | 0.0726 |
| Avian | 0.0969 | 0.0868 | 0.0943 | 0.0671 | 0.0597 | 0.0770 | 0.0943 | 0.0935 | 0.0943 | 0.0918 | 0.0715 | 0.0727 |
| Mosquito | 0.0957 | 0.0858 | 0.0995 | 0.0679 | 0.0594 | 0.0799 | 0.0967 | 0.0834 | 0.0903 | 0.0971 | 0.0724 | 0.0718 |
| Interhost Substitution Ratio | ||||||||||||
| # A - U | # A - C | # A - G | # U - A | # U - G | # U - C | # G - A | # G - C | # G - U | # C - U | # C - G | # C - A | |
| All | 0.0462 | 0.0151 | 0.1087 | 0.0325 | 0.0038 | 0.2592 | 0.1169 | 0.0031 | 0.0151 | 0.3879 | 0.0013 | 0.0100 |
| Avian | 0.0549 | 0.0092 | 0.1187 | 0.0359 ns | 0.0049 | 0.2701 | 0.1104 | 0.0023 | 0.0131 | 0.3716 | 0.0009 | 0.0076 |
| Mosquito | 0.0398 * | 0.0195 | 0.1018 | 0.0300 | 0.0030 | 0.2508 | 0.1219 | 0.0037 | 0.0167 | 0.3998 | 0.0016 | 0.0116 |
| Interhost | Intrahost | ||
|---|---|---|---|
| Mutation | Significance | Mutation | Significance |
| # A - U | a **** | # A - U | ns |
| # A - C | m **** | # A - C | ns |
| # A - G | a **** | # A - G | m **** |
| # U - A | a ** | # U - A | ns |
| # U - G | a * | # U - G | ns |
| # U - C | a **** | # U - C | m ** |
| # G - A | a **** | # G - A | m * |
| # G - C | ns | # G - C | a **** |
| # G - U | m * | # G - U | a *** |
| # C - U | m **** | # C - U | m **** |
| # C - G | ns | # C - G | ns |
| # C - A | m ** | # C - A | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldwell, H.S.; Lasek-Nesselquist, E.; Follano, P.; Kramer, L.D.; Ciota, A.T. Divergent Mutational Landscapes of Consensus and Minority Genotypes of West Nile Virus Demonstrate Host and Gene-Specific Evolutionary Pressures. Genes 2020, 11, 1299. https://doi.org/10.3390/genes11111299
Caldwell HS, Lasek-Nesselquist E, Follano P, Kramer LD, Ciota AT. Divergent Mutational Landscapes of Consensus and Minority Genotypes of West Nile Virus Demonstrate Host and Gene-Specific Evolutionary Pressures. Genes. 2020; 11(11):1299. https://doi.org/10.3390/genes11111299
Chicago/Turabian StyleCaldwell, Haley S., Erica Lasek-Nesselquist, Paisley Follano, Laura D. Kramer, and Alexander T. Ciota. 2020. "Divergent Mutational Landscapes of Consensus and Minority Genotypes of West Nile Virus Demonstrate Host and Gene-Specific Evolutionary Pressures" Genes 11, no. 11: 1299. https://doi.org/10.3390/genes11111299