The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Genetic Analysis and Pathogenic Interpretation
2.3. Haplotype Analysis of the c.5597C>T Variant
3. Results
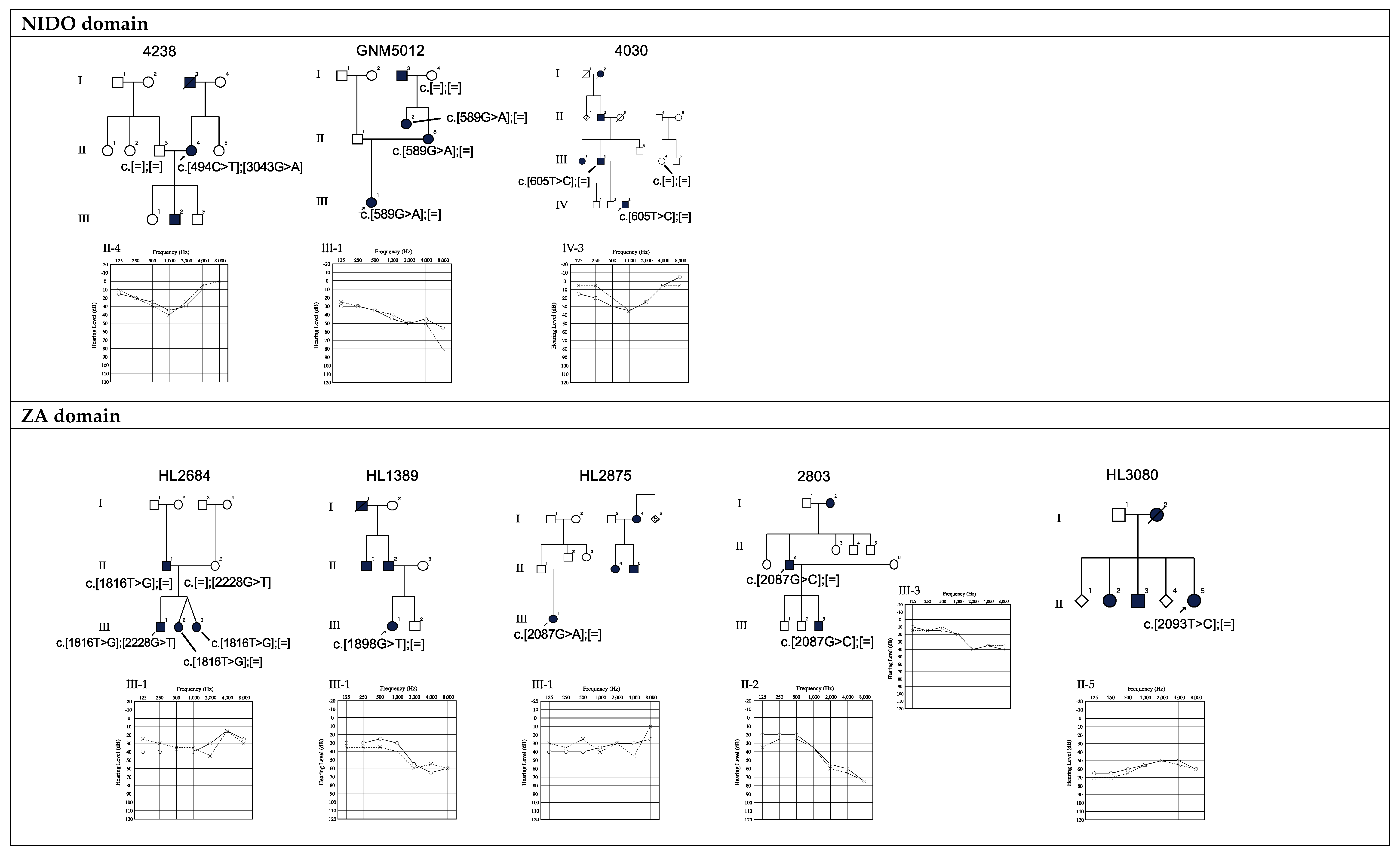
3.1. The Prevalence of TECTA Mutations in Japanese ADSNHL Patients
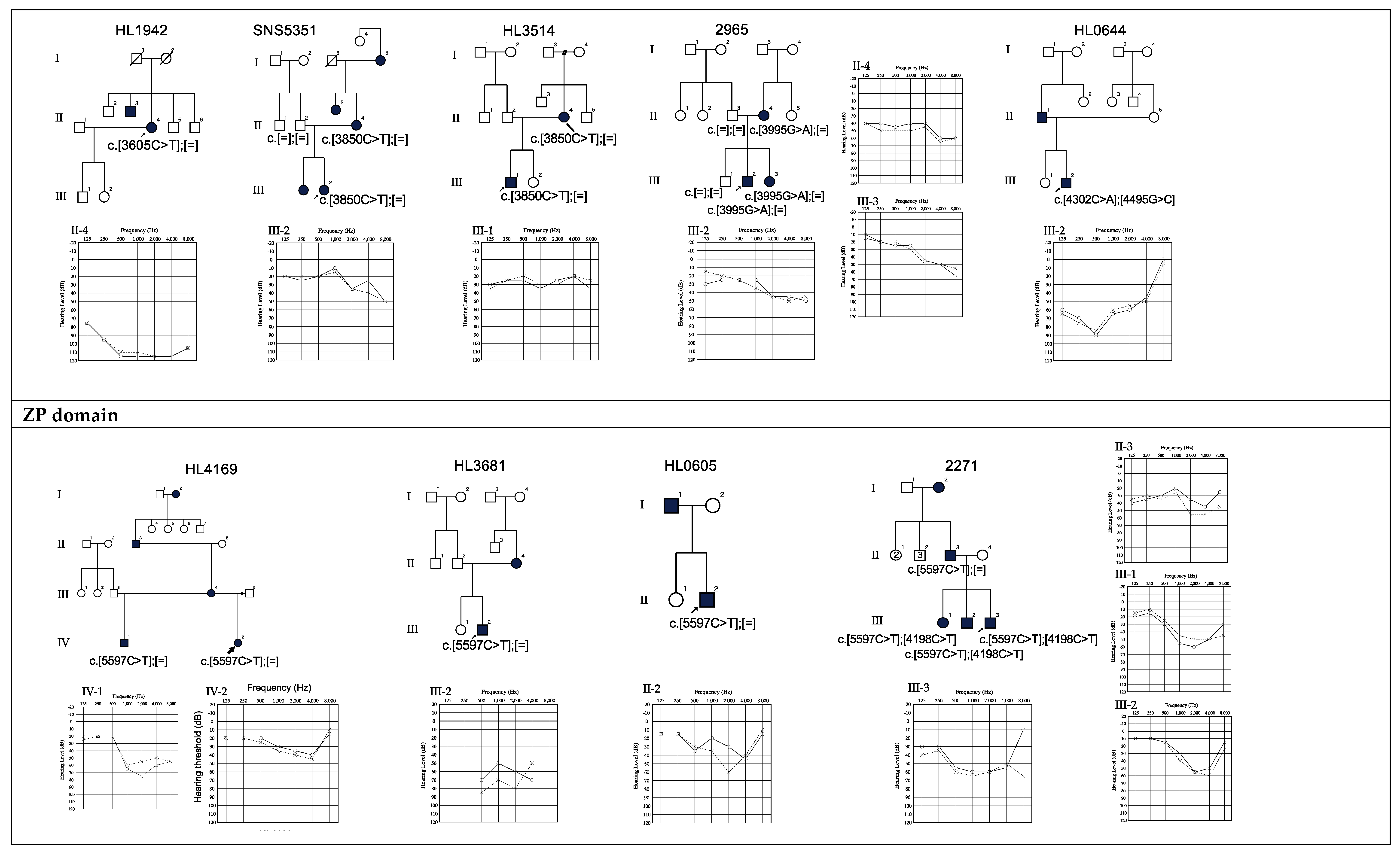
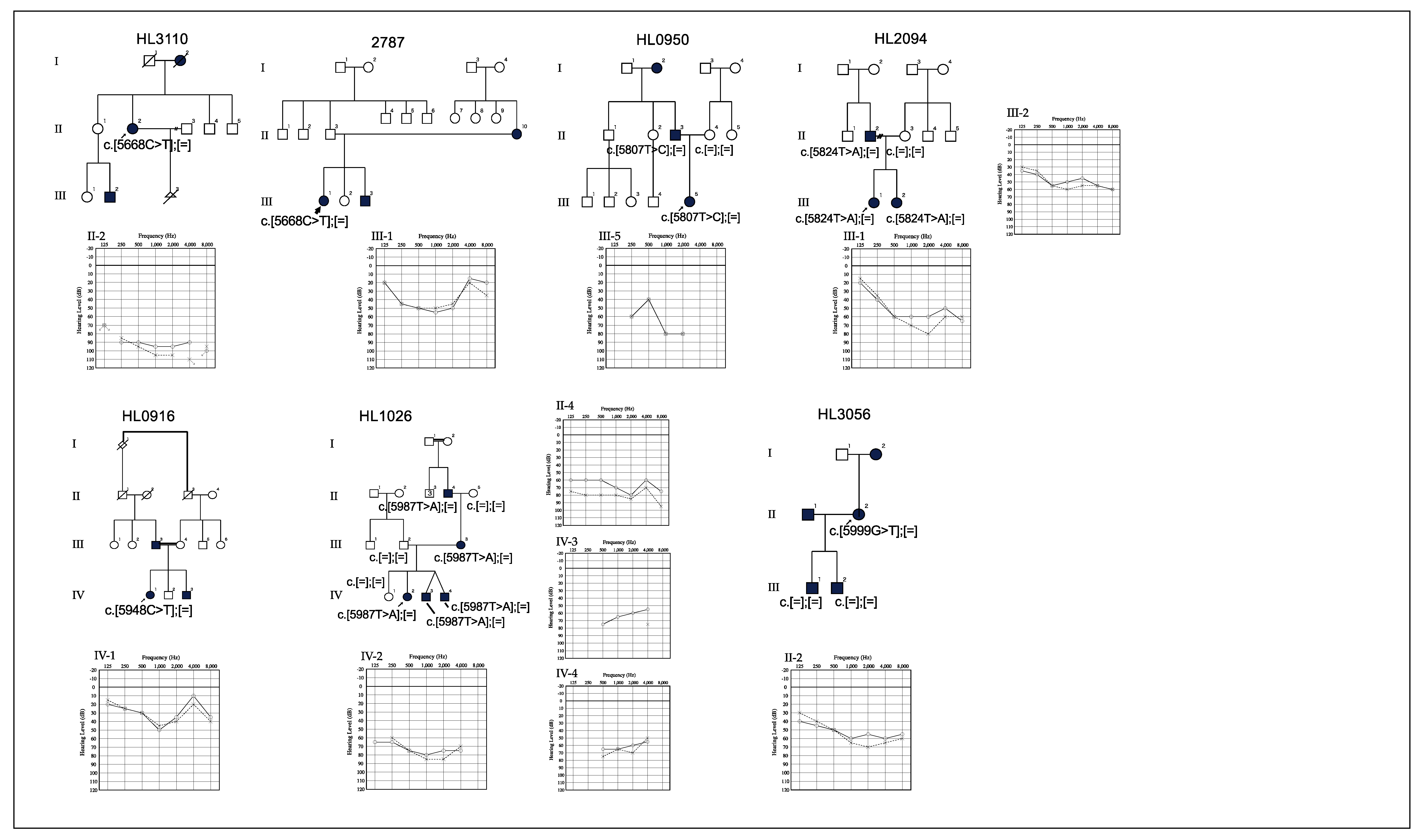
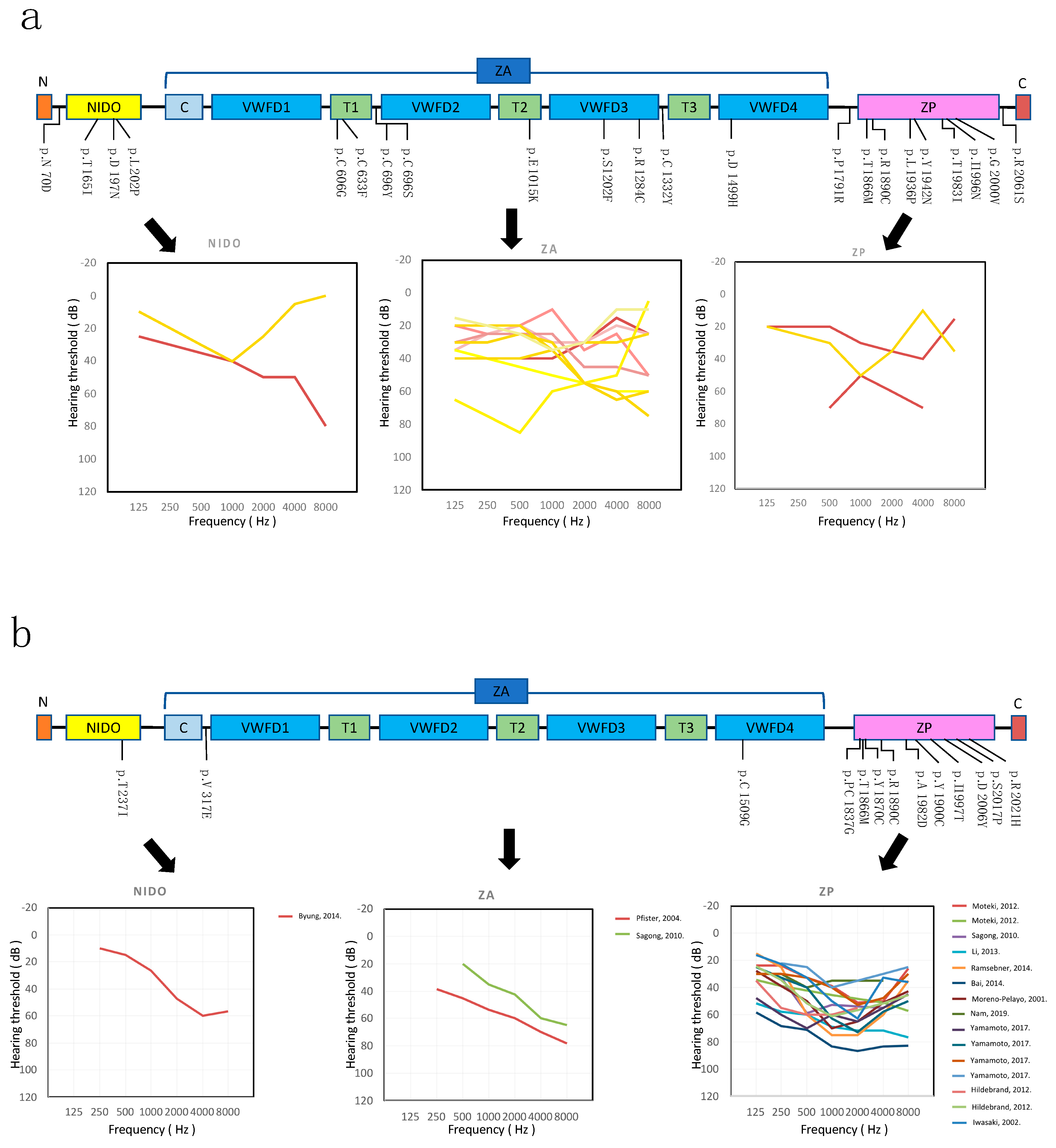
3.2. Genotype-Phenotype Correlation
3.3. Relationship between Age and Hearing Levels
3.4. The c.5597C>T (p.Thr1866Met) Variant in the ZP Domain
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Hereditary Hearing Loss Homepage. Available online: http://hereditaryhearingloss.org/ (accessed on 20 July 2019).
- Schrijver, I. Hereditary non-syndromic sensorineural hearing loss: Transforming silence to sound. J. Mol. Diagn. 2004, 6, 275–284. [Google Scholar] [CrossRef]
- Verhoeven, K.; Van Laer, L.; Kirschhofer, K.; Legan, P.K.; Hughes, D.C.; Schatteman, I.; Verstreken, M.; Van Hauwe, P.; Coucke, P.; Chen, A.; et al. Mutations in the human α-tectorin gene cause autosomal dominant non- syndromic hearing impairment. Nat. Genet. 1998, 19, 60–62. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, R.F.; De Brouwer, A.P.M.; Huygen, P.L.M.; Kunst, H.P.M.; Kremer, H.; Cremers, C.W.R.J. A novel TECTA mutation in a Dutch DFNA8/12 family confirms genotype-phenotype correlation. J. Assoc. Res. Otolaryngol. 2006, 7, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Legan, P.K.; Lukashkina, V.A.; Goodyear, R.J.; Kössl, M.; Russell, I.J.; Richardson, G.P. A targeted deletion in α-tectorin reveals that the tectorial membrane is required for the gain and timing of cochlear feedback. Neuron 2000, 28, 273–285. [Google Scholar] [CrossRef]
- Plantinga, R.F.; Cremers, C.W.R.J.; Huygen, P.L.M.; Kunst, H.P.M.; Bosman, A.J. Audiological evaluation of affected members from a Dutch DFNA8/12 (TECTA) family. J. Assoc. Res. Otolaryngol. 2007, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Moteki, H.; Usami, S. Simple and efficient germline copy number variant visualization method for the Ion AmpliSeqTM custom panel. Mol. Genet. Genom. Med. 2018, 6, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Auton, A.; Brooks, L.D.; DePristo, M.A.; Durbin, R.M.; Handsaker, R.E.; Kang, H.M.; Marth, G.T.; McVean, G.A. An integrated map of genetic variation from 1,092 human genomes. Nature 2012, 491, 56–65. [Google Scholar] [Green Version]
- NHLBI Exome Sequencing Project (ESP) Exome Variant Server. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 15 May 2018).
- Nakahara, M.; Higasa, K.; Nakamura, S.; Tabata, Y.; Kawaguchi, T.; Ishii, M.; Matsubara, K.; Matsuda, F.; Yamada, R. Large-scale East-Asian eQTL mapping reveals novel candidate genes for LD mapping and the genomic landscape of transcriptional effects of sequence variants. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Rödelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SNPinfo Web Server. Available online: https://snpinfo.niehs.nih.gov/ (accessed on 15 May 2018).
- Hildebrand, M.S.; Morín, M.; Meyer, N.C.; Mayo, F.; Modamio-Hoybjor, S.; Mencía, A.; Olavarrieta, L.; Morales-Angulo, C.; Nishimura, C.J.; Workman, H.; et al. DFNA8/12 caused by TECTA mutations is the most identified subtype of nonsyndromic autosomal dominant hearing loss. Hum. Mutat. 2011, 32, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Sagong, B.; Park, R.; Kim, Y.H.; Lee, K.Y.; Baek, J.I.; Cho, H.J.; Cho, I.J.; Kim, U.K.; Lee, S.H. Two novel missense mutations in the TECTA gene in Korean families with autosomal dominant nonsyndromic hearing loss. Ann. Clin. Lab. Sci. 2010, 40, 380–385. [Google Scholar]
- Moteki, H.; Nishio, S.; Hashimoto, S.; Takumi, Y.; Iwasaki, S.; Takeichi, N.; Fukuda, S.; Usami, S. TECTA mutations in Japanese with mid-frequency hearing loss affected by zona pellucida domain protein secretion. J. Hum. Genet. 2012, 57, 587–592. [Google Scholar] [CrossRef] [PubMed]
- ExAC Browser (Beta)-Exome Aggregation Consortium. Available online: http://exac.broadinstitute.org/ (accessed on 1 July 2019).
- 5KJPN-Integrative Japanese Genome Variation Database. Available online: https://jmorp.megabank.tohoku.ac.jp/201905/ (accessed on 1 July 2019).
- Mustapha, M. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum. Mol. Genet. 1999, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.R.; Chang, M.Y.; Koo, J.W.; Oh, S.H.; Choi, B.Y. Novel TECTA mutations identified in stable sensorineural hearing loss and their clinical implications. Audiol. Neurotol. 2015, 20, 17–25. [Google Scholar] [CrossRef] [PubMed]
- UniProt. Available online: https://www.uniprot.org/uniprot/O75443 (accessed on 25 July 2019).
- Sloan-Heggen, C.M.; Bierer, A.O.; Shearer, A.E.; Kolbe, D.L.; Nishimura, C.J.; Frees, K.L.; Ephraim, S.S.; Shibata, S.B.; Booth, K.T.; Campbell, C.A.; et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 2016, 135, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, T.; Nishio, S.; Iwasa, Y.; Yano, T.; Kumakawa, K.; Abe, S.; Ishikawa, K.; Kojima, H.; Namba, A.; Oshikawa, C.; et al. Comprehensive Genetic Screening of KCNQ4 in a Large Autosomal Dominant Nonsyndromic Hearing Loss Cohort: Genotype-Phenotype Correlations and a Founder Mutation. PLoS ONE 2013, 8, e63231. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.; Chung, J.; Kim, A.R.; Mun, S.J.; Kang, S.I.L.; Lee, S.H.; Kim, N.; Oh, S.H. Whole-exome sequencing identifies a novel genotype-phenotype correlation in the entactin domain of the known deafness gene TECTA. PLoS ONE 2014, 9, e97040. [Google Scholar] [CrossRef] [PubMed]
- Kitano, T.; Miyagawa, M.; Nishio, S.; Moteki, H.; Oda, K.; Ohyama, K.; Miyazaki, H.; Hidaka, H.; Nakamura, K.; Murata, T.; et al. POU4F3 mutation screening in Japanese hearing loss patients: Massively parallel DNA sequencing-34based analysis identified novel variants associated with autosomal dominant hearing loss. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Liu, F.; Ma, D. Research progress in pathogenic genes of hereditary non-syndromic mid-frequency deafness. Front. Med. 2016, 10, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Mutai, H.; Namba, K.; Morita, N.; Masuda, S.; Nishi, Y.; Nakano, A.; Masuda, S.; Fujioka, M.; Kaga, K.; et al. Prevalence of TECTA mutation in patients with mid-frequency sensorineural hearing loss. Orphanet J. Rare Dis. 2017, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Behlouli, A.; Bonnet, C.; Abdi, S.; Hasbellaoui, M.; Boudjenah, F.; Hardelin, J.P.; Louha, M.; Makrelouf, M.; Ammar-Khodja, F.; Zenati, A.; et al. A novel biallelic splice site mutation of TECTA causes moderate to severe hearing impairment in an Algerian family. Int. J. Pediatr. Otorhinolaryngol. 2016, 87, 28–33. [Google Scholar] [CrossRef]
- Su, Y.; Tang, W.X.; Gao, X.; Yu, F.; Dai, Z.Y.; Zhao, J.D.; Lu, Y.; Ji, F.; Huang, S.S.; Yuan, Y.Y.; et al. A novel mutation in the TECTA gene in a Chinese family with autosomal dominant nonsyndromic hearing loss. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sample ID | Gender | Onset Age | Age at Genetic Testing | Fluctuation | Progression | Base Change | AA Change | Domain | Type of HL | Severity | CADD Phred | ACMG Criteria | ACMG Category | MAF in ExAC | MAF in ToMMo | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
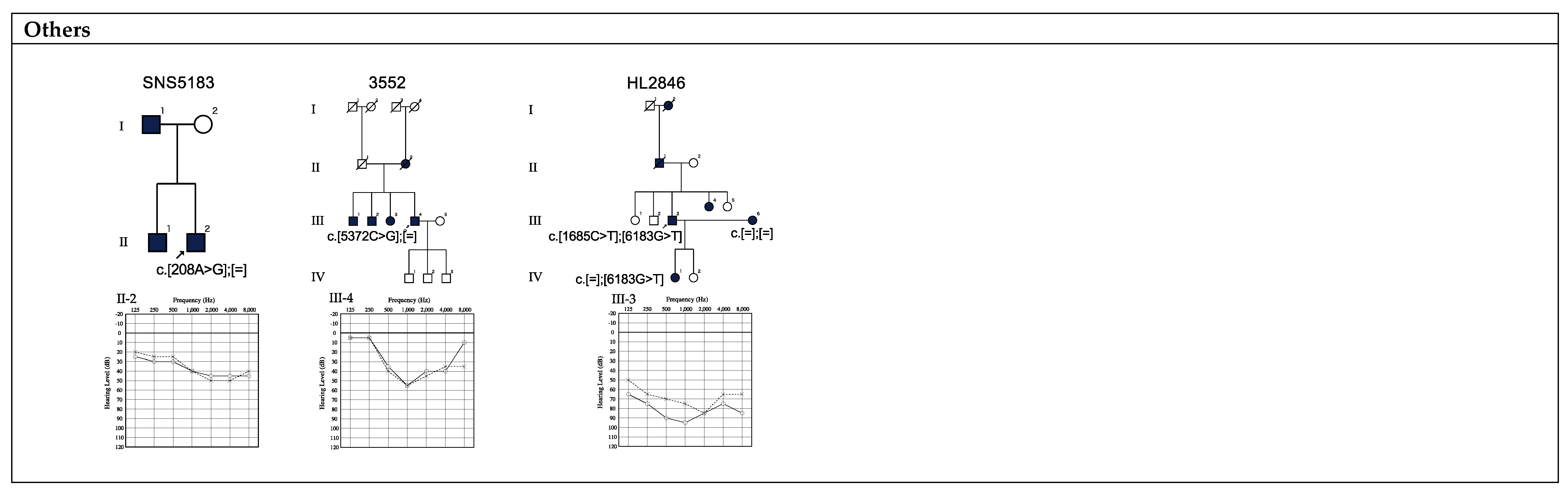
| 1 | SNS5183 | M | 16 | 24 | Yes | No | c.208A>G | p.N70D | Flat | Mild | 24.0 | VUS | PM2, PP3 | - | - | This study | |
| 2 | 4238 | F | Unknown | 49 | No | No | c.494C>T | p.T165I | NIDO | Mid | Mild | 32.0 | VUS | PM1, PM2, PP3 | 0.0000331 | 0.0004 | This study |
| 3 | GNM5012 | F | 5 | 7 | Unknown | Yes | c.589G>A | p.D197N | NIDO | High | Moderate | 27.8 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | Hildebrand, 2011 [22]. |
| 4 | 4030 | M | 0 | 10 | No | No | c.605T>C | p.L202P | NIDO | Mid | Mild | 27.0 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 5 | HL2684 | F | 6 | 10 | No | Yes | c.1816T>G | p.C606G | ZA (TIL1) | Mid | Mild | 22.3 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 6 | HL1389 | F | 6 | 34 | No | No | c.1898G>T | p.C633F | ZA (TIL1) | High | Moderate | 27.6 | VUS | PM1, PM2, PP3 | - | - | This study |
| 7 | HL2875 | F | 7 | 23 | Yes | No | c.2087G>A | p.C696Y | ZA | Flat | Mild | 26.7 | VUS | PM1, PM2, PP3 | - | - | This study |
| 8 | 2803 | M | 25 | 46 | No | No | c.2087G>C | p.C696S | ZA | High | Moderate | 25.6 | VUS | PM1,PM2,PP3 | - | - | This study |
| 9 | 4238 | F | Unknown | 49 | No | No | c.3043G>A | p.E1015K | ZA (TIL2) | Mid | Mild | 26.4 | VUS | PM1,PM2,PP3 | 0.0000165 | 0.0004 | This study |
| 10 | HL1942 | F | 59 | 77 | No | Yes | c.3605C>T | p.S1202F | ZA (VWD3) | Flat | Profound | 24.1 | VUS | PM1, PM2, PP3, BS2 | 0.0000741 | 0.0001 | This study |
| 11 | SNS5351 | F | 12 | 16 | No | No | c.3850C>T | p.R1284C | ZA (VWD3) | High | Mild | 26.1 | Likely pathogenic | PM1, PM2, PM7, PP3 | - | - | This study |
| 12 | HL3514 | M | 6 | 8 | No | No | c.3850C>T | p.R1284C | ZA (VWD3) | Flat | Mild | 26.1 | Likely pathogenic | PM1, PM2, PM7, PP3 | - | - | This study |
| 13 | 2965 | M | 6 | 10 | No | No | c.3995G>A | p.C1332Y | ZA | High | Mild | 24.4 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 14 | HL0644 | M | 0 | 16 | No | Yes | c.4495G>C | p.D1499H | ZA (VWD4) | Mid | Moderate | 29.7 | VUS | PM1, PM2, PP3, BS2 | - | 0.0006 | This study |
| 15 | 3552 | M | 62 | 64 | Yes | Yes | c.5372C>G | p.P1791R | Mid | Moderate | 22.5 | VUS | PM2, PP5, BP4 | 0.0002 | - | Hildebrand, 2011 [22]. | |
| 16 | HL4169 | F | 6 | 13 | No | No | c.5597C>T | p.T1866M | ZP | Flat | Mild | 35.0 | Likely pathogenic | PM1, PM2, PM7, PP3, PP5 | 0.00000824 | - | Sagong, 2010 [23]. |
| 17 | HL3681 | M | 0 | 0 | No | No | c.5597C>T | p.T1866M | ZP | Mid | Moderate | 35.0 | Likely pathogenic | PM1, PM2, PM7, PP3, PP5 | 0.00000824 | - | Sagong, 2010 [23]. |
| 18 | HL0605 | M | 0 | 10 | No | No | c.5597C>T | p.T1866M | ZP | Mid | Mild | 35.0 | Likely pathogenic | PM1, PM2, PM7, PP3, PP5 | 0.00000824 | - | Sagong, 2010 [23]. |
| 19 | 2271 | M | Unknown | 6 | Unknown | Unknown | c.5597C>T | p.T1866M | ZP | Mid | Moderate | 35.0 | Likely pathogenic | PM1, PM2, PM7, PP3, PP5 | 0.00000824 | - | Sagong, 2010 [23]. |
| 20 | HL3110 | F | Unknown | 82 | No | Yes | c.5668C>T | p.R1890C | ZP | Flat | Severe | 34.0 | Likely pathogenic | PM1, PM2, PP3, PP5 | - | - | Plantinga, 2006 [5]. |
| 21 | 2787 | F | 15 | 27 | Unknown | Yes | c.5668C>T | p.R1890C | ZP | Mid | Mild | 34.0 | Likely pathogenic | PM1, PM2, PP3, PP5 | - | - | Plantinga, 2006 [5]. |
| 22 | HL0950 | F | 3 | 4 | No | No | c.5807T>C | p.L1936P | ZP | unspecified | Moderate | 25.4 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 23 | HL2094 | F | 3 | 8 | No | No | c.5824T>A | p.Y1942N | ZP | Mid | Moderate | 32.0 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 24 | HL0916 | F | 6 | 38 | No | No | c.5948C>T | p.T1983I | ZP | Mid | Mild | 33.0 | VUS | PM1, PM2, PP3 | - | - | This study |
| 25 | HL1026 | F | 0 | 4 | No | No | c.5987T>A | p.I1996N | ZP | Mid | Severe | 34.0 | Likely pathogenic | PM1, PM2, PP1, PP3 | - | - | This study |
| 26 | HL3056 | F | 4 | 32 | Yes | Yes | c.5999G>T | p.G2000V | ZP | Flat | Moderate | 26.2 | VUS | PM1, PM2, PP3 | - | - | This study |
| 27 | HL2846 | M | 2 | 71 | No | No | c.6183G>T | p.R2061S | Mid | Severe | 24.4 | VUS | PM2, PP1, PP3 | - | - | This study |
| No. | Sample ID | Gender | Onset Age | Age at Genetic Testing | Fluctuation | Progression | Base Change | AA Change | Domain | Type of HL | Severity | CADD Phred | ACMG Criteria | ACMG Category | MAF in ExAC | MAF in ToMMo | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | HL0150 | M | Unknown | 60 | No | Yes | c.842T>C | p.V281A | ZA (VWC) | High | Moderate | 10.5 | VUS | PM1, PM2, BP4 | - | - | This study |
| 2 | SNS5496 | F | 50 | 57 | No | Yes | c.1049G>A | p.R350Q | ZA (VWD1) | Flat | Mild | 25.9 | VUS * | PM1, PM2, PP3 | 0.00000824 | - | This study |
| 3 | HL0133 | F | 0 | 2 | No | No | c.1049G>A | p.R350Q | ZA (VWD1) | Flat | Moderate | 25.9 | VUS * | PM1, PM2, PP3, BP4 | 0.00000824 | - | This study |
| 4 | HL0770 | F | 0 | 33 | No | Yes | c.1424C>T | p.P475L | ZA (VWD1) | Mid | Severe | 23.4 | VUS * | PM1, PM2, PP3, BS4, BP5 | 0.00000824 | - | This study |
| 5 | HL2846 | M | 2 | 71 | No | No | c.1685C>T | p.T562M | ZA | Mid | Severe | 25.6 | VUS * | PM1, PP3, PP5 | 0.000099 | 0.0001 | Hildebrand, 2011 [22]. |
| 6 | HL3080 | F | 60 | 73 | No | Yes | c.2093T>C | p.V698A | ZA | Flat | Moderate | 14.1 | VUS | PM1, PM2, BP4 | - | 0.0001 | This study |
| 7 | HL2684 | F | 6 | 10 | No | Yes | c.2228G>T | p.C743F | ZA (VWD2) | Mid | Mild | 24.4 | VUS * | PM1, PM2, PP3, BS2, BS4 | - | 0.0003 | This study |
| 8 | HL1091 | F | 5 | 42 | No | Yes | c.2228G>T | p.C743F | ZA (VWD2) | High | Profound | 24.4 | VUS * | PM1, PM2, PP3, BS2, BS4 | - | 0.0003 | This study |
| 9 | HL4176 | F | 0 | 45 | No | Yes | c.2228G>T | p.C743F | ZA (VWD2) | Flat | Profound | 24.4 | VUS * | PM1, PM2, PP3, BS2, BS4 | - | 0.0003 | This study |
| 10 | HL1937 | F | 45 | 77 | No | Yes | c.3556C>T | p.R1186W | ZA (VWD3) | High | Severe | 33.0 | VUS * | PM1, PP3, BS1, BS4 | 0.0009 | 0.0001 | This study |
| 11 | 2271 | M | Unknown | 6 | Unknown | Unknown | c.4198C>T | p.H1400Y | ZA (TIL3) | Mid | Moderate | 25.0 | VUS * | PM1, PP3, BS2 | 0.0002 | 0.0016 | Moteki,2012 [24]. |
| 12 | HL0644 | M | 0 | 16 | No | Yes | c.4302C>A | p.Y1434X | ZA | Mid | Moderate | 36.0 | VUS * | PM2 | - | - | This study |
| 13 | HL0280 | M | 0 | 9 | No | No | c.5908G>A | p.A1970T | ZP | Flat | Mild | 13.5 | Likely Benign | BS2, BP4 | 0.0000906 | 0.001 | This study |
| Distance from the c.5597C>T Mutation (bp) | Marker | Fm1 | Fm2 | Fm3 | Fm4 | Allele Frequency (HapMap-JPT) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HL3681 | HL0605 | 2271 | * 6 | * 7 | * 5 | HL4169 | * 4 | ||||||||||||||
| A | U | A | U | A | U | A | U | A | U | A | U | ||||||||||
| 429809 | rs752979 | T | C | T | C | T | T | T | T | T | T | C | C | T | C | T | C | C | 0.46 | T | 0.54 |
| 315340 | rs4430518 | T | G | T | T | G | G | T | T | T | T | T | G | T | T | T | T | G | 0.36 | T | 0.64 |
| 236767 | rs4936565 | G | G | A | G | A | G | A | A | A | G | A | G | G | G | G | G | A | 0.34 | G | 0.66 |
| 0 | c.5597C>T | - | - | - | - | - | - | - | - | ||||||||||||
| 589752 | rs7941422 | G | G | G | G | G | A | G | G | G | G | G | G | G | G | G | G | A | 0.17 | G | 0.83 |
| 649183 | rs621360 | T | C | T | T | T | C | T | T | T | T | T | T | C | T | C | C | C | 0.24 | T | 0.76 |
| 822346 | rs2852833 | T | T | G | T | G | T | G | G | G | G | G | G | G | G | G | G | G | 0.63 | T | 0.37 |
| 1008518 | rs515449 | G | G | A | A | A | G | A | G | A | A | A | A | A | A | A | A | A | 0.65 | G | 0.35 |
| 1089754 | rs489877 | A | A | A | C | A | A | A | A | A | A | A | A | A | C | A | A | A | 0.72 | C | 0.28 |
| 1145749 | rs528219 | C | C | C | T | C | C | C | C | C | C | C | C | C | T | C | C | C | 0.69 | T | 0.31 |
| 1292838 | rs3906964 | A | A | G | G | G | A | G | A | G | A | G | G | G | G | G | A | A | 0.26 | G | 0.74 |
| 1495732 | rs7117842 | T | T | T | T | T | C | T | C | T | C | T | C | T | C | T | C | C | 0.37 | T | 0.63 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasukawa, R.; Moteki, H.; Nishio, S.-y.; Ishikawa, K.; Abe, S.; Honkura, Y.; Hyogo, M.; Mihashi, R.; Ikezono, T.; Shintani, T.; et al. The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss. Genes 2019, 10, 744. https://doi.org/10.3390/genes10100744
Yasukawa R, Moteki H, Nishio S-y, Ishikawa K, Abe S, Honkura Y, Hyogo M, Mihashi R, Ikezono T, Shintani T, et al. The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss. Genes. 2019; 10(10):744. https://doi.org/10.3390/genes10100744
Chicago/Turabian StyleYasukawa, Rika, Hideaki Moteki, Shin-ya Nishio, Kotaro Ishikawa, Satoko Abe, Yohei Honkura, Misako Hyogo, Ryota Mihashi, Tetsuo Ikezono, Tomoko Shintani, and et al. 2019. "The Prevalence and Clinical Characteristics of TECTA-Associated Autosomal Dominant Hearing Loss" Genes 10, no. 10: 744. https://doi.org/10.3390/genes10100744