Improving Species Identification of Ancient Mammals Based on Next-Generation Sequencing Data

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Data Resource
2.2. Sample Preparation and DNA Extraction
2.3. Library Preparation and Sequencing
2.4. Processing of Sequencing Reads
2.5. Burrows-Wheeler Aligner Mapping and DNA Damage Analysis
2.6. Exploring the Species Identification Pipeline
3. Results
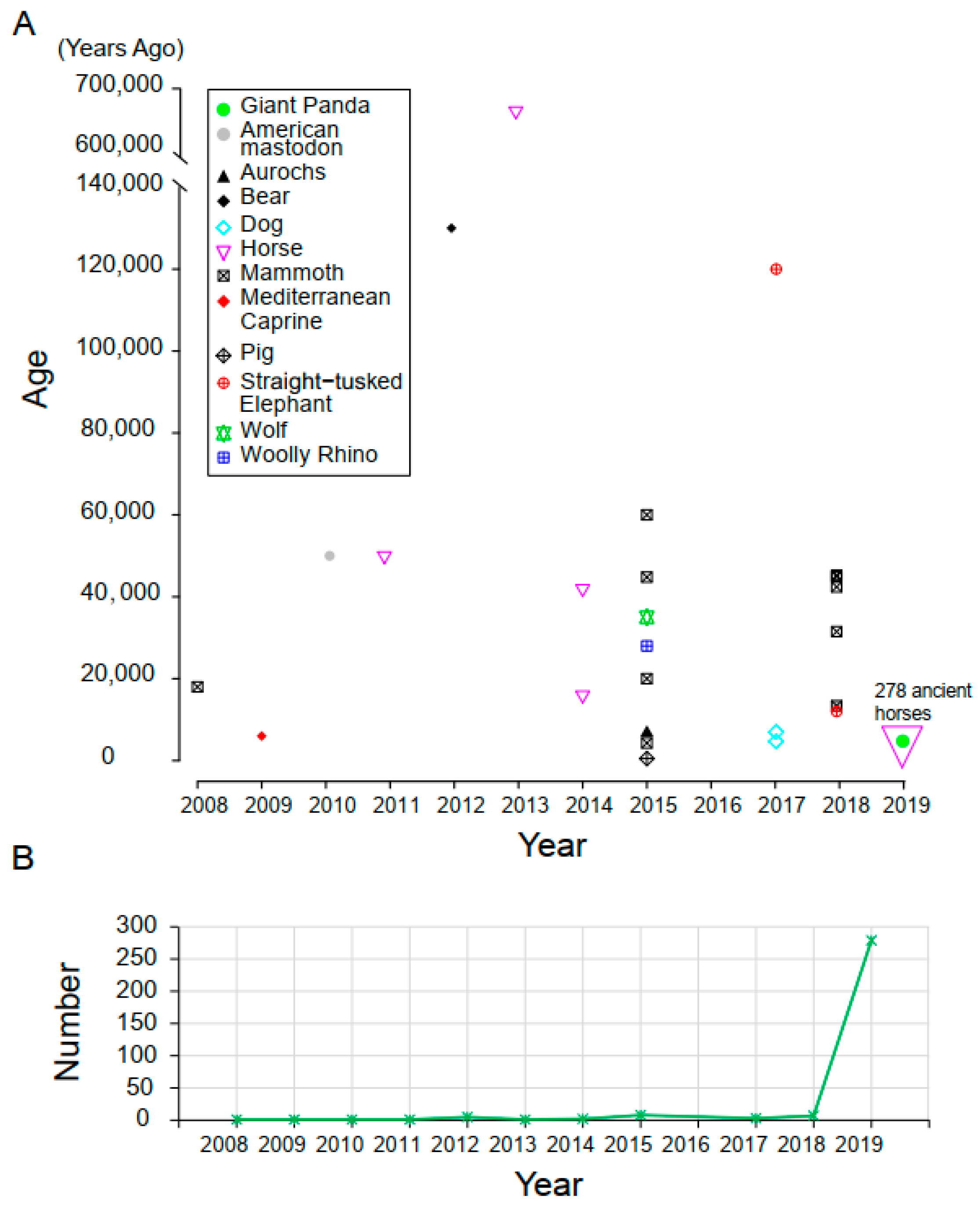
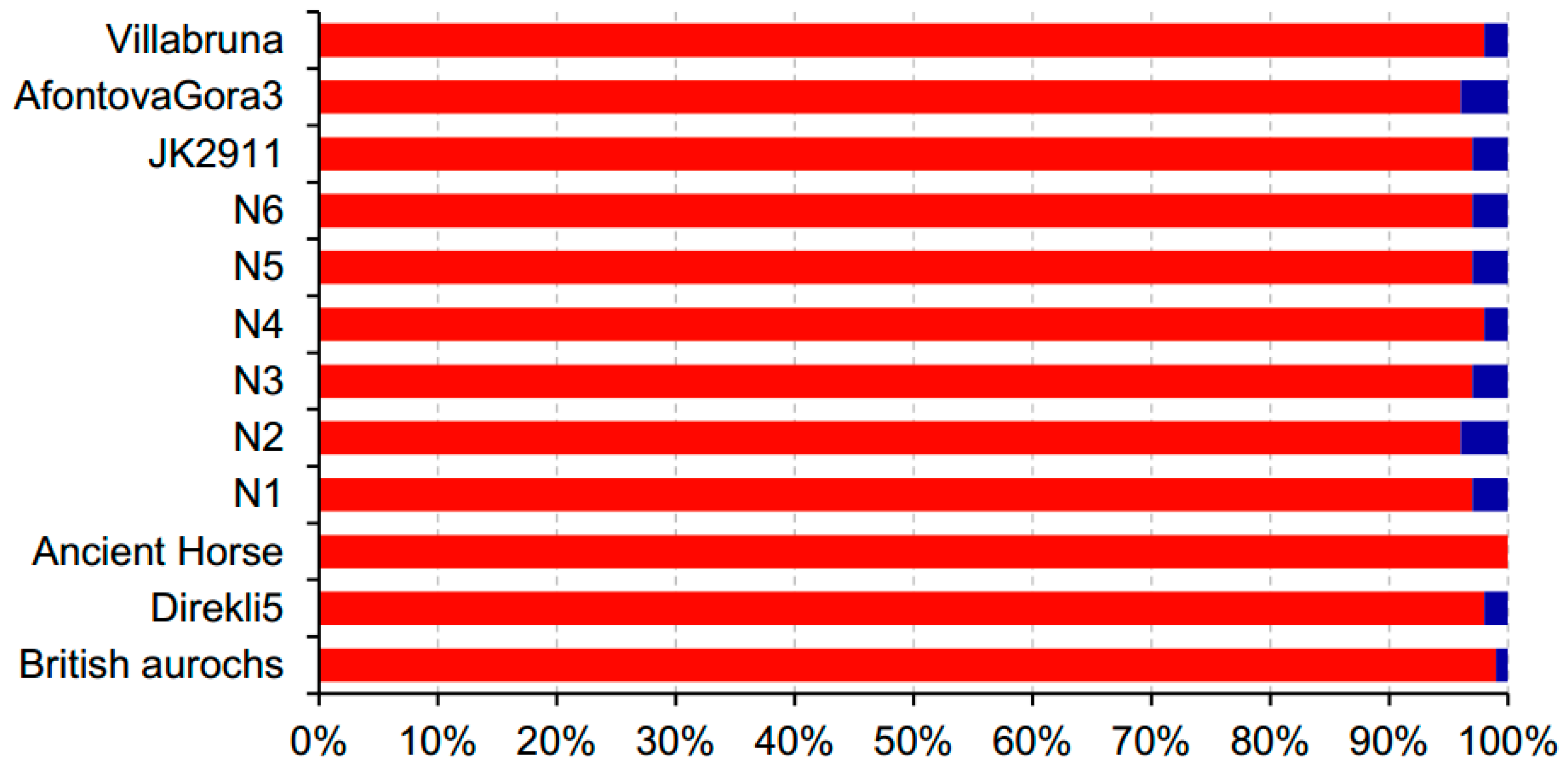
3.1. Samples and Data Description
3.2. Basic Local Alignment Search Tool Search Using Nucleotide (nt) and Mitochondrial DNA (mtDNA) Databases
3.3. Screening the Mapping Results by Sequence Similarity
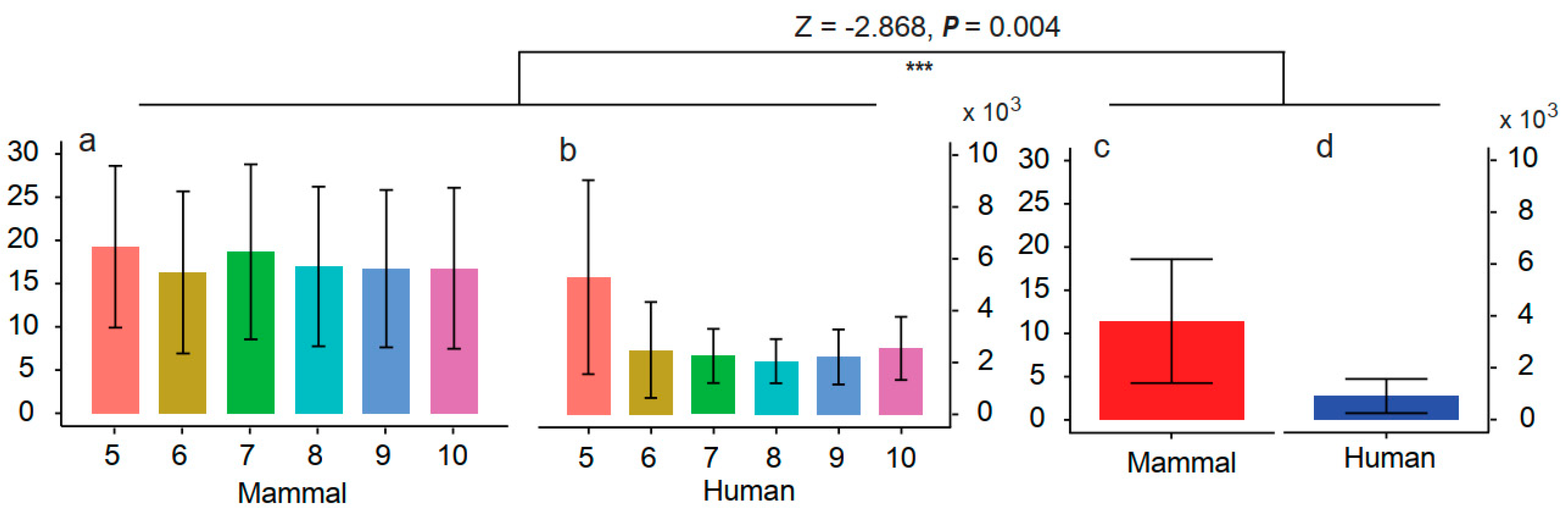
3.4. Screening the Mapping Results by Query Coverages
3.5. Mapping Using the BWA Functions “aln” and “mem”.
3.6. Testing the Recommended Method and Parameters for Ancient Mammal Species Identification
3.7. Screening the Mapping Results Using Deamination Characteristics
4. Discussion
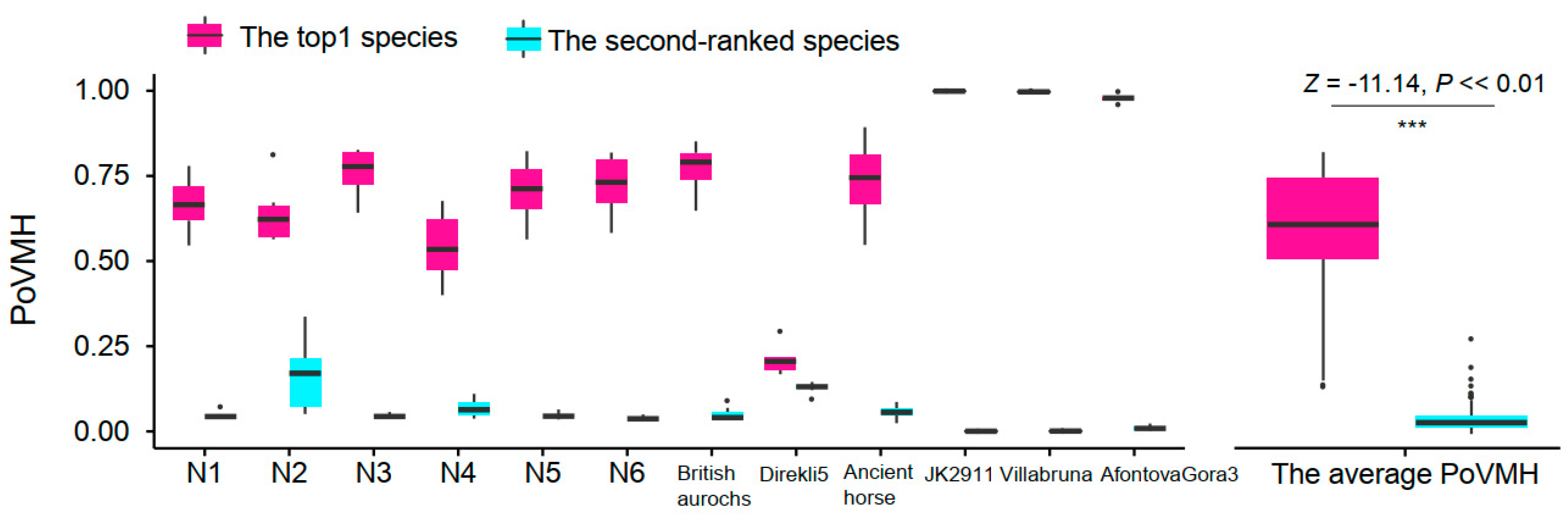
4.1. Comparing the nt and mtDNA Databases to Improve the Identification Accuracy
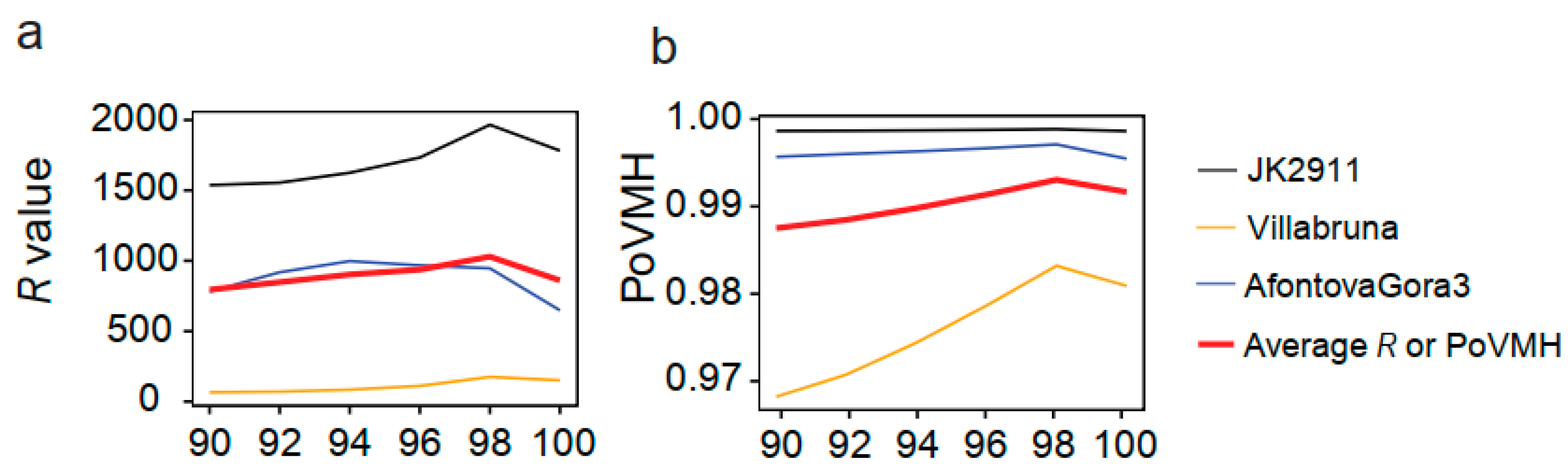
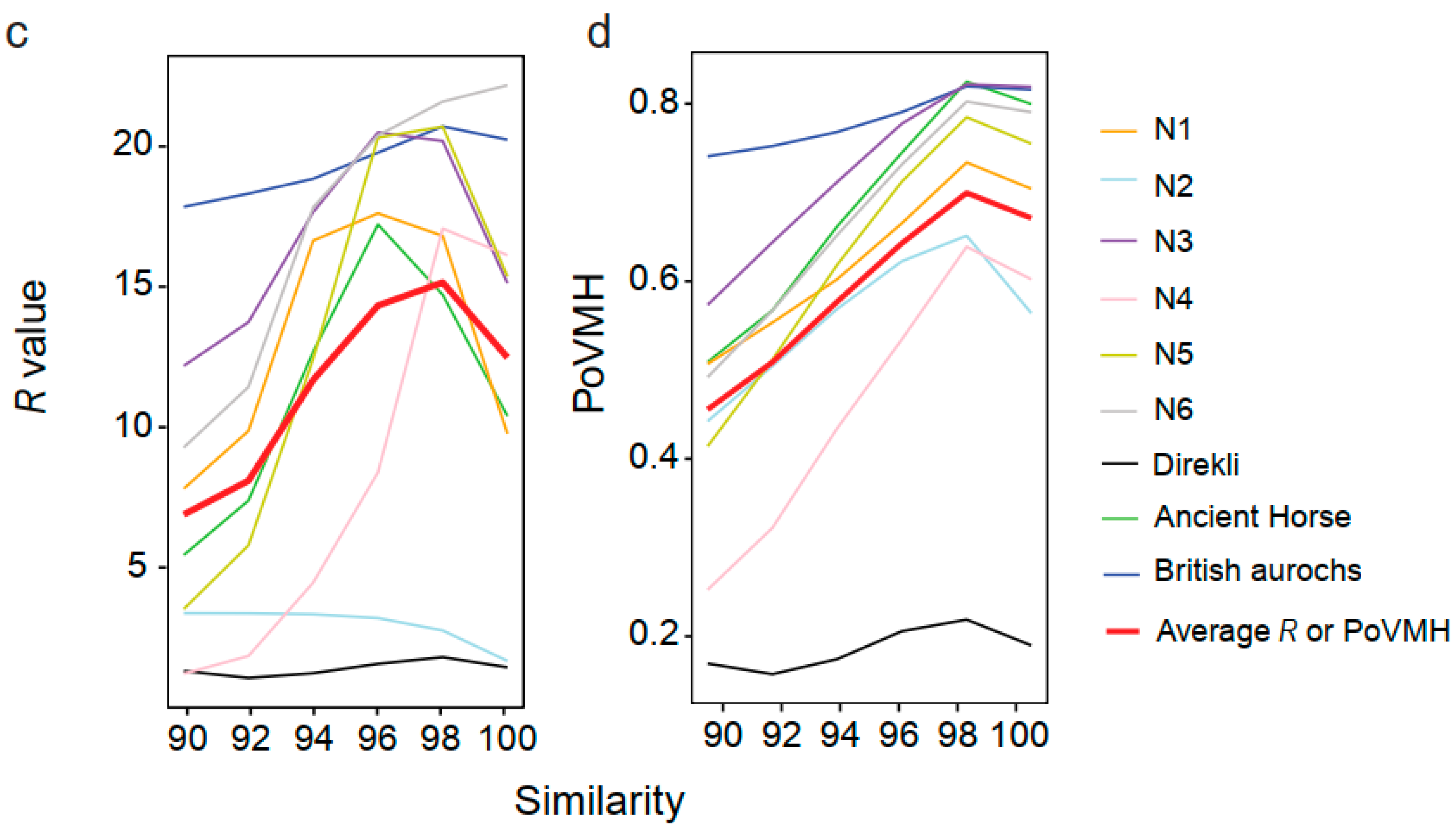
4.2. Evaluating the Influence of Sequencing Similarity and Coverage on Identification Accuracy
4.3. Comparison of Different Mapping Algorithms to Improve Identification Accuracy
4.4. Improving Identification Accuracy by Using Deamination Characteristics
4.5. Influence of the Sample Age, Fragment Length, Proportion of Endogenous DNA, and Sequencing Platform on the Identification Accuracy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Higuchi, R.; Bowman, B.; Freiberger, M.; Ryder, O.A.; Wilson, A.C. DNA Sequences from the Quagga, an extinct member of the horse family. Nature 1984, 312, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Hagelberg, E.; Hofreiter, M.; Keyser, C. Introduction. Ancient DNA: The first three decades. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20130371. [Google Scholar] [CrossRef] [PubMed]
- Palkopoulou, E.; Lipson, M.; Mallick, S.; Nielsen, S.; Rohland, N.; Baleka, S.; Karpinski, E.; Ivancevic, A.M.; To, T.H.; Kortschak, R.D.; et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl. Acad. Sci. USA 2018, 115, E2566–E2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early middle pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Allentoft, M.E.; Sikora, M.; Sjogren, K.G.; Rasmussen, S.; Rasmussen, M.; Stenderup, J.; Damgaard, P.B.; Schroeder, H.; Ahlstrom, T.; Vinner, L.; et al. Population genomics of bronze age Eurasia. Nature 2015, 522, 167–172. [Google Scholar] [CrossRef]
- Damgaard, P.B.; Marchi, N.; Rasmussen, S.; Peyrot, M.; Renaud, G.; Korneliussen, T.; Moreno-Mayar, J.V.; Pedersen, M.W.; Goldberg, A.; Usmanova, E.; et al. 137 ancient human genomes from across the Eurasian steppes. Nature 2018, 577, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Olalde, I.; Brace, S.; Allentoft, M.E.; Armit, I.; Kristiansen, K.; Booth, T.; Rohland, N.; Mallick, S.; Szecsenyi-Nagy, A.; Mittnik, A.; et al. The beaker phenomenon and the genomic transformation of Northwest Europe. Nature 2018, 555, 190–196. [Google Scholar] [CrossRef]
- Mathieson, I.; Alpaslan-Roodenberg, S.; Posth, C.; Szecsenyi-Nagy, A.; Rohland, N.; Mallick, S.; Olalde, I.; Broomandkhoshbacht, N.; Candilio, F.; Cheronet, O.; et al. The genomic history of Southeastern Europe. Nature 2018, 555, 197–203. [Google Scholar] [CrossRef]
- McCutcheon, J.P.; Moran, N.A. Parallel genomic evolution and metabolic interdependence in an ancient symbiosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19392–19397. [Google Scholar] [CrossRef] [Green Version]
- Hofman, C.A.; Rick, T.C.; Fleischer, R.C.; Maldonado, J.E. Conservation archaeogenomics: ancient DNA and biodiversity in the Anthropocene. Trends Ecol. Evol. 2015, 30, 540–549. [Google Scholar] [CrossRef]
- Muhlemann, B.; Jones, T.C.; Damgaard, P.B.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient hepatitis B viruses from the bronze age to the medieval period. Nature 2018, 577, 418–423. [Google Scholar] [CrossRef] [PubMed]
- MacHugh, D.E.; Larson, G.; Orlando, L. Taming the past: ancient DNA and the study of animal domestication. Annu. Rev. Anim. Biosci. 2017, 5, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Linacre, A.; Tobe, S.S. An Overview to the investigative approach to species testing in wildlife forensic science. Investig. Genet. 2011, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Dalen, L.; Lagerholm, V.K.; Nylander, J.A.A.; Barton, N.; Bochenski, Z.M.; Tomek, T.; Rudling, D.; Ericson, P.G.P.; Irestedt, M.; Stewart, J.R. Identifying bird remains using ancient DNA barcoding. Genes (Basel) 2017, 8, 169. [Google Scholar] [CrossRef] [PubMed]
- de Flamingh, A.; Mallott, E.K.; Roca, A.L.; Boraas, A.S.; Malhi, R.S. Species identification and mitochondrial genomes of ancient fish bones from the Riverine Kachemak tradition of the Kenai peninsula, Alaska. Mitochondrial DNA Part B 2018, 3, 409–411. [Google Scholar] [CrossRef]
- Schröder, O.; Wagner, M.; Wutke, S.; Zhang, Y.; Ma, Y.; Xu, D.; Goslar, T.; Neef, R.; Tarasov, P.E.; Ludwig, A. Ancient DNA identification of domestic animals used for leather objects in central Asia during the bronze age. Holocene 2016, 26, 1722–1729. [Google Scholar] [CrossRef]
- Dove, C.J.; Rotzel, N.C.; Heacker, M.; Weigt, L.A. Using DNA barcodes to identify bird species involved in birdstrikes. J. Wildl. Manag. 2008, 72, 1231–1236. [Google Scholar] [CrossRef]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, S.; Krause, J.; Guschanski, K.; Savolainen, V.; Paabo, S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS ONE 2012, 7, e34131. [Google Scholar] [CrossRef]
- Foran, D.R.; Fischer, A.B.; Stoloff, M.E. A comparison of mitochondrial DNA amplification strategies for species identification. J. Forensic Investig. 2015, 3, 7. [Google Scholar]
- Yang, D.Y.; Cannon, A.; Saunders, S.R. DNA Species identification of archaeological Salmon bone from the Pacific Northwest coast of North America. J. Archaeol. Sci. 2004, 31, 619–631. [Google Scholar] [CrossRef]
- Han, L.; Zhu, S.; Ning, C.; Cai, D.; Wang, K.; Chen, Q.; Hu, S.; Yang, J.; Shao, J.; Zhu, H.; et al. Ancient DNA provides new insight into the maternal lineages and domestication of Chinese donkeys. BMC Evol. Biol. 2014, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Blow, M.J.; Zhang, T.; Woyke, T.; Speller, C.F.; Krivoshapkin, A.; Yang, D.Y.; Derevianko, A.; Rubin, E.M. Identification of ancient remains through genomic sequencing. Genome Res. 2008, 18, 1347–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoglund, P.; Northoff, B.H.; Shunkov, M.V.; Derevianko, A.P.; Paabo, S.; Krause, J.; Jakobsson, M. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc. Natl. Acad. Sci. USA 2014, 111, 2229–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuenemann, V.J.; Peltzer, A.; Welte, B.; van Pelt, W.P.; Molak, M.; Wang, C.C.; Furtwangler, A.; Urban, C.; Reiter, E.; Nieselt, K.; et al. Ancient Egyptian mummy genomes suggest an increase of Sub-Saharan African ancestry in post-roman periods. Nat. Commun. 2017, 8, 15694. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwangler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Daly, K.G.; Delser, P.M.; Mullin, V.E.; Scheu, A.; Mattiangeli, V.; Teasdale, M.D.; Hare, A.J.; Burger, J.; Verdugo, M.P.; Collins, M.J.; et al. Ancient goat genomes reveal mosaic domestication in the fertile crescent. Science 2018, 361, 85–88. [Google Scholar] [CrossRef]
- Park, S.D.; Magee, D.A.; McGettigan, P.A.; Teasdale, M.D.; Edwards, C.J.; Lohan, A.J.; Murphy, A.; Braud, M.; Donoghue, M.T.; Liu, Y.; et al. Genome sequencing of the extinct Eurasian wild aurochs, Bos Primigenius, illuminates the phylogeography and evolution of cattle. Genome Biol. 2015, 16, 234. [Google Scholar] [CrossRef]
- Ngatia, J.N.; Lan, T.M.; Dinh, T.D.; Zhang, L.; Ahmed, A.K.; Xu, Y.C. Signals of positive selection in mitochondrial protein-coding genes of Woolly mammoth: adaptation to extreme environments? Ecol. Evol. 2019, 9, 6821–6832. [Google Scholar] [CrossRef]
- Rohland, N.; Hofreiter, M. Ancient DNA extraction from bones and teeth. Nat. Protoc. 2007, 2, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Gansauge, M.T.; Gerber, T.; Glocke, I.; Korlevic, P.; Lippik, L.; Nagel, S.; Riehl, L.M.; Schmidt, A.; Meyer, M. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 2017, 45, e79. [Google Scholar] [PubMed]
- Schubert, M.; Lindgreen, S.; Orlando, L. Adapterremoval V2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Picard Toolkit. Available online: https://broadinstitute.github.io/picard/ (accessed on 29 July 2013).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 genome project data processing subgroup. The sequence alignment/map format and Samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.; Orlando, L. mapDamage2.0: Fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. The nucleotide database. Available online: ftp://ftp.ncbi.nlm.nih.gov/blast/db/ (accessed on 29 March 2019).
- National Center for Biotechnology Information. BLAST+. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=Download (accessed on 20 October 2015).
- Sheng, G.L.; Basler, N.; Ji, X.P.; Paijmans, J.L.A.; Alberti, F.; Preick, M.; Hartmann, S.; Westbury, M.V.; Yuan, J.X.; Jablonski, N.G.; et al. Paleogenome reveals genetic contribution of extinct giant panda to extant populations. Curr. Biol. 2019, 29, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, O.; Burgos-Paz, W.; Casas, E.; Ballester, M.; Bianco, E.; Olalde, I.; Santpere, G.; Novella, V.; Gut, M.; Lalueza-Fox, C.; et al. Genome data from a sixteenth century pig illuminate modern breed relationships. Heredity 2015, 114, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Fages, A.; Hanghoj, K.; Khan, N.; Gaunitz, C.; Seguin-Orlando, A.; Leonardi, M.; Constantz, C.M.; Gamba, C.; Al-Rasheid, K.A.S.; Albizuri, S.; et al. Tracking five millennia of horse management with extensive ancient genome time series. Cell 2019, 177, 1419–1435. [Google Scholar] [CrossRef]
- Ebihara, A.; Nitta, J.H.; Ito, M. Molecular species identification with rich floristic sampling: DNA barcoding the Pteridophyte flora of Japan. PLoS ONE 2010, 5, e15136. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Genome Information by Organism. Available online: https://www.ncbi.nlm.nih.gov/genome/browse/#!/overview/ (accessed on 30 April 2019).
- Mora, C.; Tittensor, D.P.; Adl, S.; Simpson, A.G.; Worm, B. How many species are there on Earth and in the Ocean? PLoS Biol. 2011, 9, e1001127. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [PubMed] [Green Version]
- Smith, D.R. The past, present and future of mitochondrial genomics: Have we sequenced enough mtDNAs? Brief. Funct. Genomics 2016, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Paabo, S. DNA Sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.D.; Ngatia, J.N.; Cui, L.Y.; Ma, Y.; Dhamer, T.D.; Xu, Y.C. Influence of pairwise genetic distance computation and reference sample size on the reliability of species identification using Cyt b and COI gene fragments in a group of native passerines. Forensic Sci. Int. Genet. 2019, 40, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Hosein, F.N.; Austin, N.; Maharaj, S.; Johnson, W.; Rostant, L.; Ramdass, A.C.; Rampersad, S.N. Utility of DNA barcoding to identify rare endemic vascular plant species in trinidad. Ecol. Evol. 2017, 7, 7311–7333. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.; Fu, Q.; Aximu-Petri, A.; Glocke, I.; Nickel, B.; Arsuaga, J.L.; Martinez, I.; Gracia, A.; de Castro, J.M.; Carbonell, E. A mitochondrial genome sequence of a hominin from Sima de los Huesos. Nature 2014, 505, 403–406. [Google Scholar] [CrossRef]
- Orlando, L.; Ginolhac, A.; Raghavan, M.; Vilstrup, J.; Rasmussen, M.; Magnussen, K.; Steinmann, K.E.; Kapranov, P.; Thompson, J.F.; Zazula, G.; et al. True single-molecule DNA sequencing of a pleistocene horse bone. Genome Res. 2011, 21, 1705–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.; Green, R.E.; Kelso, J.; Prufer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prufer, K.; de Filippo, C.; et al. A high-coverage genome sequence from an archaic Denisovan individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef]
- Mak, S.S.T.; Gopalakrishnan, S.; Caroe, C.; Geng, C.; Liu, S.; Sinding, M.S.; Kuderna, L.F.K.; Zhang, W.; Fu, S.; Vieira, F.G.; et al. Comparative performance of the BGISEQ-500 vs Illumina HiSeq2500 sequencing platforms for palaeogenomic sequencing. Gigascience 2017, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample ID | Age (kyr BP) | Data Sources | Sequencing Platform | Reads Number | Bases Number | Average Length (bp) | Proportion of Endogenous DNA |
|---|---|---|---|---|---|---|---|---|
| Mammuthus primigenius | N1 | 26 | Sequencing | BGISEQ-500 | 1.00 x 107 | 9.32 x 108 | 93.17 | 59.51% |
| Mammuthus primigenius | N2 | 28 | Sequencing | BGISEQ-500 | 1.00 x 107 | 8.76 x 108 | 87.57 | 0.93% |
| Mammuthus primigenius | N3 | >43.5 | Sequencing | BGISEQ-500 | 1.00 x 107 | 8.70 x 108 | 87.03 | 34.50% |
| Mammuthus primigenius | N6 | >43.5 | Sequencing | BGISEQ-500 | 1.00 x 107 | 9.07 x 108 | 90.69 | 1.51% |
| Mammuthus primigenius | N9 | >43.5 | Sequencing | BGISEQ-500 | 1.00 x 107 | 8.99 x 108 | 89.87 | 0.54% |
| Mammuthus primigenius | N12 | 17 | Sequencing | BGISEQ-500 | 1.00 x 107 | 9.10 x 108 | 91.02 | 21.90% |
| Homo sapiens | JK2911 | 2.7 | Schuenemann et al. | Illumina HiSeq 2500 | 9.74 x 105 | 7.08 x 107 | 72.62 | 39.20% |
| Homo sapiens | AfontovaGora3 | 17 | Fu et al. | Illumina HiSeq 2500 | 8.88 x 105 | 5.17 x 107 | 58.18 | 44.64% |
| Homo sapiens | Villabruna | 14 | Fu et al. | Illumina HiSeq 2500 | 1.22 x 107 | 6.69 x 107 | 55.02 | 41.13% |
| Capra aegagrus hircus | Direkli5 | 11.5 | Daly et al. | Illumina HiSeq 2000 | 3.04 x 107 | 1.40 x 107 | 45.94 | 5.29% |
| Bos primigenius | British aurochs | 6.7 | Park et al. | Illumina Genome Analyzer IIx | 7.51 x 107 | 3.48 x 109 | 46.29 | 5.91% |
| Ancient horse | Ancient horse | 560-780 | Orlando et al. | Illumina HiSeq 2000 | 6.27 x 106 | 3.34 x 108 | 53.23 | 0.43% |
| Conditions | nt Database | mtDNA Database | ||||
|---|---|---|---|---|---|---|
| Animal mtDNA Database (Whole) | Animal mtDNA Database (Partial) | |||||
| BLASTall | BLASTall | BWA aln | BWA mem | BLASTall | ||
| Similarity levels (L, %) | 90 ≤ L ≤ 100 | 4/12 | 11/12 | 9/12 | 9/12 | 5/12 |
| 92 ≤ L ≤ 100 | 4/12 | 11/12 | 9/12 | 9/12 | 5/12 | |
| 94 ≤ L ≤ 100 | 4/12 | 12/12 | 9/12 | 9/12 | 6/12 | |
| 96 ≤ L ≤ 100 | 4/12 | 12/12 | 9/12 | 9/12 | 6/12 | |
| 98 ≤ L ≤ 100 | 4/12 | 12/12 | 9/12 | 9/12 | 8/12 | |
| L = 100 | 4/12 | 12/12 | 9/12 | 9/12 | 5/12 | |
| 90 ≤ L < 100 | 4/12 | 10/12 | 7/12 | 7/12 | 3/12 | |
| 92 ≤ L < 100 | 4/12 | 11/12 | 7/12 | 7/12 | 3/12 | |
| 94 ≤ L < 100 | 4/12 | 12/12 | 8/12 | 9/12 | 4/12 | |
| 96 ≤ L < 100 | 4/12 | 12/12 | 9/12 | 9/12 | 5/12 | |
| 98 ≤ L < 100 | 4/12 | 12/12 | 10/12 | 10/12 | 8/12 | |
| Query coverage (C, %) | C < 90 | -- | 7/12 | -- | -- | -- |
| C ≥ 90-92 | -- | 7/12 | -- | -- | -- | |
| C ≥ 92-94 | -- | 11/12 | -- | -- | -- | |
| C ≥ 94-96 | -- | 11/12 | -- | -- | -- | |
| C ≥ 96-98 | -- | 12/12 | -- | -- | -- | |
| C ≥ 98-100 | -- | 12/12 | -- | -- | -- | |
| C = 100 | -- | 12/12 | -- | -- | -- | |
| The first and last X bases for screening reads with C-to-T and/or G-to-A changes | X = 5 | -- | 11/12 | -- | -- | -- |
| X = 6 | -- | 11/12 | -- | -- | -- | |
| X = 7 | -- | 11/12 | -- | -- | -- | |
| X = 8 | -- | 12/12 | -- | -- | -- | |
| X = 9 | -- | 12/12 | -- | -- | -- | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lan, T.M.; Lin, Y.; Njaramba-Ngatia, J.; Guo, X.S.; Li, R.G.; Li, H.M.; Kumar-Sahu, S.; Wang, X.; Yang, X.J.; Guo, H.B.; et al. Improving Species Identification of Ancient Mammals Based on Next-Generation Sequencing Data. Genes 2019, 10, 509. https://doi.org/10.3390/genes10070509
Lan TM, Lin Y, Njaramba-Ngatia J, Guo XS, Li RG, Li HM, Kumar-Sahu S, Wang X, Yang XJ, Guo HB, et al. Improving Species Identification of Ancient Mammals Based on Next-Generation Sequencing Data. Genes. 2019; 10(7):509. https://doi.org/10.3390/genes10070509
Chicago/Turabian StyleLan, Tian Ming, Yu Lin, Jacob Njaramba-Ngatia, Xiao Sen Guo, Ren Gui Li, Hai Meng Li, Sunil Kumar-Sahu, Xie Wang, Xiu Juan Yang, Hua Bing Guo, and et al. 2019. "Improving Species Identification of Ancient Mammals Based on Next-Generation Sequencing Data" Genes 10, no. 7: 509. https://doi.org/10.3390/genes10070509