
Disulfide Bond Engineering for Enhancing the Thermostability of the Maltotetraose-Forming Amylase from Pseudomonas saccharophila STB07


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Construction of Site-Directed Mutants
2.3. Expression and Purification of Wild-Type MFAps and Its Mutants
2.4. Measurement of Activity and Kinetics
2.5. Temperature Inactivation to Wild-Type MFAps and Its Mutants
2.6. Measurement of Thermal Transition Mid-Point (Tm)
2.7. Homology Modeling
2.8. Location of the Disulfide Bridge Sites
2.9. ΔΔG Calculations and Mutational Scanning
2.10. Molecular Dynamics Simulations
2.11. Product Analysis Using Corn Starch
2.12. Statistical Analysis
3. Results and Discussion
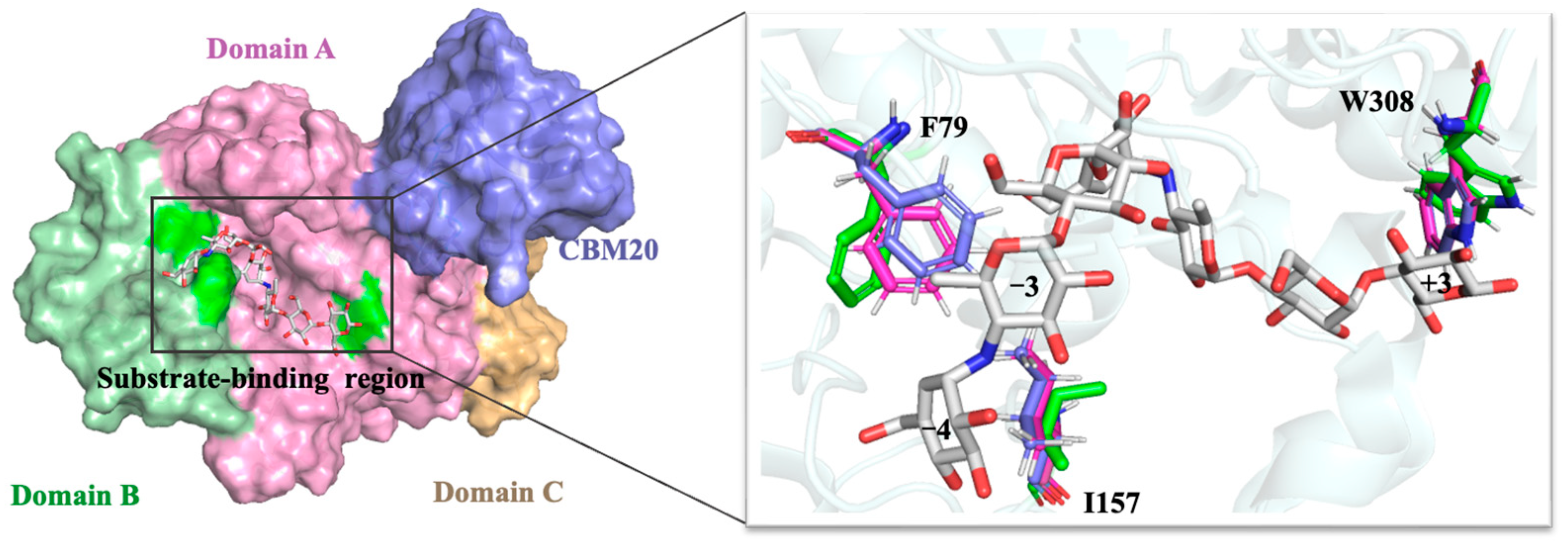
3.1. Structural Modeling
3.2. Computational Design and Screening of Disulfide Bonds
3.3. Expression, Purification and Biochemical Characterization of MFAps and Its Mutants
3.4. Effect of Mutations on Thermostability
3.5. Analysis of Structural Stability
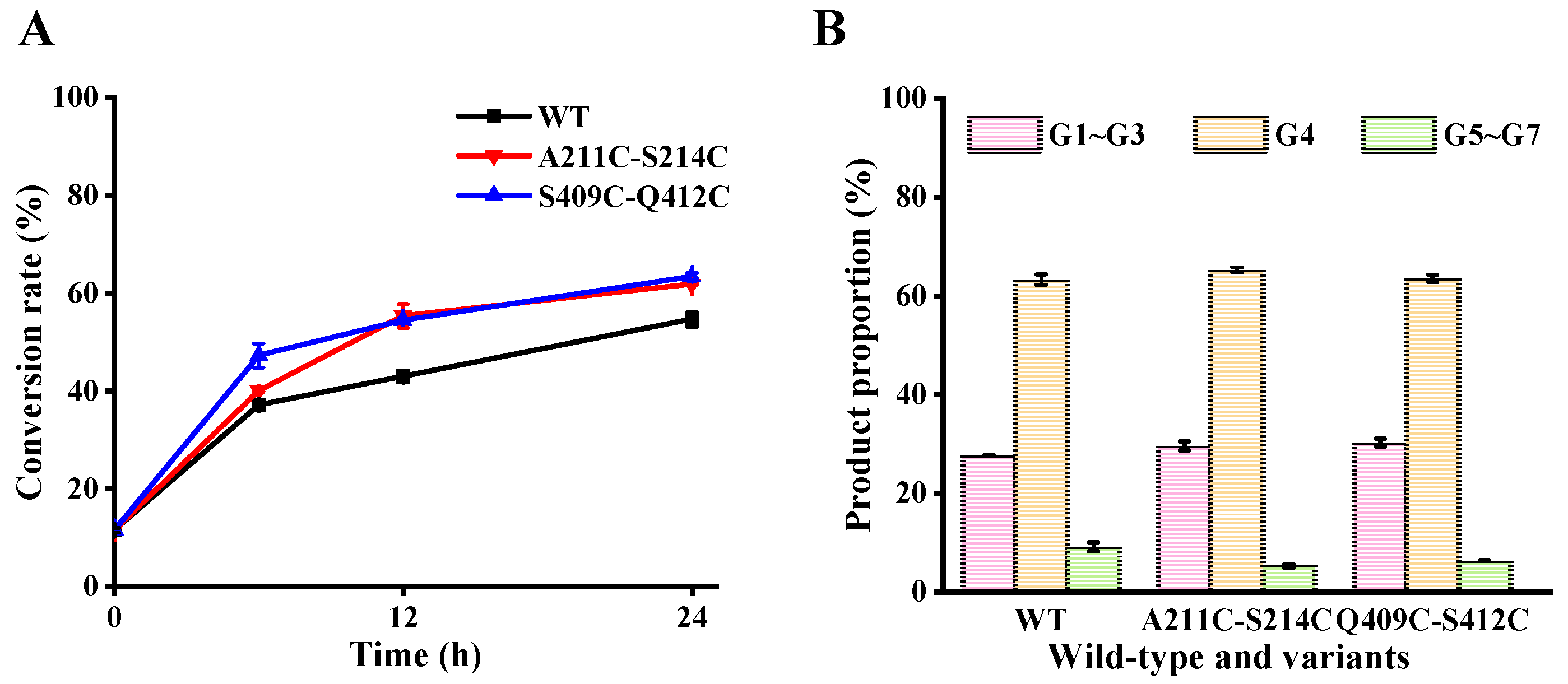
3.6. Effect of Mutations on Maltodextrin Hydrolysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Min, B.C.; Yoon, S.H.; Kim, J.W.; Lee, Y.W.; Kim, Y.B.; Park, K.H. Cloning of Novel Maltooligosaccharide-Producing Amylases as Antistaling Agents for Bread. J. Agric. Food Chem. 1998, 46, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Ding, N.; Ren, J.; Gu, Z.; Li, C.; Hong, Y.; Cheng, L.; Holler, T.P.; Li, Z. Maltooligosaccharide-forming amylase: Characteristics, preparation, and application. Biotechnol. Adv. 2017, 35, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Luan, S.; Duan, X. A Novel Thermal-Activated β-Galactosidase from Bacillus aryabhattai GEL-09 for Lactose Hydrolysis in Milk. Foods 2022, 11, 372. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Shen, L.; Mu, W.; Shi, Y.; Wu, J. Enzymatic Preparation of Gentiooligosaccharides by a Thermophilic and Thermostable β-Glucosidase at a High Substrate Concentration. Foods 2022, 11, 357. [Google Scholar] [CrossRef]
- Bashirova, A.; Pramanik, S.; Volkov, P.; Rozhkova, A.; Nemashkalov, V.; Zorov, I.; Gusakov, A.; Sinitsyn, A.; Schwaneberg, U.; Davari, M. Disulfide Bond Engineering of an Endoglucanase from Penicillium verruculosum to Improve Its Thermostability. Int. J. Mol. Sci. 2019, 20, 1602. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Vinayagam, A.; Pugalenthi, G.; Rajesh, R.; Sowdhamini, R. DSDBASE: A consortium of native and modelled disulphide bonds in proteins. Nucleic Acids Res. 2004, 32, 200–202. [Google Scholar] [CrossRef] [Green Version]
- Badieyan, S.; Bevan, D.R.; Zhang, C. Study and design of stability in GH5 cellulases. Biotechnol. Bioeng. 2012, 109, 31–44. [Google Scholar] [CrossRef]
- Cambillau, C. Structure and Topology Prediction of Phage Adhesion Devices Using AlphaFold2: The Case of Two Oenococcus oeni Phages. Microorganisms 2021, 9, 2151–2164. [Google Scholar]
- Guex, N.; Peitsch, M.C. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 16, 296–303. [Google Scholar]
- Kim, D.E.; Dylan, C.; David, B. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, 526–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Zhang, Y. The I-TASSER suite: Protein structure and function prediction. Nat. Methods 2014, 12, 7–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Liu, L.; Li, J.; Chen, J.; Du, G. Rational design to improve protein thermostability: Recent advances and prospects. Chembioeng Rev. 2015, 2, 87–94. [Google Scholar] [CrossRef]
- Borg, J.; Nys, R.; Schymkoitz, J.; Stricher, F. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, 382–388. [Google Scholar]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant: Predicting protein stability upon mutation. Bioinformatics 2004, 20, 163–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasigroch, J.M.; Gilis, D.; Dehouck, Y.; Rooman, M. PoPMuSiC, rationally designing point mutations in protein structures. Bioinformatics 2002, 12, 1701–1702. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Le, Q.; Yong, H.K. Development of thermostable lipase B from Candida antarctica (CalB) through in silico design employing B-factor and RosettaDesign. Enzym. Microb. Technol. 2010, 47, 1–5. [Google Scholar] [CrossRef]
- Khan, S.; Vihinen, M. Performance of protein stability predictors. Hum. Mutat. 2010, 31, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Wang, S.; Xu, Y.; Yu, X. Engineering of a thermo-alkali-stable lipase from Rhizopus chinensis by rational design of a buried disulfide bond and combinatorial mutagenesis. J. Ind. Microbiol. Biotechnol. 2020, 12, 1019–1030. [Google Scholar] [CrossRef]
- Navone, L.; Vogl, T.; Luangthongkam, P.; Blinco, J.A.; Speight, R. Disulfide bond engineering of AppA phytase for increased thermostability requires co-expression of protein disulfide isomerase in Pichia pastoris. Biotechnol. Biofuels 2021, 14, 1754–6834. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Y.; Li, X.; Bai, Y.; Xia, W. Impact of disulfide bonds on the folding and refolding capability of a novel thermostable GH45 cellulase. Appl. Microbiol. Biotechnol. 2018, 102, 9183–9192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ting, T.; Xie, X.; Ban, X.; Li, C.; Hong, Y.; Cheng, L.; Li, Z. Structure of maltotetraose-forming amylase from Pseudomonas saccharophila STB07 provides insights into its product specificity. Int. J. Biol. Macromol. 2020, 154, 1303–1313. [Google Scholar] [CrossRef]
- Xie, X.; Ban, X.; Gu, Z.; Li, C.; Li, Z. Structure-based engineering of a maltooligosaccharide-forming amylase to enhance product specificity. J. Agric. Food Chem. 2020, 68, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Gu, Z.; Ding, N.; Zhang, Z.; Chen, D.; Li, C.; Hong, Y.; Cheng, L.; Li, Z. Calcium and sodium ions synergistically enhance the thermostability of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Food Chem. 2019, 283, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Xie, X.; Qiu, G.; Zhang, Z.; Ban, X.; Li, Z. Importance of Trp139 in the product specificity of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Appl. Microbiol. Biotechnol. 2019, 103, 9433–9442. [Google Scholar] [CrossRef]
- Miller, G.L. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Biochem. 1959, 31, 426–428. [Google Scholar]
- Bu, O.; Rudat, J.; Ochsenreither, K. FoldX as Protein Engineering Tool: Better Than Random Based Approaches. Comput. Struct. Biotechnol. J. 2018, 16, 25–33. [Google Scholar]
- Yu, H.; Dalby, P.A. Coupled molecular dynamics mediate long- and short-range epistasis between mutations that affect stability and aggregation kinetics. Proc. Natl. Acad. Sci. USA 2018, 115, 11043–11052. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Xun, G.; Feng, Y. The state-of-the-art strategies of protein engineering for enzyme stabilization. Biotechnol. Adv. 2019, 37, 530–537. [Google Scholar] [CrossRef]
- Jürgens, B.; Jürgens, C.; Wilmanns, M.; Sterner, R. Stability, catalytic versatility and evolution of the (β/α)8-barrel fold. Curr. Opin. Biotechnol. 2001, 12, 376–381. [Google Scholar]
- Mok, S.C.; Teh, A.H.; Saito, J.A.; Najimudin, N.; Alam, M. Crystal structure of a compact alpha-amylase from Geobacillus Thermoleovorans. Enzym. Microb. Technol. 2013, 53, 46–54. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, J.; Zhang, W.; Guang, C.; Zhang, T.; Mu, W. Improving Thermostability and Catalytic Behavior of L-Rhamnose Isomerase from Caldicellulosiruptor obsidiansis OB47 toward D-Allulose by Site-Directed Mutagenesis. J. Agric. Food Chem. 2018, 66, 12017–12024. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Ban, X.; Zhang, Z.; Li, Z. Crystal structure of a maltooligosaccharide-forming amylase from Bacillus stearothermophilus STB04. Int. J. Biol. Macromol. 2019, 138, 394–402. [Google Scholar] [CrossRef]
- Kanai, R.; Haga, K.; Akiba, T.; Yamane, K.; Harata, K. Biochemical and crystallographic analyses of maltohexaose-producing amylase from alkalophilic Bacillus sp. 707. Biochemistry 2004, 43, 14047–14056. [Google Scholar] [CrossRef]
- Yu, H.; Dalby, P.A. Exploiting correlated molecular-dynamics networks to counteract enzyme activity-stability trade-off. Proc. Natl. Acad. Sci. USA 2018, 115, 12192–12200. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, K.S. Defying the activity-stability trade-off in enzymes: Taking advantage of entropy to enhance activity and thermostability. Crit. Rev. Biotechnol. 2017, 37, 309–322. [Google Scholar] [CrossRef]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced enzyme kinetic stability by increasing rigidity within the active site. J. Biol. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, G.; Zhang, Y.; Xie, Y.; Withers, S.; Feng, Y. A general and efficient strategy for generating the stable enzymes. Sci. Rep. 2016, 6, 33797. [Google Scholar] [CrossRef]








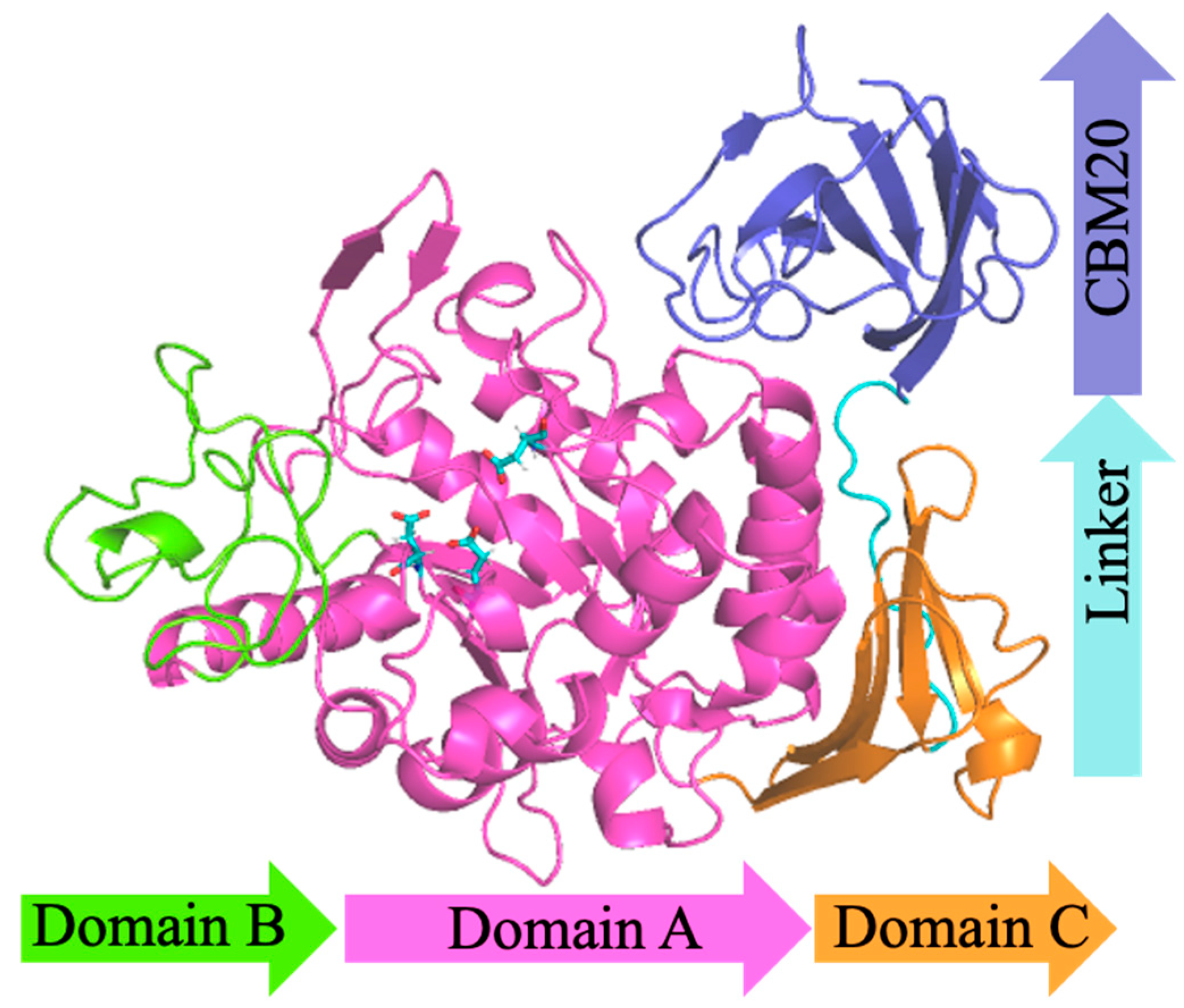{kind=link}
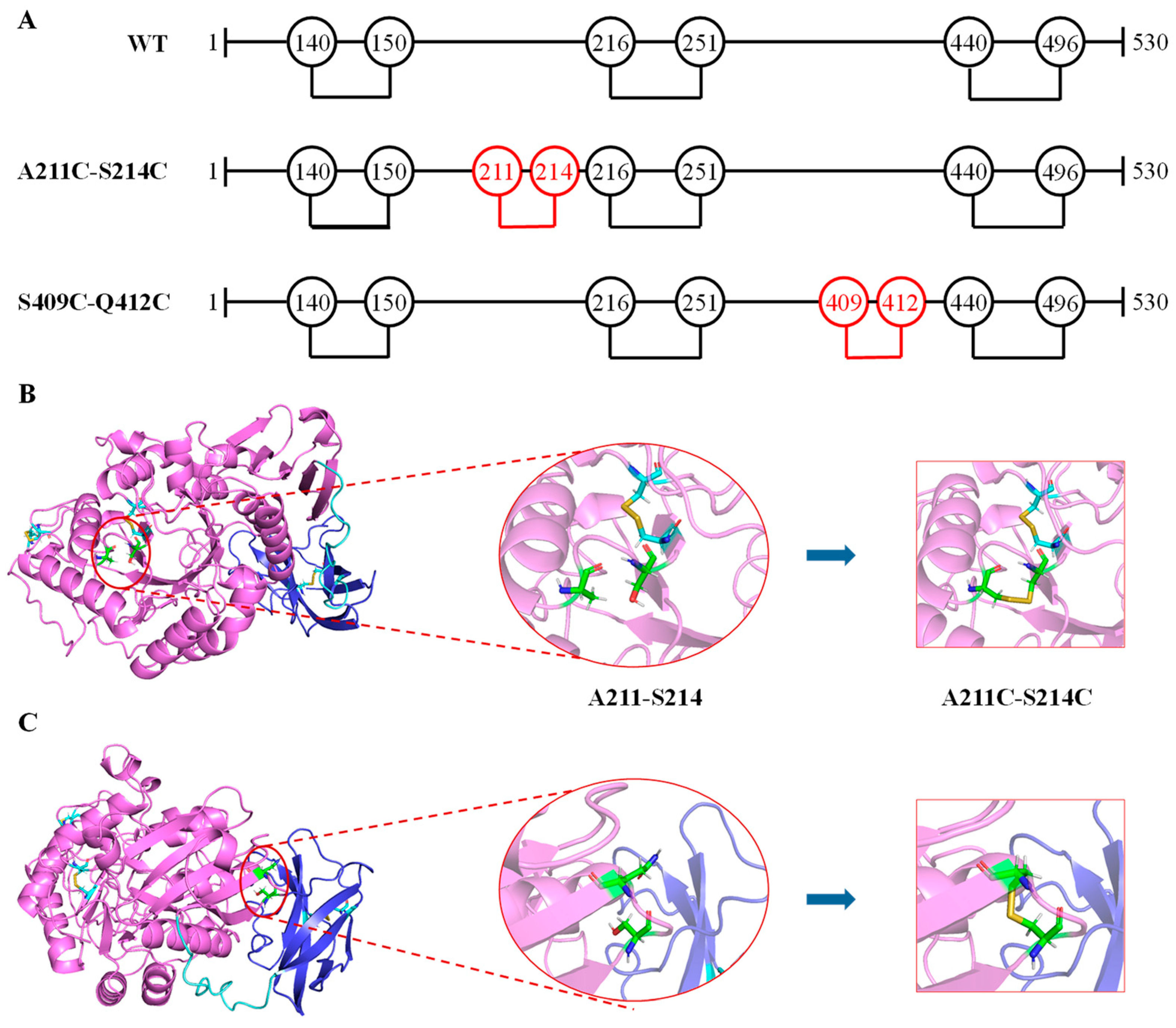{kind=link}
{kind=link}
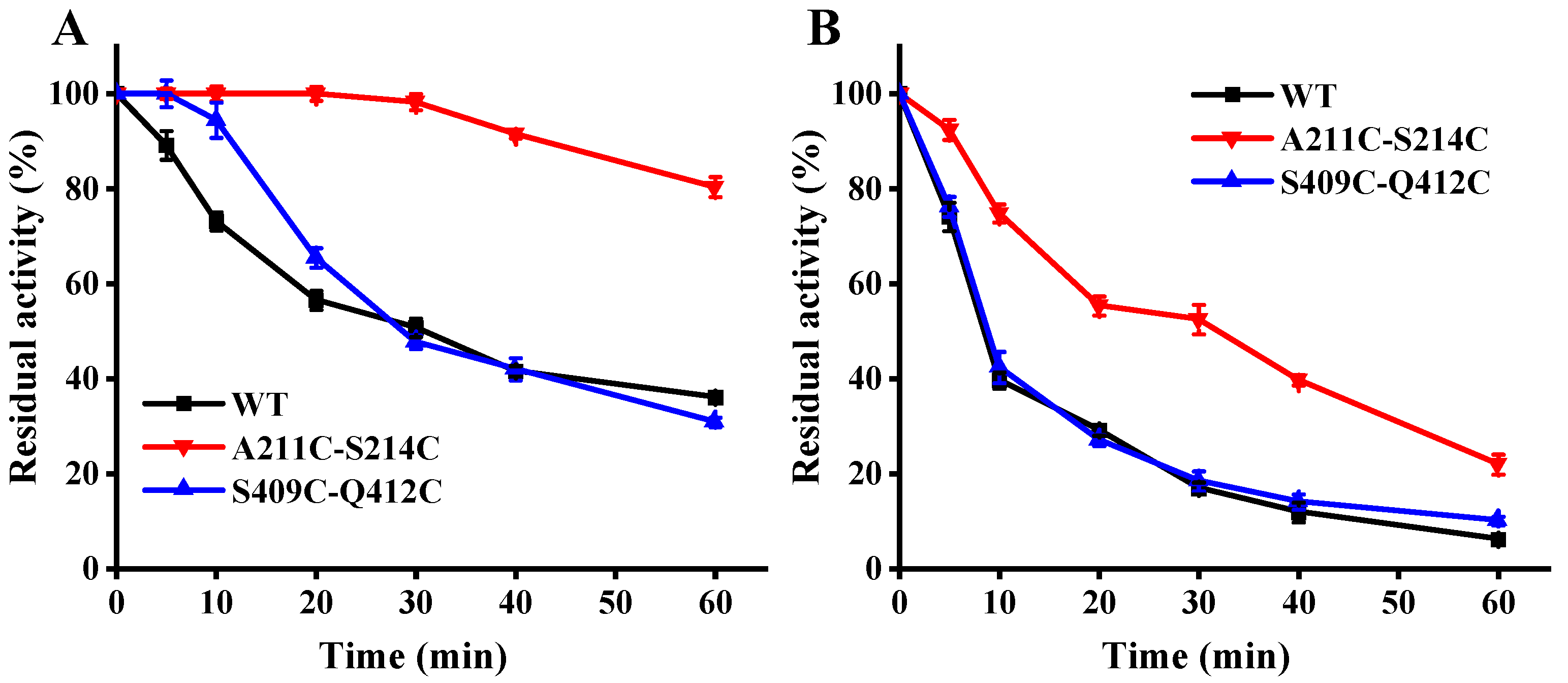{kind=link}
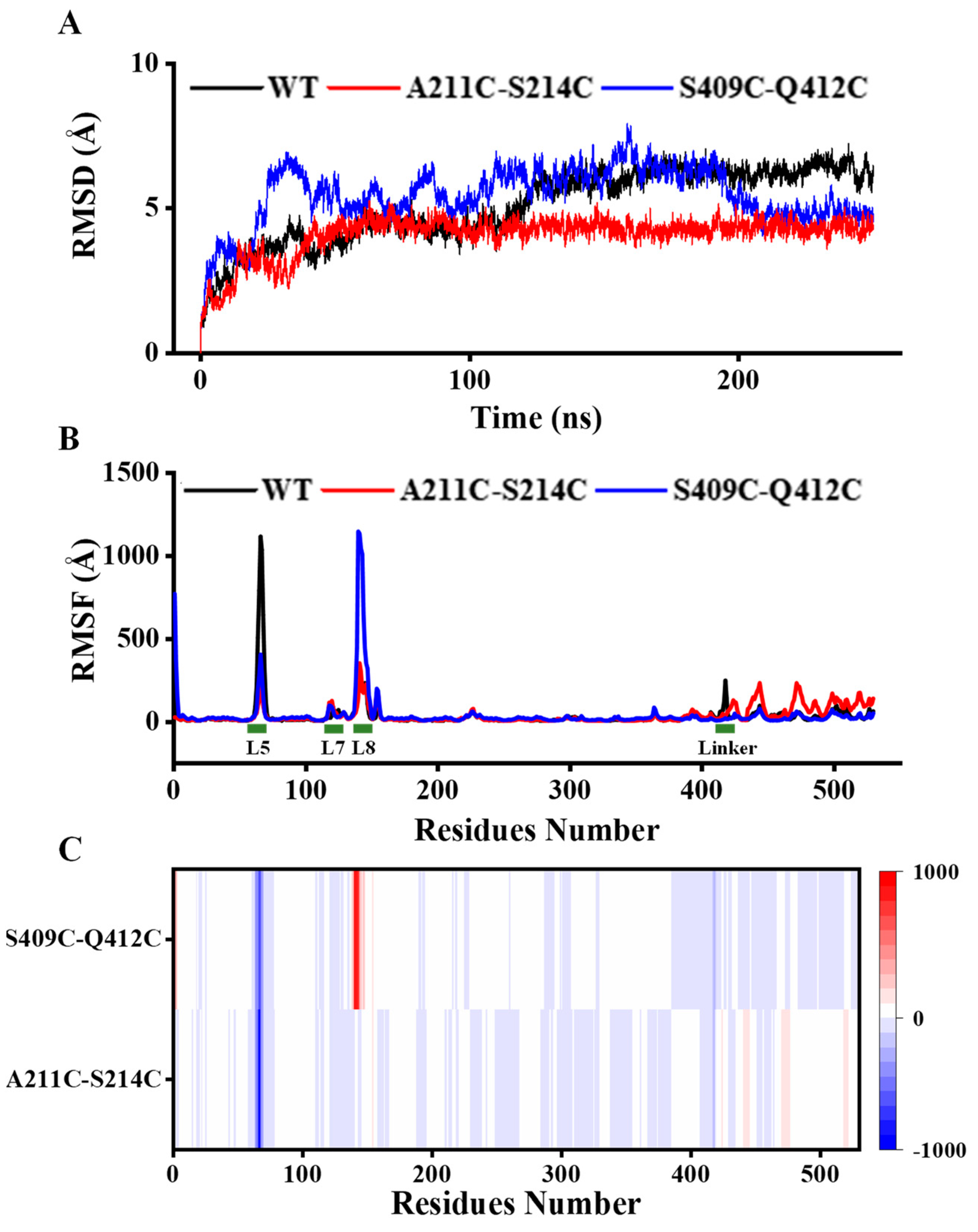{kind=link}
{kind=link}
{kind=link}
| Mutants | Primer Sequence A |
|---|---|
| A211C-S214C | R1 5′-tgtGACAGCtgtTTCTGCGTTGGCGAGCTGTGG-3′ R2 5′-acaGCTGTCacaGCTGTCGCTCATCCAGCTG-3′ |
| S409C-Q412C | R1 5′-tgtAACGGCtgtGTGCGCGTCTGGCGCAGC-3′ |
| R1 5′-acaGCCGTTacaGGCGTTGACCGCCTCGCT-3′ | |
| All mutants B | F1 5′-AAGTAGCGAAAACTCGTATCCTTCT-3′ F2 5′-AGAAGGATACGAGTTTTCGCTACTT-3′ |
| Enzyme | Relative Activity A,B (%) | Specific Activity B (U/mg) | KmB (mg/mL) | VmaxB (μmol·min−1·mL−1) | kcatB (×103 sec−1) | kcat/KmB (×103 mL·mg−1·sec−1) |
|---|---|---|---|---|---|---|
| Wild-type | 100 c | 427.5 ± 24.6 b | 2.48 ± 0.08 b | 6.90 ± 0.37 a | 21.90 ± 0.49 b | 8.83 ± 0.10 a |
| A211C-S214C | 68 a | 349.3 ± 12.7 a | 2.29 ± 0.17 a | 6.43 ± 0.10 a | 21.05 ± 0.54 a | 9.19 ± 0.18 b |
| S409C-Q412C | 76 b | 415.2 ± 14.8 b | 2.23 ± 0.13 a | 6.29 ± 0.42 a | 22.13 ± 0.68 b | 9.91 ± 0.09 c |
| Enzyme | Half-live at 60 °C (min) A | Tm (°C) A |
|---|---|---|
| Wild-type | 10.6 ± 0.4 a | 57.6 ± 0.2 a |
| A211C-S214C | 28.2 ± 0.2 c | 59.2 ± 0.1 c |
| S409C-Q412C | 11.6 ± 0.3 b | 58.0 ± 0.1 b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, C.; Ban, X.; Gu, Z.; Hong, Y.; Cheng, L.; Li, Z. Disulfide Bond Engineering for Enhancing the Thermostability of the Maltotetraose-Forming Amylase from Pseudomonas saccharophila STB07. Foods 2022, 11, 1207. https://doi.org/10.3390/foods11091207
Wang Y, Li C, Ban X, Gu Z, Hong Y, Cheng L, Li Z. Disulfide Bond Engineering for Enhancing the Thermostability of the Maltotetraose-Forming Amylase from Pseudomonas saccharophila STB07. Foods. 2022; 11(9):1207. https://doi.org/10.3390/foods11091207
Chicago/Turabian StyleWang, Yinglan, Caiming Li, Xiaofeng Ban, Zhengbiao Gu, Yan Hong, Li Cheng, and Zhaofeng Li. 2022. "Disulfide Bond Engineering for Enhancing the Thermostability of the Maltotetraose-Forming Amylase from Pseudomonas saccharophila STB07" Foods 11, no. 9: 1207. https://doi.org/10.3390/foods11091207