
Comparative Transcriptome and Pigment Analyses Reveal Changes in Gene Expression Associated with Flavonol Metabolism in Yellow Camellia

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sample Preparation
2.2. Flavonol Content Measurement
2.3. RNA Extraction and RNA-seq
2.4. Read Filtering and De Novo Assembly
2.5. Functional Annotation and Differential Expression Analysis
2.6. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.7. Statistical Analysis
3. Results
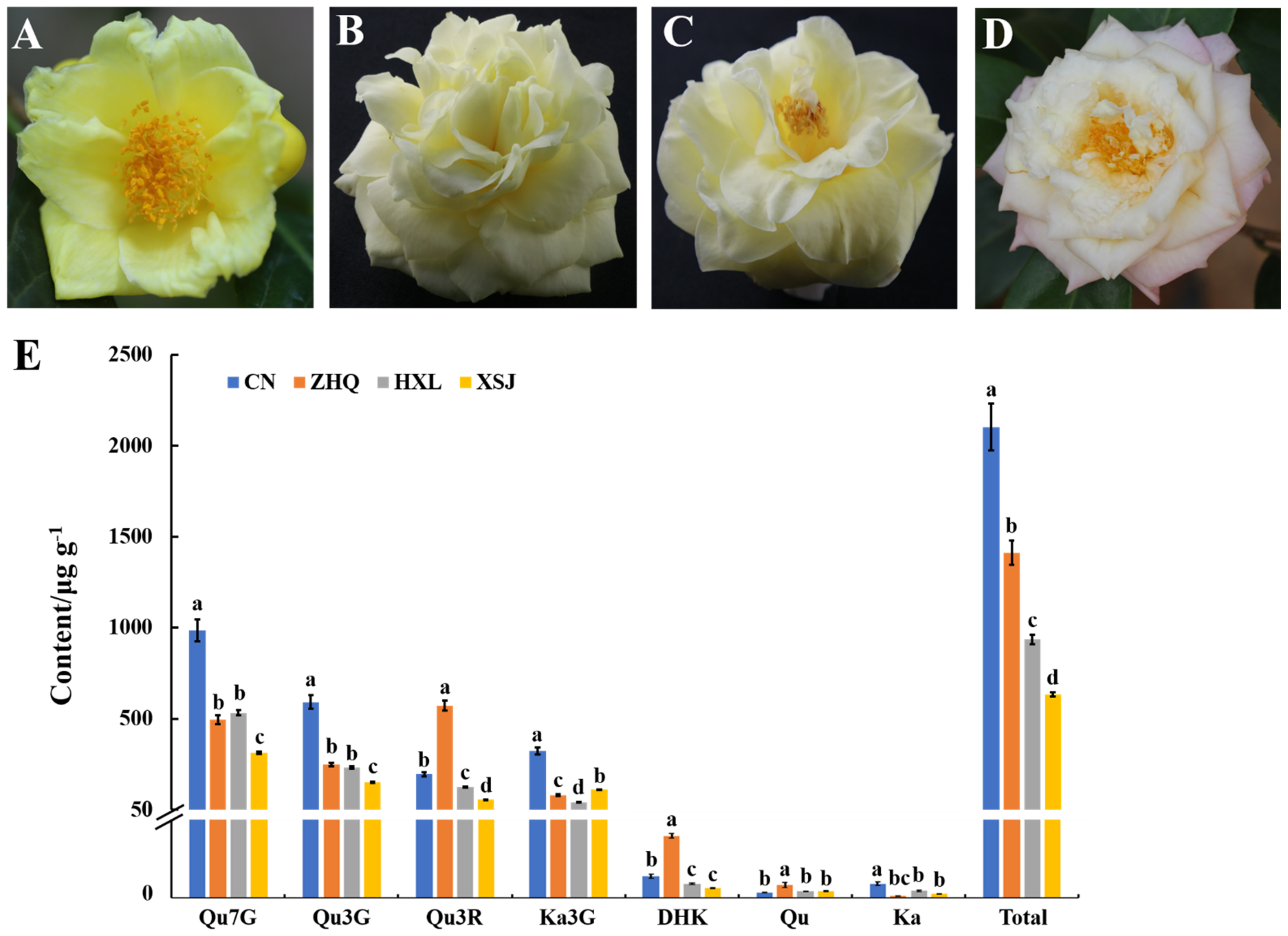
3.1. Morphology Analysis and Pigment Accumulation among Camellia with Different Colors
3.2. Transcriptome Sequencing and Assembly
3.3. Functional Annotation of Unigenes
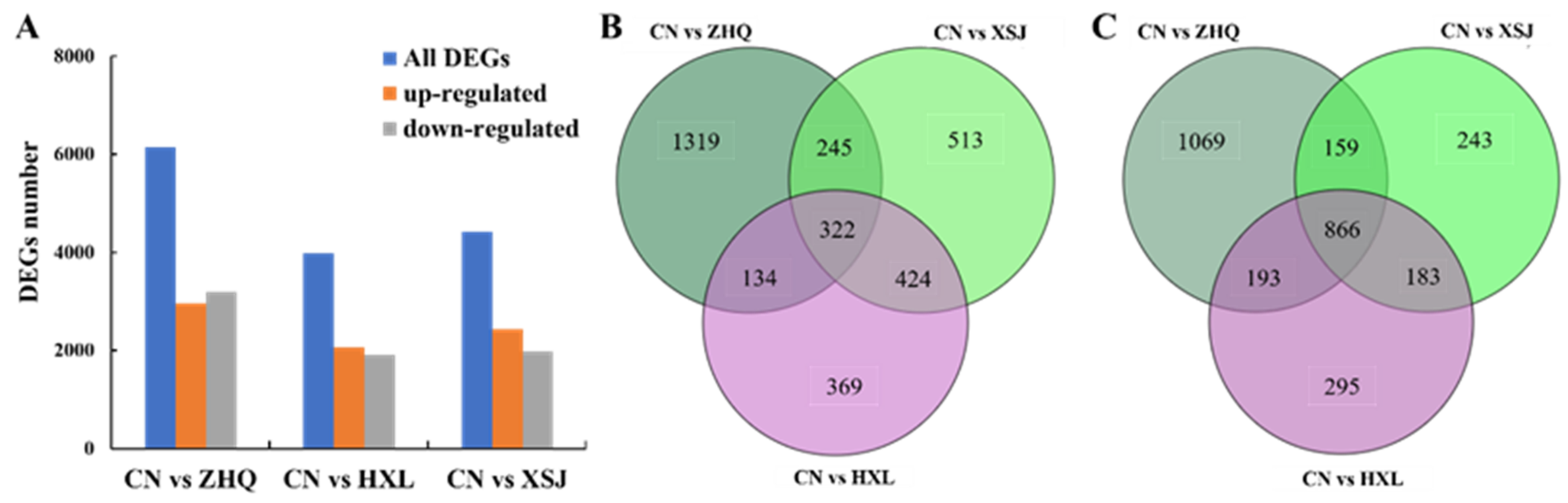
3.4. Differential Expression Analysis
3.5. Gene Ontology (GO) Classification and Functional Enrichment Analysis of DEGs
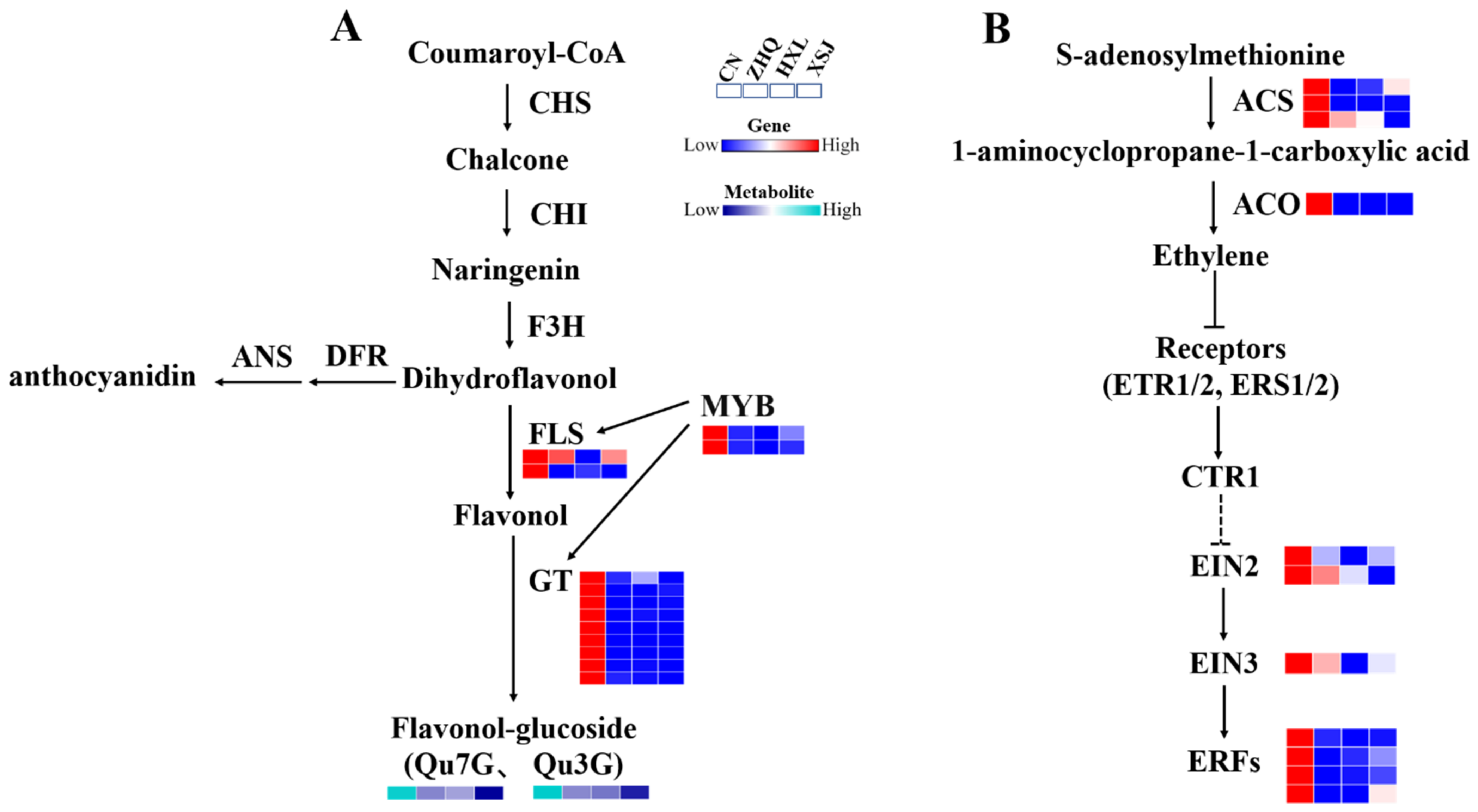
3.6. Flavonol Metabolism and Regulation
3.7. Ethylene May Be Involved in Flower Color Formation in Yellow Camellia
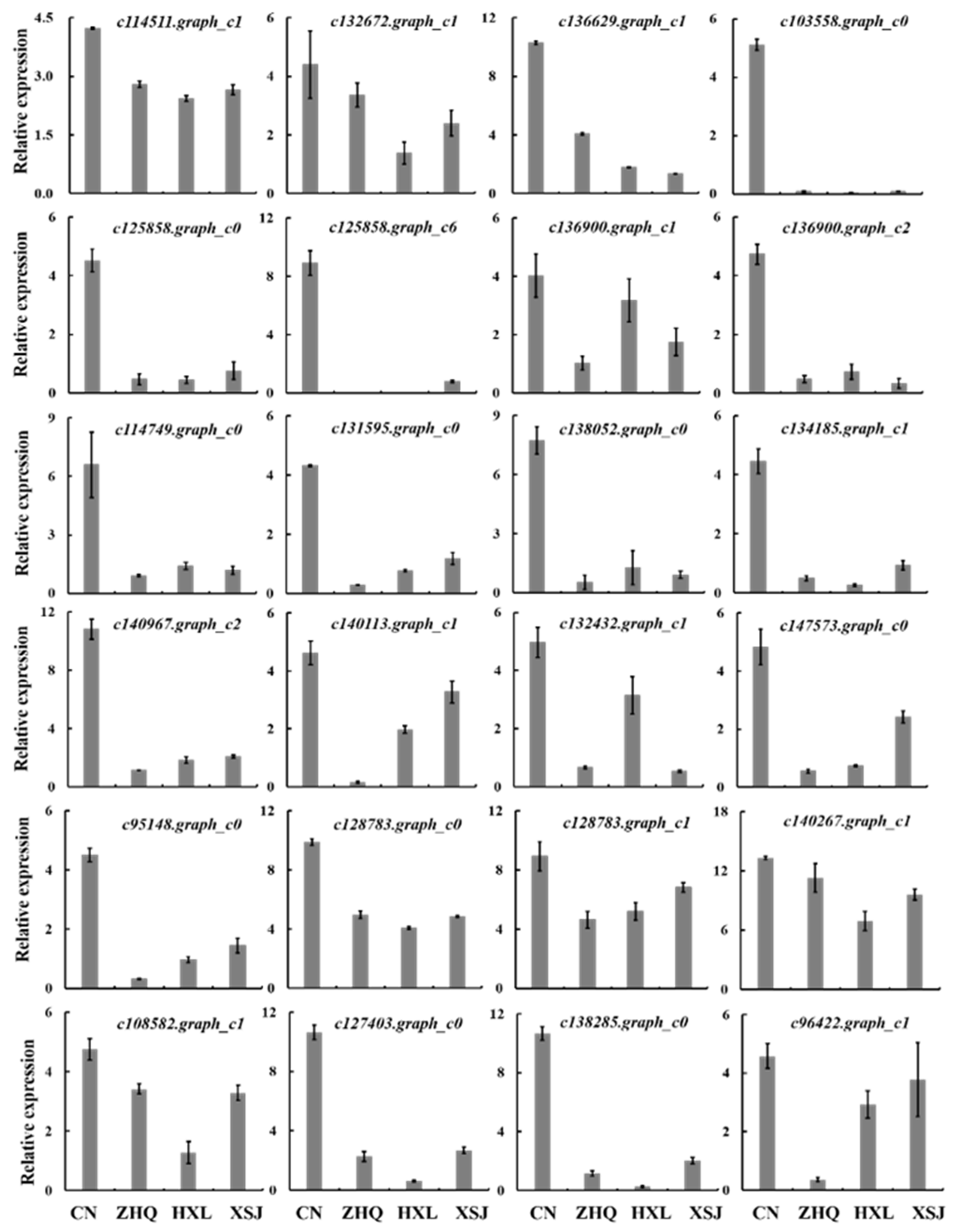
3.8. Validation of Candidate Genes of Flavonol and Ethylene with Quantitative Real-Time PCR
4. Discussion
4.1. Flavonol Metabolism in Flower Color Formation of Yellow Camellia
4.2. Ethylene May Be Involved in the Regulation of Yellow Flower Formation in Camellia
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sasaki, N.; Nakayama, T. Achievements and Perspectives in Biochemistry Concerning Anthocyanin Modification for Blue Flower Coloration. Plant Cell Physiol. 2015, 56, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Fan, Z.; Guo, H.; Ye, N.; Lyu, T.; Yang, W.; Wang, J.; Wang, J.T.; Wu, B.; Li, J.; et al. Comparative Genomics Analysis Reveals Gene Family Expansion and Changes of Expression Patterns Associated with Natural Adaptations of Flowering Time and Secondary Metabolism in Yellow Camellia. Funct. Integr. Genom. 2018, 18, 659–671. [Google Scholar] [CrossRef] [PubMed]
- He, D.; Li, X.; Sai, X.; Wang, L.; Li, S.; Xu, Y. Camellia nitidissima C.W. Chi: A Review of Botany, Chemistry, and Pharmacology. Phytochem. Rev. 2017, 17, 327–349. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Sun, Z.; Wang, J.; Yin, H.; Fan, Z.; Li, J.; Jiang, C.; Huang, X. Flavonoid Components and Their Relationship with Flower Colors in Five Species of Camellia Section Chrysantha. Chin. J. Ecol. 2019, 38, 961–966. [Google Scholar]
- Guan, K.; Li, J.; Wang, Z. Camellias of China. Hangzhou Zhejiang Sci. Technol. Publ. House 2014, 444, 395–406. [Google Scholar]
- Gao, J.; Liu, X.; Zhao, Q. Illustrations of the New Camellia Hybirds that Bloom Year-Round; Zhejiang Science and Technology Press: Hangzhou, China, 2016. [Google Scholar]
- Grotewold, E. The Genetics and Biochemistry of Floral Pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef]
- Yang, Y.; Li, B.; Feng, C.; Wu, Q.; Wang, Q.; Li, S.; Yu, X.; Wang, L. Chemical Mechanism of Flower Color Microvariation in Paeonia with Yellow Flowers. Hortic. Plant J. 2020, 6, 179–190. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, H.; Wu, R. Recent Advances on Blue Flower Formation. Chin. Bull. Bot. 2020, 55, 216–227. [Google Scholar]
- Tanaka, Y.; Sasaki, N.; Ohmiya, A. Biosynthesis of Plant Pigments: Anthocyanins, Betalains and Carotenoids. Plant J. 2008, 54, 733–749. [Google Scholar] [CrossRef]
- Dong, N.Q.; Lin, H.X. Contribution of Phenylpropanoid Metabolism to Plant Development and Plant-environment Interactions. J. Integr. Plant Biol. 2020, 63, 180–209. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Noda, N. Recent Advances in the Research and Development of Blue Flowers. Breed. Sci. 2018, 68, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.M.; Šamec, D.; Tomczyk, M.; Milella, L.; Russo, D.; Habtemariam, S.; Suntar, I.; Rastrelli, L.; Daglia, M.; Xiao, J.; et al. Flavonoid Biosynthetic Pathways in Plants: Versatile Targets for Metabolic Engineering. Biotechnol. Adv. 2018, 38, 107316. [Google Scholar] [CrossRef]
- Liu, W.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J.; Yin, H. The Flavonoid Biosynthesis Network in Plants. Int. J. Mol. Sci. 2021, 22, 12824. [Google Scholar] [CrossRef]
- Zhou, X.W.; Fan, Z.Q.; Chen, Y.; Zhu, Y.L.; Li, J.Y.; Yin, H.F. Functional Analyses of a Flavonol Synthase-like Gene from Camellia nitidissima Reveal Its Roles in Flavonoid Metabolism during Floral Pigmentation. J. Biosci. 2013, 38, 593–604. [Google Scholar] [CrossRef]
- Li, X.; Yin, H.; Fan, Z.; Li, J. The Relationship Between Anthocyanins and Flower Colors of Bud Mutation in Camellia japonica. Sci. Agric. Sin. 2019, 52, 1961–1969. [Google Scholar]
- Tanikawa, N.; Kashiwabara, T.; Hokura, A.; Abe, T.; Shibata, M.; Nakayama, M. A Peculiar Yellow Flower Coloration of Camellia Using Aluminum-flavonoid Interaction. J. Jpn. Soc. Hortic. Sci. 2008, 77, 402–407. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ren, C.; Dong, S.; Chen, C.; Xian, B.; Wu, Q.; Wang, J.; Pei, J.; Chen, J. Integrated Metabolomics and Transcriptome Analysis of Flavonoid Biosynthesis in Safflower (Carthamus tinctorius L.) With Different Colors. Front. Plant Sci. 2021, 12, 712038. [Google Scholar] [CrossRef]
- Yang, F.; Li, C.H.; Das, D.; Zheng, Y.H.; Song, T.; Wang, L.X.; Chen, M.X.; Li, Q.Z.; Zhang, J. Comprehensive Transcriptome and Metabolic Profiling of Petal Color Development in Lycoris sprengeri. Front. Plant Sci. 2021, 12, 747131. [Google Scholar] [CrossRef]
- Li, X.; Wang, J.; Sun, Z.; Wang, J.; Yin, H.; Fan, Z.; Li, J. Flavonoid Components and Their Effects on Flower Colors in Camellia nitidissima, White C. japonica and Their Three Hybrid Cultivars. Acta Hortic. Sin. 2019, 46, 1145–1154. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Yin, H.; Feng, Y.; Yu, S.; Fan, Z.; Li, X.; Li, J. Comparative Transcriptome Analysis of Flower Senescence of Camellia lutchuensis. Curr. Genom. 2022, 23, 66–76. [Google Scholar] [CrossRef]
- Pan, L.; Li, J.; Yin, H.; Fan, Z.; Li, X. Integrated Physiological and Transcriptomic Analyses Reveal a Regulatory Network of Anthocyanin Metabolism Contributing to the Ornamental Value in a Novel Hybrid Cultivar of Camellia Japonica. Plants 2020, 9, 1724. [Google Scholar] [CrossRef]
- Fang, F.; Wang, F.Z. Research Progress on Key Genes of Flavonol Biosynthesis in Plants. Sci. Technol. Food Ind. 2018, 39, 335–340. [Google Scholar]
- Yu, Z.; Dong, W.; Teixeira da Silva, J.A.; He, C.; Si, C.; Duan, J. Ectopic Expression of DoFLS1 from Dendrobium officinale Enhances Flavonol Accumulation and Abiotic Stress Tolerance in Arabidopsis thaliana. Protoplasma 2021, 258, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Shi, Y.; Fu, Z.; Li, W.W.; Lai, S.; Wu, Y.; Wang, Y.; Liu, Y.; Gao, L.; Xia, T. Functional Characterization of Three Flavonol Synthase Genes from Camellia sinensis: Roles in Flavonol Accumulation. Plant Sci. 2020, 300, 110632. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ren, C.; Zhang, B.; Xu, C.; Li, X. Plant UDP-glycosyltransferases in Flavonoids Biosynthesis. Acta Hortic. Sin. 2019, 46, 1655–1669. [Google Scholar]
- Zhang, F.; Guo, H.; Huang, J.; Yang, C.; Li, Y.; Wang, X.; Qu, L.; Liu, X.; Luo, J. A UV-B-responsive Glycosyltransferase, OsUGT706C2, Modulates Flavonoid Metabolism in Rice. Sci. China. Life Sci. 2020, 63, 1037–1052. [Google Scholar] [CrossRef]
- Meng, X.; Li, Y.; Zhou, T.; Sun, W.; Shan, X.; Gao, X.; Wang, L. Functional Differentiation of Duplicated Flavonoid 3-O-Glycosyltransferases in the Flavonol and Anthocyanin Biosynthesis of Freesia hybrida. Front. Plant Sci. 2019, 10, 1330. [Google Scholar] [CrossRef]
- Li, J.; Luan, Q.; Han, J.; Zhang, C.; Liu, M.; Ren, Z. CsMYB60 Directly and Indirectly Activates Structural Genes to Promote the Biosynthesis of Flavonols and Proanthocyanidins in Cucumber. Hortic. Res. 2020, 7, 103. [Google Scholar] [CrossRef]
- Binder, B.M. Ethylene Signaling in Plants. J. Biol. Chem. 2020, 295, 7710–7725. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, Y.; Yan, S. Salicylic Acid and Ethylene Coordinately Promote Leaf Senescence. J. Integr. Plant Biol. 2021, 63, 823–827. [Google Scholar] [CrossRef]
- Ma, N.; Ma, C.; Liu, Y.; Shahid, M.O.; Wang, C.; Gao, J. Petal senescence: A Hormone View. J. Exp. Bot. 2018, 69, 719–732. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, C.; Wu, J.; Ouyang, J. Research Progress in Biosynthesis of Anthocyanins. Chem. Ind. For. Prod. 2020, 40, 1–11. [Google Scholar]
- Wang, Z.L.; Wang, S.; Kuang, Y.; Hu, Z.M.; Qiao, X.; Ye, M. A comprehensive review on phytochemistry, pharmacology, and flavonoid biosynthesis of Scutellaria baicalensis. Pharm. Biol. 2018, 56, 465–484. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.G.; Yu, J.Q.; Han, P.L.; Xie, X.B.; Sun, C.H.; Zhang, Q.Y.; Wang, J.H.; Hao, Y.J. The Regulatory Module MdPUB29-MdbHLH3 Connects Ethylene Biosynthesis with Fruit Quality in Apple. New Phytol. 2019, 221, 1966–1982. [Google Scholar] [CrossRef]
- Lewis, D.R.; Ramirez, M.V.; Miller, N.D.; Vallabhaneni, P.; Ray, W.K.; Helm, R.F.; Winkel, B.S.; Muday, G.K. Auxin and Ethylene Induce Flavonol Accumulation through Distinct Transcriptional Networks. Plant Physiol. 2011, 156, 144–164. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, N.; Wang, T.; Li, J.; Wen, T.; Yang, X.; Lindsey, K.; Zhang, X. The GhmiR157a-GhSPL10 Regulatory Module Controls Initial Cellular Dedifferentiation and Callus Proliferation in Cotton by Modulating Ethylene-mediated Flavonoid Biosynthesis. J. Exp. Bot. 2018, 69, 1081–1093. [Google Scholar] [CrossRef]
- Ni, J.; Bai, S.; Zhao, Y.; Qian, M.; Tao, R.; Yin, L.; Gao, L.; Teng, Y. Ethylene Response Factors Pp4ERF24 and Pp12ERF96 Regulate Blue Light-induced Anthocyanin Biosynthesis in ’Red Zaosu’ Pear Fruits by Interacting with MYB114. Plant Mol. Biol. 2019, 99, 67–78. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Yu, S.; Feng, Y.; Mo, R.; Wang, K.; Fan, M.; Fan, Z.; Yin, H.; Li, J.; Li, X. Comparative Transcriptome and Pigment Analyses Reveal Changes in Gene Expression Associated with Flavonol Metabolism in Yellow Camellia. Forests 2022, 13, 1094. https://doi.org/10.3390/f13071094
Liu W, Yu S, Feng Y, Mo R, Wang K, Fan M, Fan Z, Yin H, Li J, Li X. Comparative Transcriptome and Pigment Analyses Reveal Changes in Gene Expression Associated with Flavonol Metabolism in Yellow Camellia. Forests. 2022; 13(7):1094. https://doi.org/10.3390/f13071094
Chicago/Turabian StyleLiu, Weixin, Suhang Yu, Yi Feng, Runhong Mo, Kaineng Wang, Menglong Fan, Zhengqi Fan, Hengfu Yin, Jiyuan Li, and Xinlei Li. 2022. "Comparative Transcriptome and Pigment Analyses Reveal Changes in Gene Expression Associated with Flavonol Metabolism in Yellow Camellia" Forests 13, no. 7: 1094. https://doi.org/10.3390/f13071094