
Focus on Paediatric Restrictive Cardiomyopathy: Frequently Asked Questions
 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. How Common Is RCM?
3. Is Family History and Genetic Testing Important?
4. What Are the Main Clinical Signs and Symptoms of Paediatric RCM?
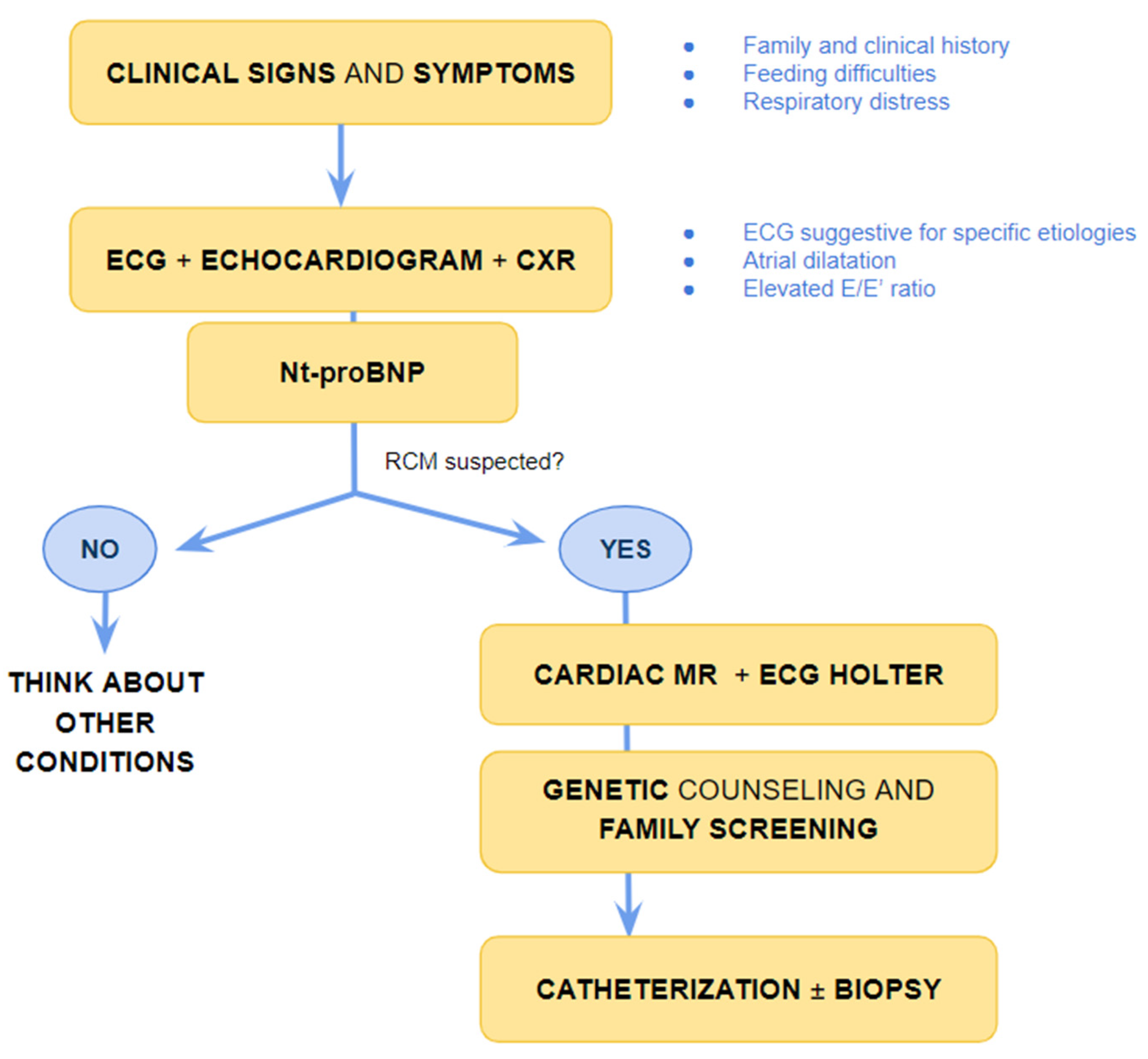
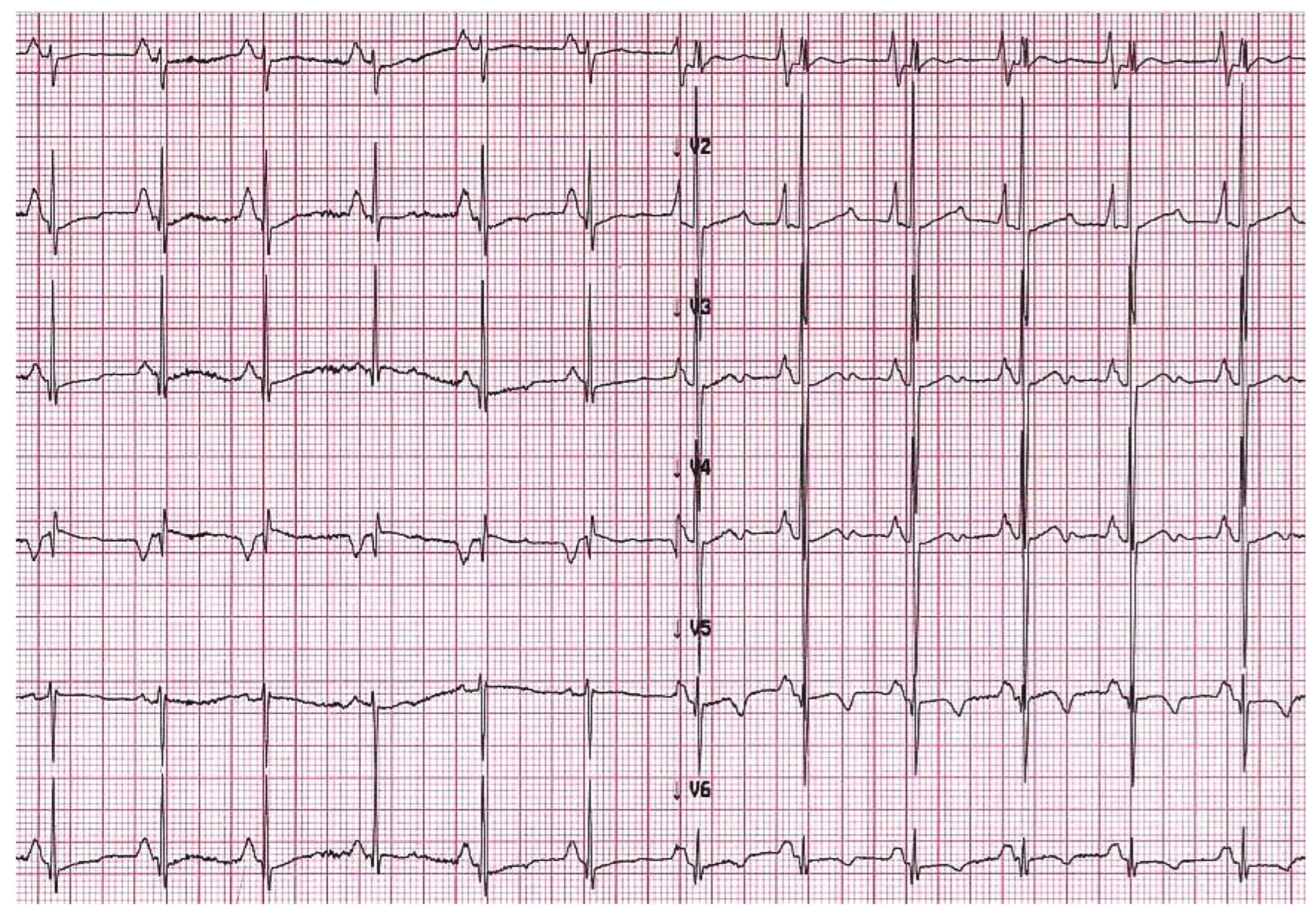
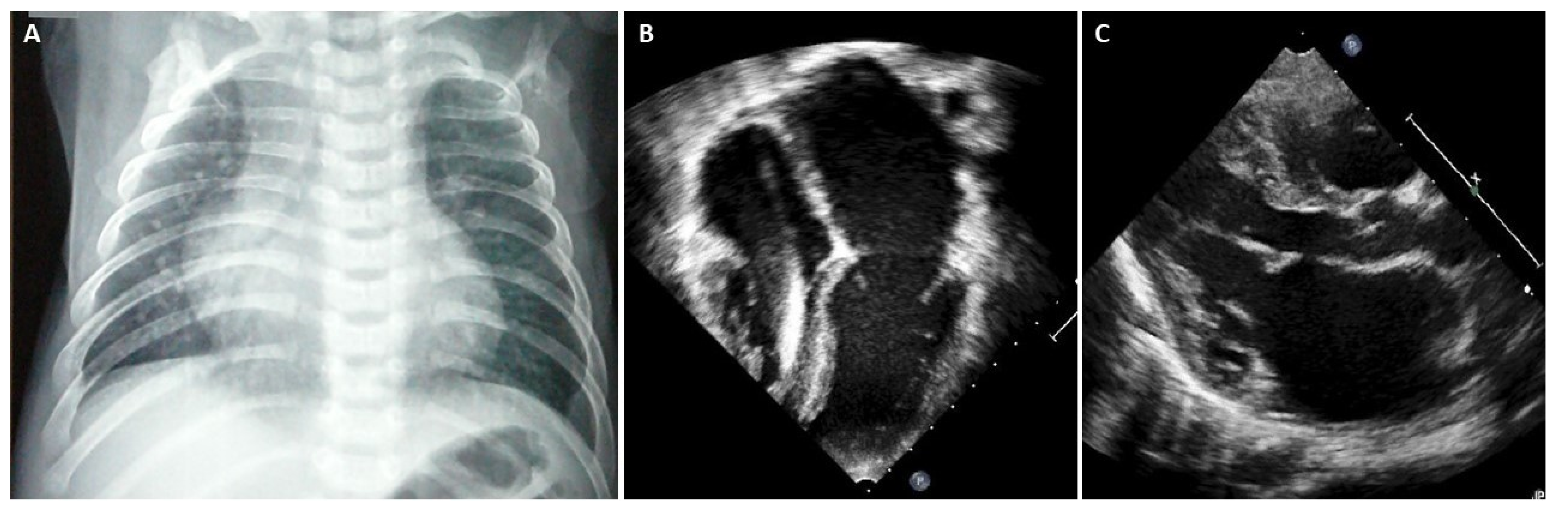
5. When Should I Suspect Restrictive Phenotype on Electrocardiogram or Imaging Techniques?
6. What Are the Limitations of ECG and Imaging Techniques?
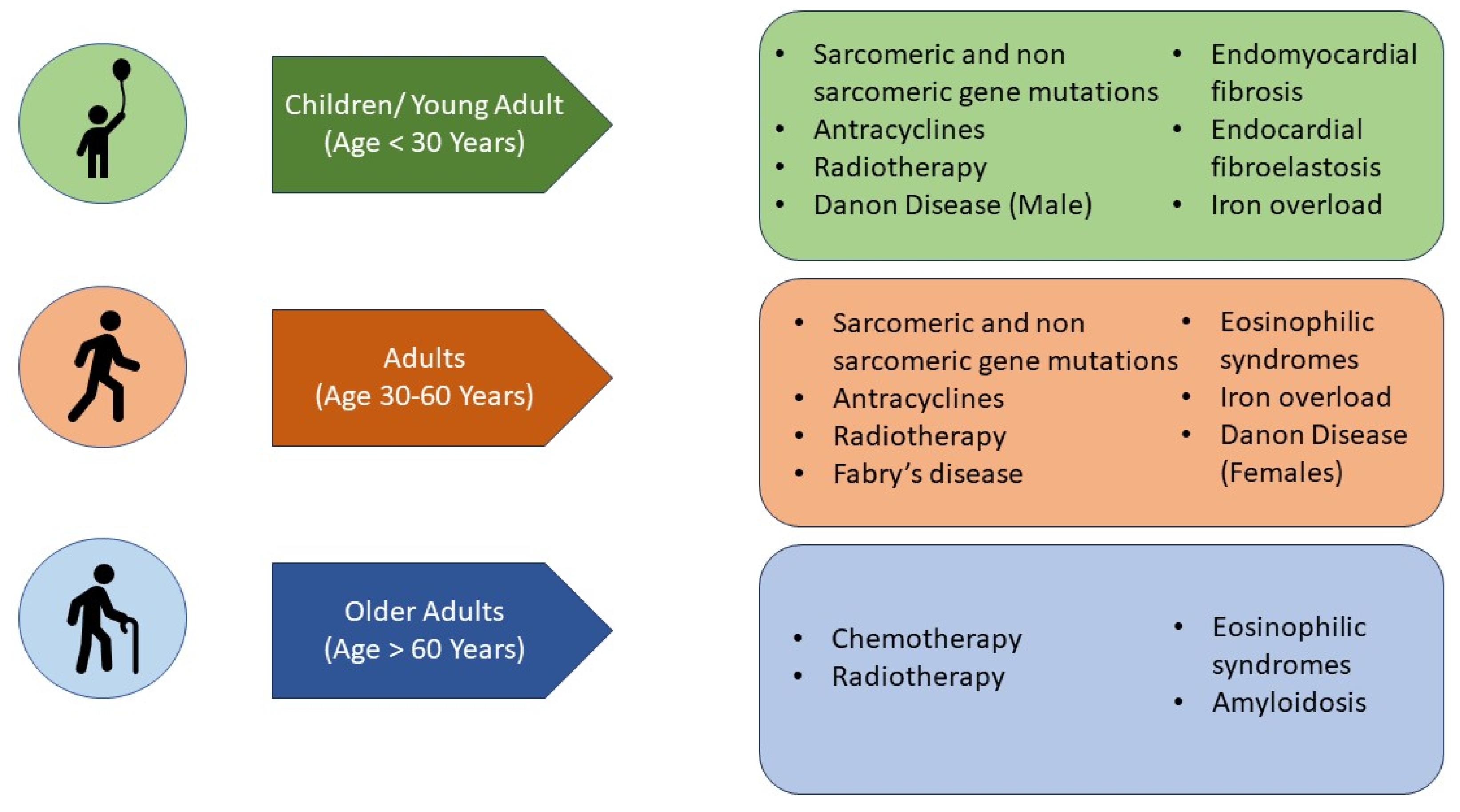
7. What Are the Aetiologies That Mainly Affect the Paediatric Population (Figure 4)?

8. What Are the Aetiologies That Can Affect Both Paediatric and Adult Populations (Figure 4)?
9. Which Causes of Adult RCM May Be Identified (Preclinically) in the Paediatric Age (Figure 4)?
10. What Are the Main Haemodynamic Features?
11. When Endomyocardial Biopsy Is Useful?
12. What Is the Prognosis of RCM?
13. What Pharmacological Treatments Are Available for the Most Common Forms?
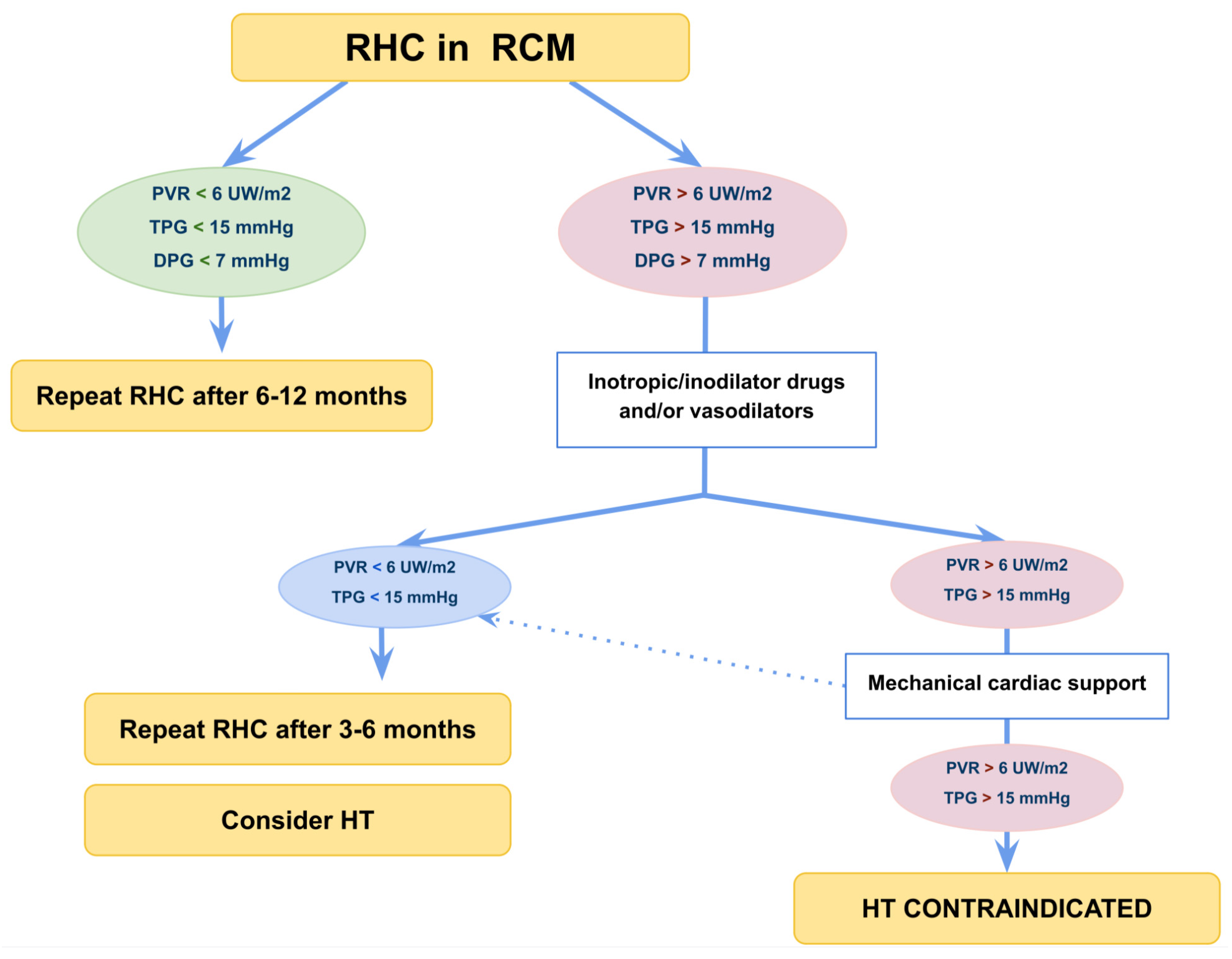
14. When Should I Refer Patients for Cardiac Hemodynamic Mechanical Support or Heart Transplantation?
15. Conclusions
Funding
Conflicts of Interest
References
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; De Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Rapezzi, C.; Aimo, A.; Barison, A.; Emdin, M.; Porcari, A.; Linhart, A.; Keren, A.; Merlo, M.; Sinagra, G. Restrictive cardiomyopathy: Definition and diagnosis. Eur. Heart J. 2022, 43, 4679–4693. [Google Scholar] [CrossRef] [PubMed]
- Denfield, S.W. Overview of pediatric restrictive cardiomyopathy—2021. Prog. Pediatr. Cardiol. 2021, 62, 101415. [Google Scholar] [CrossRef]
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef]
- Ditaranto, R.; Caponetti, A.G.; Ferrara, V.; Parisi, V.; Minnucci, M.; Chiti, C.; Baldassarre, R.; Di Nicola, F.; Bonetti, S.; Hasan, T.; et al. Pediatric Restrictive Cardiomyopathies. Front. Pediatr. 2021, 9, 745365. [Google Scholar] [CrossRef]
- Mocumbi, A.O.; Ferreira, M.B.; Sidi, D.; Yacoub, M.H. A population study of endomyocardial fibrosis in a rural area of Mozambique. N. Engl. J. Med. 2008, 359, 43–49. [Google Scholar] [CrossRef]
- Bukhman, G.; Ziegler, J.; Parry, E. Endomyocardial Fibrosis: Still a Mystery after 60 Years. PLoS Negl. Trop. Dis. 2008, 2, e97. [Google Scholar] [CrossRef] [PubMed]
- Webber, S.A.; Lipshultz, S.E.; Sleeper, L.A.; Lu, M.; Wilkinson, J.D.; Addonizio, L.J.; Canter, C.E.; Colan, S.D.; Everitt, M.D.; Jefferies, J.L.; et al. Outcomes of restrictive cardiomyopathy in childhood and the influence of phenotype: A report from the Pediatric Cardiomyopathy Registry. Circulation 2012, 126, 1237–1244. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef] [PubMed]
- Masarone, D.; Valente, F.; Rubino, M.; Vastarella, R.; Gravino, R.; Rea, A.; Russo, M.G.; Pacileo, G.; Limongelli, G. Pediatric Heart Failure: A Practical Guide to Diagnosis and Management. Pediatr. Neonatol. 2017, 58, 303–312. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Cochran, T.R.; Briston, D.A.; Brown, S.R.; Sambatakos, P.J.; Miller, T.L.; Carrillo, A.A.; Corcia, L.; Sanchez, J.E.; Diamond, M.B.; et al. Pediatric cardiomyopathies: Causes, epidemiology, clinical course, preventive strategies and therapies. Future Cardiol. 2013, 9, 817–848. [Google Scholar] [CrossRef] [PubMed]
- Quennelle, S.; Bonnet, D. Pediatric heart failure with preserved ejection fraction, a review. Front. Pediatr. 2023, 11, 1137853. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Garcia, M.; Ko, H.H.; Sharma, S.; Parness, I.A.; Srivastava, S. Applicability of published guidelines for assessment of left ventricular diastolic function in adults to children with restrictive cardiomyopathy: An observational study. Pediatr. Cardiol. 2015, 36, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Barretto, A.C.; Mady, C.; Nussbacher, A.; Ianni, B.M.; Oliveira, S.A.; Jatene, A.; Ramires, J.A. Atrial fibrillation in endomyocardial fibrosis is a marker of worse prognosis. Int. J. Cardiol. 1998, 67, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Muraji, S.; Sumitomo, N.; Imamura, T.; Yasuda, K.; Nishihara, E.; Iwamoto, M.; Tateno, S.; Doi, S.; Hata, T.; Kogaki, S.; et al. Diagnostic value of P-waves in children with idiopathic restrictive cardiomyopathy. Heart Vessel. 2021, 36, 1141–1150. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.D.; Madueme, P.C.; Jefferies, J.L.; Michelfelder, E.C.; Towbin, J.A.; Woo, J.G.; Sahay, R.D.; King, E.C.; Brown, R.; Moore, R.A.; et al. Utility of Echocardiography in the Assessment of Left Ventricular Diastolic Function and Restrictive Physiology in Children and Young Adults with Restrictive Cardiomyopathy: A Comparative Echocardiography-Catheterization Study. Pediatr. Cardiol. 2017, 38, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Denfield, S.W.; Webber, S.A. Restrictive Cardiomyopathy in Childhood. Heart Fail. Clin. 2010, 6, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Rosales, X.Q.; Moser, S.J.; Tran, T.; McCarthy, B.; Dunn, N.; Habib, P.; Simonetti, O.P.; Mendell, J.R.; Raman, S.V. Cardiovascular magnetic resonance of cardiomyopathy in limb girdle muscular dystrophy 2B and 2I. J. Cardiovasc. Magn. Reson. 2011, 13, 39. [Google Scholar] [CrossRef]
- de Ville de Goyet, M.; Brichard, B.; Robert, A.; Renard, L.; Veyckemans, F.; Vanhoutte, L.; Moniotte, S. Prospective cardiac MRI for the analysis of biventricular function in children undergoing cancer treatments. Pediatr. Blood Cancer 2015, 62, 867–874. [Google Scholar] [CrossRef]
- Axelsson Raja, A.; Farhad, H.; Valente, A.M.; Couce, J.-P.; Jefferies, J.L.; Bundgaard, H.; Zahka, K.; Lever, H.; Murphy, A.M.; Ashley, E.; et al. Prevalence and Progression of Late Gadolinium Enhancement in Children and Adolescents With Hypertrophic Cardiomyopathy. Circulation 2018, 138, 782–792. [Google Scholar] [CrossRef]
- Garcia, M.J. Constrictive Pericarditis Versus Restrictive Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 67, 2061–2076. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef]
- Habib, G.; Bucciarelli-Ducci, C.; Caforio, A.L.P.; Cardim, N.; Charron, P.; Cosyns, B.; Dehaene, A.; Derumeaux, G.; Donal, E.; Dweck, M.R.; et al. Multimodality Imaging in Restrictive Cardiomyopathies: An EACVI expert consensus document In collaboration with the “Working Group on myocardial and pericardial diseases” of the European Society of Cardiology Endorsed by The Indian Academy of Echocardiography. Eur. Heart J. Cardiovasc. Imaging 2017, 18, 1090–1121. [Google Scholar] [CrossRef]
- Rutakingirwa, M.; Ziegler, J.L.; Newton, R.; Freers, J. Poverty and eosinophilia are risk factors for endomyocardial fibrosis (EMF) in Uganda. Trop. Med. Int. Health 1999, 4, 229–235. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, F.P.; Azevedo, C.F. Comprehensive Assessment of Endomyocardial Fibrosis with Cardiac MRI: Morphology, Function, and Tissue Characterization. Radiographics 2020, 40, 336–353. [Google Scholar] [CrossRef]
- Ozdemir, D.; Cortopassi, I.O.; McNamara, R.L. An illustrative case of endocardial fibroelastosis and recalcitrant intracardiac thrombosis: A case report. Thromb. J. 2019, 17, 8. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.C.; Nghiem, Q.X. Cardiomyopathies in infants and children. Prog. Cardiovasc. Dis. 1972, 15, 255–287. [Google Scholar] [CrossRef]
- Weixler, V.; Hammer, P.E.; Marx, G.R.; Emani, S.M.; Del Nido, P.J.; Friehs, I. Flow disturbances and progression of endocardial fibroelastosis—A case report. Cardiovasc. Pathol. 2019, 42, 1–3. [Google Scholar] [CrossRef]
- Schryer, M.J.; Karnauchow, P.N. Endocardial fibroelastosis; etiologic and pathogenetic considerations in children. Am. Heart J. 1974, 88, 557–565. [Google Scholar] [CrossRef]
- Lane, K.L.; Herzberg, A.J.; Reimer, K.A.; Bradford, W.D.; Schall, S.A. Endocardial fibroelastosis with coronary artery thromboembolus and myocardial infarction. Clin. Pediatr. 1991, 30, 593–598. [Google Scholar] [CrossRef]
- Kaski, J.P.; Syrris, P.; Burch, M.; Tome-Esteban, M.-T.; Fenton, M.; Christiansen, M.; Andersen, P.S.; Sebire, N.; Ashworth, M.; Deanfield, J.E.; et al. Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 2008, 94, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Pasotti, M.; Pilotto, A.; Pellegrini, C.; Grasso, M.; Previtali, S.; Repetto, A.; Bellini, O.; Azan, G.; Scaffino, M.; et al. Desmin accumulation restrictive cardiomyopathy and atrioventricular block associated with desmin gene defects. Eur. J. Heart Fail. 2006, 8, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Ploski, R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. 2016, 170, 3241–3248. [Google Scholar] [CrossRef] [PubMed]
- Caleshu, C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rame, J.E. Furthering the link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. 2011, 155, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.R.; Parvatiyar, M.S.; Jones, M.A.; Liang, J.; Potter, J.D. A Troponin T Mutation That Causes Infantile Restrictive Cardiomyopathy Increases Ca2+ Sensitivity of Force Development and Impairs the Inhibitory Properties of Troponin. J. Biol. Chem. 2008, 283, 2156–2166. [Google Scholar] [CrossRef]
- Ware, S.; Quinn, M.; Ballard, E.; Miller, E.; Uzark, K.; Spicer, R. Pediatric restrictive cardiomyopathy associated with a mutation in β-myosin heavy chain. Clin. Genet. 2007, 73, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Bang, M.-L.; Centner, T.; Fornoff, F.; Geach, A.J.; Gotthardt, M.; McNabb, M.; Witt, C.C.; Labeit, D.; Gregorio, C.C.; Granzier, H.; et al. The Complete Gene Sequence of Titin, Expression of an Unusual ≈700-kDa Titin Isoform, and Its Interaction With Obscurin Identify a Novel Z-Line to I-Band Linking System. Circ. Res. 2001, 89, 1065–1072. [Google Scholar] [CrossRef]
- Linke, W.A.; Hamdani, N. Gigantic Business: Titin Properties and Function Through Thick and Thin. Circ. Res. 2014, 114, 1052–1068. [Google Scholar] [CrossRef]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.G.; Granzier, H.; Mestroni, L. A Review of the Giant Protein Titin in Clinical Molecular Diagnostics of Cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21. [Google Scholar] [CrossRef]
- Peled, Y.; Gramlich, M.; Yoskovitz, G.; Feinberg, M.S.; Afek, A.; Polak-Charcon, S.; Pras, E.; Sela, B.-A.; Konen, E.; Weissbrod, O.; et al. Titin Mutation in Familial Restrictive Cardiomyopathy. Int. J. Cardiol. 2014, 171, 24–30. [Google Scholar] [CrossRef]
- Herman, D.S.; Lam, L.; Taylor, M.R.G.; Wang, L.; Teekakirikul, P.; Christodoulou, D.; Conner, L.; DePalma, S.R.; McDonough, B.; Sparks, E.; et al. Truncations of titin causing dilated cardiomyopathy. N. Engl. J. Med. 2012, 366, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Purevjav, E.; Arimura, T.; Augustin, S.; Huby, A.-C.; Takagi, K.; Nunoda, S.; Kearney, D.L.; Taylor, M.D.; Terasaki, F.; Bos, J.M.; et al. Molecular basis for clinical heterogeneity in inherited cardiomyopathies due to myopalladin mutations. Hum. Mol. Genet. 2012, 21, 2039–2053. [Google Scholar] [CrossRef] [PubMed]
- Goldfarb, L.G.; Park, K.-Y.; Cervenáková, L.; Gorokhova, S.; Lee, H.-S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Hain, C.; Flottmann, F.; Ratnavadivel, S.; Gaertner, A.; Klauke, B.; Kalinowski, J.; Körperich, H.; Gummert, J.; Paluszkiewicz, L.; et al. The Desmin Mutation DES-c.735G>C Causes Severe Restrictive Cardiomyopathy by Inducing In-Frame Skipping of Exon-3. Biomedicines 2021, 9, 1400. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes 2019, 10, 918. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.-A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Santos-Mateo, J.J.; Fernández, A.; García-Hernández, S.; Ramos, K.A.; Piqueras-Flores, J.; Cabrera-Romero, E.; Barriales-Villa, R.; de la Higuera Romero, L.; et al. ROD2 domain filamin C missense mutations exhibit a distinctive cardiac phenotype with restrictive/hypertrophic cardiomyopathy and saw-tooth myocardium. Rev. Esp Cardiol. 2023, 76, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Girolami, F.; Passantino, S.; Ballerini, A.; Gozzini, A.; Porcedda, G.; Olivotto, I.; Favilli, S. Clinical Exome Sequencing Revealed a De Novo FLNC Mutation in a Child with Restrictive Cardiomyopathy. Cardiogenetics 2022, 12, 206–211. [Google Scholar] [CrossRef]
- Celeghin, R.; Cipriani, A.; Bariani, R.; Bueno Marinas, M.; Cason, M.; Bevilacqua, M.; De Gaspari, M.; Rizzo, S.; Rigato, I.; Da Pozzo, S.; et al. Filamin-C variant-associated cardiomyopathy: A pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022, 19, 235–243. [Google Scholar] [CrossRef]
- Gigli, M.; Stolfo, D.; Graw, S.L.; Merlo, M.; Gregorio, C.; Nee Chen, S.; Dal Ferro, M.; PaldinoMD, A.; De Angelis, G.; Brun, F.; et al. Phenotypic Expression, Natural History, and Risk Stratification of Cardiomyopathy Caused by Filamin C Truncating Variants. Circulation 2021, 144, 1600–1611. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Arad, M.; Maron, B.J.; Gorham, J.M.; Johnson, W.H.; Saul, J.P.; Perez-Atayde, A.R.; Spirito, P.; Wright, G.B.; Kanter, R.J.; Seidman, C.E.; et al. Glycogen Storage Diseases Presenting as Hypertrophic Cardiomyopathy. N. Engl. J. Med. 2005, 352, 362–372. [Google Scholar] [CrossRef]
- Rigolli, M.; Kahn, A.M.; Brambatti, M.; Contijoch, F.J.; Adler, E.D. Cardiac Magnetic Resonance Imaging in Danon Disease Cardiomyopathy. JACC Cardiovasc. Imaging 2021, 14, 514–516. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.J.; Oudit, G.Y. Iron-Overload Cardiomyopathy: Pathophysiology, Diagnosis, and Treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Khamseekaew, J.; Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. Effects of Iron Overload on Cardiac Calcium Regulation: Translational Insights Into Mechanisms and Management of a Global Epidemic. Can. J. Cardiol. 2016, 32, 1009–1016. [Google Scholar] [CrossRef]
- Walter, P.B.; Fung, E.B.; Killilea, D.W.; Jiang, Q.; Hudes, M.; Madden, J.; Porter, J.; Evans, P.; Vichinsky, E.; Harmatz, P. Oxidative stress and inflammation in iron-overloaded patients with ?-thalassaemia or sickle cell disease. Br. J. Haematol. 2006, 135, 254–263. [Google Scholar] [CrossRef]
- Oudit, G.Y.; Trivieri, M.G.; Khaper, N.; Liu, P.P.; Backx, P.H. Role of L-type Ca2+ channels in iron transport and iron-overload cardiomyopathy. J. Mol. Med. 2006, 84, 349–364. [Google Scholar] [CrossRef]
- Kremastinos, D.T.; Farmakis, D.; Aessopos, A.; Hahalis, G.; Hamodraka, E.; Tsiapras, D.; Keren, A. Beta-thalassemia cardiomyopathy: History, present considerations, and future perspectives. Circ. Heart Fail. 2010, 3, 451–458. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Scully, R.E.; Stevenson, K.E.; Franco, V.I.; Neuberg, D.S.; Colan, S.D.; Silverman, L.B.; Moslehi, J.J.; Cheng, S.; Sallan, S.E. Hearts too small for body size after doxorubicin for childhood ALL: Grinch syndrome. JCO 2014, 32 (Suppl. S15), 10021. [Google Scholar] [CrossRef]
- Trachtenberg, B.H.; Landy, D.C.; Franco, V.I.; Henkel, J.M.; Pearson, E.J.; Miller, T.L.; Lipshultz, S.E. Anthracycline-Associated Cardiotoxicity in Survivors of Childhood Cancer. Pediatr. Cardiol. 2011, 32, 342–353. [Google Scholar] [CrossRef]
- Lipshultz, S.E.; Lipsitz, S.R.; Sallan, S.E.; Dalton, V.M.; Mone, S.M.; Gelber, R.D.; Colan, S.D. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J. Clin. Oncol. 2005, 23, 2629–2636. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Amdani, S.; Lipshultz, E.R.; Lipshultz, S.E. Chemotherapy-induced cardiotoxicity in children. Expert. Opin. Drug Metab. Toxicol. 2017, 13, 817–832. [Google Scholar] [CrossRef] [PubMed]
- Kamphuis, J.A.M.; Linschoten, M.; Cramer, M.J.; Doevendans, P.A.; Asselbergs, F.W.; Teske, A.J. Early- and late anthracycline-induced cardiac dysfunction: Echocardiographic characterization and response to heart failure therapy. Cardiooncology 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Franco, V.I.; Miller, T.L.; Colan, S.D.; Sallan, S.E. Cardiovascular Disease in Adult Survivors of Childhood Cancer. Annu. Rev. Med. 2015, 66, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, P.A.; Hancock, S.L.; Lee, B.K.; Mariscal, C.S.; Schnittger, I. Asymptomatic cardiac disease following mediastinal irradiation. J. Am. Coll. Cardiol. 2003, 42, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Darby, S.C.; Cutter, D.J.; Boerma, M.; Constine, L.S.; Fajardo, L.F.; Kodama, K.; Mabuchi, K.; Marks, L.B.; Mettler, F.A.; Pierce, L.J.; et al. Radiation-related heart disease: Current knowledge and future prospects. Int. J. Radiat. Oncol. Biol. Phys. 2010, 76, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Velensek, V.; Mazic, U.; Krzisnik, C.; Demšar, D.; Jazbec, J.; Jereb, B. Cardiac damage after treatment of childhood cancer: A long-term follow-up. BMC Cancer 2008, 8, 141. [Google Scholar] [CrossRef]
- Erven, K.; Florian, A.; Slagmolen, P.; Sweldens, C.; Jurcut, R.; Wildiers, H.; Voigt, J.-U.; Weltens, C. Subclinical cardiotoxicity detected by strain rate imaging up to 14 months after breast radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1172–1178. [Google Scholar] [CrossRef]
- Hallahan, D.E.; Virudachalam, S. Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc. Natl. Acad. Sci. USA 1997, 94, 6432–6437. [Google Scholar] [CrossRef]
- Heidenreich, P.A.; Hancock, S.L.; Vagelos, R.H.; Lee, B.K.; Schnittger, I. Diastolic dysfunction after mediastinal irradiation. Am. Heart J. 2005, 150, 977–982. [Google Scholar] [CrossRef]
- Chang, H.-M.; Okwuosa, T.M.; Scarabelli, T.; Moudgil, R.; Yeh, E.T.H. Cardiovascular Complications of Cancer Therapy. J. Am. Coll. Cardiol. 2017, 70, 2552–2565. [Google Scholar] [CrossRef]
- Linhart, A.; Elliott, P.M. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F. Anderson-Fabry Disease and Other Lysosomal Storage Disorders. Circulation 2014, 130, 1081–1090. [Google Scholar] [CrossRef]
- Takenaka, T.; Teraguchi, H.; Yoshida, A.; Taguchi, S.; Ninomiya, K.; Umekita, Y.; Yoshida, H.; Horinouchi, M.; Tabata, K.; Yonezawa, S.; et al. Terminal stage cardiac findings in patients with cardiac Fabry disease: An electrocardiographic, echocardiographic, and autopsy study. J. Cardiol. 2008, 51, 50–59. [Google Scholar] [CrossRef]
- Linhart, A.; Germain, D.P.; Olivotto, I.; Akhtar, M.M.; Anastasakis, A.; Hughes, D.; Namdar, M.; Pieroni, M.; Hagège, A.; Cecchi, F.; et al. An expert consensus document on the management of cardiovascular manifestations of Fabry disease. Eur. J. Heart Fail. 2020, 22, 1076–1096. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Ortiz, A.; Abiose, A.; Bichet, D.G.; Cabrera, G.; Charrow, J.; Germain, D.P.; Hopkin, R.J.; Jovanovic, A.; Linhart, A.; Maruti, S.S.; et al. Time to treatment benefit for adult patients with Fabry disease receiving agalsidase β: Data from the Fabry Registry. J. Med. Genet. 2016, 53, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Séguéla, P.-E.; Iriart, X.; Acar, P.; Montaudon, M.; Roudaut, R.; Thambo, J.-B. Eosinophilic cardiac disease: Molecular, clinical and imaging aspects. Arch. Cardiovasc. Dis. 2015, 108, 258–268. [Google Scholar] [CrossRef]
- Zampieri, M.; Emmi, G.; Beltrami, M.; Fumagalli, C.; Urban, M.L.; Dei, L.-L.; Marchi, A.; Berteotti, M.; Tomberli, A.; Baldini, K.; et al. Cardiac involvement in eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): Prospective evaluation at a tertiary referral centre. Eur. J. Intern. Med. 2021, 85, 68–79. [Google Scholar] [CrossRef]
- Slungaard, A.; Vercellotti, G.M.; Tran, T.; Gleich, G.J.; Key, N.S. Eosinophil cationic granule proteins impair thrombomodulin function. A potential mechanism for thromboembolism in hypereosinophilic heart disease. J. Clin. Investig. 1993, 91, 1721–1730. [Google Scholar] [CrossRef]
- Ogbogu, P.U.; Rosing, D.R.; Horne, M.K. Cardiovascular Manifestations of Hypereosinophilic Syndromes. Immunol. Allergy Clin. N. Am. 2007, 27, 457–475. [Google Scholar] [CrossRef]
- Gottdiener, J.S.; Maron, B.J.; Schooley, R.T.; Harley, J.B.; Roberts, W.C.; Fauci, A.S. Two-dimensional echocardiographic assessment of the idiopathic hypereosinophilic syndrome. Anatomic basis of mitral regurgitation and peripheral embolization. Circulation 1983, 67, 572–578. [Google Scholar] [CrossRef]
- Anderson, L.; Pennell, D. The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Eur. Heart J. 2008, 29, 1696, author reply 1696–1697. [Google Scholar] [CrossRef]
- Limongelli, G.; Adorisio, R.; Baggio, C.; Bauce, B.; Biagini, E.; Castelletti, S.; Favilli, S.; Imazio, M.; Lioncino, M.; Merlo, M.; et al. Diagnosis and Management of Rare Cardiomyopathies in Adult and Paediatric Patients. A Position Paper of the Italian Society of Cardiology (SIC) and Italian Society of Paediatric Cardiology (SICP). Int. J. Cardiol. 2022, 357, 55–71. [Google Scholar] [CrossRef]
- Callegari, A.; Quandt, D.; Schmitz, A.; Klingel, K.; Balmer, C.; Dave, H.; Kretschmar, O.; Knirsch, W. Findings and Outcome of Transcatheter Right Ventricular Endomyocardial Biopsy and Hemodynamic Assessment in Children with Suspected Myocarditis or Cardiomyopathy. Int. J. Environ. Res. Public Health 2022, 19, 10406. [Google Scholar] [CrossRef] [PubMed]
- Mueller, G.C.; Michel-Behnke, I.; Knirsch, W.; Haas, N.A.; Abdul-Khaliq, H.; Gitter, R.; Dittrich, S.; Dähnert, I.; Uhlemann, F.; Schubert, S.; et al. Feasibility, safety and diagnostic impact of endomyocardial biopsies for the diagnosis of myocardial disease in children and adolescents. EuroIntervention 2018, 14, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Pophal, S.G.; Sigfusson, G.; Booth, K.L.; Bacanu, S.-A.; Webber, S.A.; Ettedgui, J.A.; Neches, W.H.; Park, S.C. Complications of endomyocardial biopsy in children. J. Am. Coll. Cardiol. 1999, 34, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Brighenti, M.; Donti, A.; Giulia Gagliardi, M.; Maschietto, N.; Marini, D.; Lombardi, M.; Vairo, U.; Agnoletti, G.; Milanesi, O.; Pongiglione, G.; et al. Endomyocardial biopsy safety and clinical yield in pediatric myocarditis: An Italian perspective: EMB in Pediatric Myocarditis. Cathet. Cardiovasc. Intervent. 2016, 87, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, N.; Spertus, J.A.; Kennedy, K.F.; Vincent, R.; Martin, G.R.; Curtis, J.P.; Nykanen, D.; Moore, P.M.; Bergersen, L. Modeling Major Adverse Outcomes of Pediatric and Adult Patients With Congenital Heart Disease Undergoing Cardiac Catheterization: Observations From the NCDR IMPACT Registry (National Cardiovascular Data Registry Improving Pediatric and Adult Congenital Treatment). Circulation 2017, 136, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kogaki, S.; Ishida, H.; Yoshikawa, T.; Shindo, T.; Inuzuka, R.; Furutani, Y.; Ishido, M.; Nakanishi, T. Outcomes of Restrictive Cardiomyopathy in Japanese Children―A Retrospective Cohort Study. Circ. J. 2022, 86, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Brunet-Garcia, L.; Roses-Noguer, F.; Betrián, P.; Balcells, J.; Gran, F. Restrictive cardiomyopathy: Importance of early diagnosis. An. Pediatría 2021, 95, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Wittekind, S.G.; Ryan, T.D.; Gao, Z.; Zafar, F.; Czosek, R.J.; Chin, C.W.; Jefferies, J.L. Contemporary Outcomes of Pediatric Restrictive Cardiomyopathy: A Single-Center Experience. Pediatr. Cardiol. 2019, 40, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Rivenes, S.M.; Kearney, D.L.; Smith, E.O.; Towbin, J.A.; Denfield, S.W. Sudden Death and Cardiovascular Collapse in Children With Restrictive Cardiomyopathy. Circulation 2000, 102, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Newland, D.M.; Law, Y.M.; Albers, E.L.; Friedland-Little, J.M.; Ahmed, H.; Kemna, M.S.; Hong, B.J. Early Clinical Experience with Dapagliflozin in Children with Heart Failure. Pediatr. Cardiol. 2023, 44, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Bogle, C.; Colan, S.D.; Miyamoto, S.D.; Choudhry, S.; Baez-Hernandez, N.; Brickler, M.M.; Feingold, B.; Lal, A.K.; Lee, T.M.; Canter, C.E.; et al. Treatment Strategies for Cardiomyopathy in Children: A Scientific Statement From the American Heart Association. Circulation 2023, 148, 174–195. [Google Scholar] [CrossRef] [PubMed]
- Rath, A.; Weintraub, R. Overview of Cardiomyopathies in Childhood. Front. Pediatr. 2021, 9, 708732. [Google Scholar] [CrossRef]
- Mehra, M.R.; Canter, C.E.; Hannan, M.M.; Semigran, M.J.; Uber, P.A.; Baran, D.A.; Danziger-Isakov, L.; Kirklin, J.K.; Kirk, R.; Kushwaha, S.S.; et al. The 2016 International Society for Heart Lung Transplantation listing criteria for heart transplantation: A 10-year update. J. Heart Lung Transplant. 2016, 35, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zangwill, S.D.; Naftel, D.; L’Ecuyer, T.; Rosenthal, D.; Robinson, B.; Kirklin, J.K.; Stendahl, G.; Dipchand, A.I. Pediatric Heart Transplant Study Investigators Outcomes of children with restrictive cardiomyopathy listed for heart transplant: A multi-institutional study. J. Heart Lung Transpl. 2009, 28, 1335–1340. [Google Scholar] [CrossRef]
- Fenton, M.J.; Chubb, H.; McMahon, A.M.; Rees, P.; Elliott, M.J.; Burch, M. Heart and heart-lung transplantation for idiopathic restrictive cardiomyopathy in children. Heart 2006, 92, 85–89. [Google Scholar] [CrossRef]
- Bograd, A.J.; Mital, S.; Schwarzenberger, J.C.; Mosca, R.S.; Quaegebeur, J.M.; Addonizio, L.J.; Hsu, D.T.; Lamour, J.M.; Chen, J.M. Twenty-year experience with heart transplantation for infants and children with restrictive cardiomyopathy: 1986–2006. Am. J. Transpl. 2008, 8, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Murtuza, B.; Fenton, M.; Burch, M.; Gupta, A.; Muthialu, N.; Elliott, M.J.; Hsia, T.-Y.; Tsang, V.T.; Kostolny, M. Pediatric heart transplantation for congenital and restrictive cardiomyopathy. Ann. Thorac. Surg. 2013, 95, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Hetzer, R.; Javier, M.F.D.M.; Delmo Walter, E.M. Role of paediatric assist device in bridge to transplant. Ann. Cardiothorac. Surg. 2018, 7, 82–98. [Google Scholar] [CrossRef] [PubMed]
- Su, J.A.; Menteer, J. Outcomes of Berlin Heart EXCOR® pediatric ventricular assist device support in patients with restrictive and hypertrophic cardiomyopathy. Pediatr. Transpl. 2017, 21, e13048. [Google Scholar] [CrossRef]
- Maeda, K.; Nasirov, T.; Yarlagadda, V.; Hollander, S.A.; Navarathnum, M.; Rosenthal, D.N.; Chen, S.; Almond, C.S.; Kaufman, B.D.; Reinhartz, O.; et al. Novel Trans-Septal Left Atrial VAD Cannulation Technique for Hypertrophic/Restrictive Cardiomyopathy. J. Heart Lung Transplant. 2019, 38, S479. [Google Scholar] [CrossRef]
- Lorts, A.; Conway, J.; Schweiger, M.; Adachi, I.; Amdani, S.; Auerbach, S.R.; Barr, C.; Bleiweis, M.S.; Blume, E.D.; Burstein, D.S.; et al. ISHLT consensus statement for the selection and management of pediatric and congenital heart disease patients on ventricular assist devices Endorsed by the American Heart Association. J. Heart Lung Transplant. 2021, 40, 709–732. [Google Scholar] [CrossRef] [PubMed]
- Weller, R.J.; Weintraub, R.; Addonizio, L.J.; Chrisant, M.R.K.; Gersony, W.M.; Hsu, D.T. Outcome of idiopathic restrictive cardiomyopathy in children. Am. J. Cardiol. 2002, 90, 501–506. [Google Scholar] [CrossRef]
- Hoskote, A.; Carter, C.; Rees, P.; Elliott, M.; Burch, M.; Brown, K. Acute right ventricular failure after pediatric cardiac transplant: Predictors and long-term outcome in current era of transplantation medicine. J. Thorac. Cardiovasc. Surg. 2010, 139, 146–153. [Google Scholar] [CrossRef]
- Salzberg, S.P.; Lachat, M.L.; Von Harbou, K.; Zünd, G.; Turina, M.I. Normalization of high pulmonary vascular resistance with LVAD support in heart transplantation candidates. Eur. J. Cardio-Thorac. Surg. 2005, 27, 222–225. [Google Scholar] [CrossRef]
- Gulati, G.; Ruthazer, R.; Denofrio, D.; Vest, A.R.; Kent, D.; Kiernan, M.S. Understanding Longitudinal Changes in Pulmonary Vascular Resistance After Left Ventricular Assist Device Implantation. J. Card. Fail. 2021, 27, 552–559. [Google Scholar] [CrossRef]
- Schlein, J.; Riebandt, J.; Laufer, G.; Zimpfer, D. Reversal of pulmonary hypertension in paediatric patients with restrictive cardiomyopathy. Interact. Cardiovasc. Thorac. Surg. 2021, 33, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Thangappan, K.; Morales, D.L.S.; Vu, Q.; Lehenbauer, D.; Villa, C.; Wittekind, S.; Hirsch, R.; Lorts, A.; Zafar, F. Impact of mechanical circulatory support on pediatric heart transplant candidates with elevated pulmonary vascular resistance. Artif. Organs 2021, 45, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zampieri, M.; Di Filippo, C.; Zocchi, C.; Fico, V.; Golinelli, C.; Spaziani, G.; Calabri, G.; Bennati, E.; Girolami, F.; Marchi, A.; et al. Focus on Paediatric Restrictive Cardiomyopathy: Frequently Asked Questions. Diagnostics 2023, 13, 3666. https://doi.org/10.3390/diagnostics13243666
Zampieri M, Di Filippo C, Zocchi C, Fico V, Golinelli C, Spaziani G, Calabri G, Bennati E, Girolami F, Marchi A, et al. Focus on Paediatric Restrictive Cardiomyopathy: Frequently Asked Questions. Diagnostics. 2023; 13(24):3666. https://doi.org/10.3390/diagnostics13243666
Chicago/Turabian StyleZampieri, Mattia, Chiara Di Filippo, Chiara Zocchi, Vera Fico, Cristina Golinelli, Gaia Spaziani, Giovanni Calabri, Elena Bennati, Francesca Girolami, Alberto Marchi, and et al. 2023. "Focus on Paediatric Restrictive Cardiomyopathy: Frequently Asked Questions" Diagnostics 13, no. 24: 3666. https://doi.org/10.3390/diagnostics13243666