
Mucopolysaccharidosis: What Pediatric Rheumatologists and Orthopedics Need to Know

1
Pediatric Rheumatology Unit, Gaetano Pini Hospital, 20122 Milan, Italy
2
Department of Clinical Sciences and Community Health, Research Center for Pediatric and Adult Rheumatic Diseases (RECAP.RD), University of Milan, 20122 Milan, Italy
*
Author to whom correspondence should be addressed.
Diagnostics 2023, 13(1), 75; https://doi.org/10.3390/diagnostics13010075
Submission received: 30 November 2022
/
Revised: 23 December 2022
/
Accepted: 23 December 2022
/
Published: 27 December 2022
(This article belongs to the Section Pathology and Molecular Diagnostics)
Abstract
:Mucopolysaccharidosis (MPS) is a group of disorders caused by the reduced or absent activity of enzymes involved in the glycosaminoglycans (GAGs) degradation; the consequence is the progressive accumulation of the substrate (dermatan, heparan, keratan or chondroitin sulfate) in the lysosomes of cells belonging to several tissues. The rarity, the broad spectrum of manifestations, the lack of strict genotype-phenotype association, and the progressive nature of MPS make diagnosing this group of conditions challenging. Musculoskeletal involvement represents a common and prominent feature of MPS. Joint and bone abnormalities might be the main clue for diagnosing MPS, especially in attenuated phenotypes; therefore, it is essential to increase the awareness of these conditions among the pediatric rheumatology and orthopedic communities since early diagnosis and treatment are crucial to reduce the disease burden of these patients. Nowadays, enzyme replacement therapy (ERT) and hematopoietic stem cell transplantation (HSCT) are available for some MPS types. We describe the musculoskeletal characteristics of MPS patients through a literature review of MPS cases misdiagnosed as having rheumatologic or orthopedic conditions.
1. Introduction
Mucopolysaccharidosis (MPS) are inherited lysosomal storage diseases caused by the deficiency of enzymes necessary for the degradation of glycosaminoglycans (GAGs) such as dermatan, heparan, keratan or chondroitin sulphate [1]. The incomplete degradation of GAGs leads to accumulation of these substrates in lysosomes in different tissues and organs in the body resulting in multi-organ overload and dysfunction [1,2]. The severity can range from disability in early childhood to milder forms with late onset [3]. Depending on residual enzymatic activity, the same mutation may lead to different phenotypes. MPS are commonly grouped in neuronopathic and somatic forms, based on the central nervous system (CNS). The type of accumulated GAG affects clinical manifestations; for example, accumulation of heparan sulfate (HS) induces CNS manifestations, while accumulation of keratan sulfate (KS) and dermatan sulfate (DS) induces corneal opacities, bone abnormalities and heart disease without neurological impairment [4]. All MPS are transmitted with an autosomal recessive trait, except for MPS type II, which is X-Linked recessive transmitted [1]. Genetic and demographic features of MPS are summarized in Table 1. The global incidence is estimated around 3.5 per 100,000 live births [5], but the prevalence varies between different countries. Nowadays, the real incidence of MPS is difficult to assess, also due to frequent misdiagnosis. Indeed, in the first phases of the disease the clinical picture may be incomplete and the development of the child is frequently normal [6]. In this scenario, early diagnosis is mandatory because the effectiveness of therapy depends not only on the type of enzyme deficiency but also on the stage of the disease [7]. Early intervention can improve outcomes for MPS patients [8], especially for patients eligible for hematopoietic stem cell transplantation (HSCT) or enzyme replacement therapy (ERT).
The aim of this review is to elucidate the characteristics of MPS and offer an overview of the possible differential diagnosis from a rheumatological and orthopedic point of view.
2. Pathogenesis and the Role of Inflammation
GAGs are linear sulfated chains, typically associated with proteins to form proteoglycans and represent a crucial component of the extracellular matrix (ECM). GAGs not only have a structural function, but they are also involved in many cellular processes, including cells adhesion, signal transduction and activation of specific inflammatory pathways [9,10]. Dysfunction of these enzymes leads to the accumulation of GAGs in the lysosome, and in extracellular tissues affecting cellular homeostasis and cross-talk [11]. In MPS I-II and VII, the defective enzyme leads to accumulation of both HS and DS, while in MPS III only HS catabolism is involved [12]. HS chains act as a cofactor in chemokines-ligands binding and sulphation state [13,14], and chain length [15], which can be impaired in MPS [16], seems to play a key role in regulating these interactions. Conversely, DS seems to induce chondrocyte apoptosis directly [9]. Finally, the etiology of KS accumulation damage in MPS IV is very poorly understood. In recent years, the hypothesis of an inflammatory mediated state consequent to GAGs accumulation has been extensively studied, opening up new treatment options. First of all, autophagy is a process mediated by lysosomes. A study on MPS mice demonstrated impaired pH homeostasis in lysosomes with a lower content of H+ associated with an increased cytosolic Ca+ suggesting an increase in lysosome membrane permeability and a secondary dysfunction of autophagy [17,18]. In this setting, a secondary mitochondrial dysfunction may cause a release of reactive oxygen species and reactive nitrogen species producing oxidative stress (Figure 1) that can contribute to the inflammatory process [19,20,21,22,23]. In MPS I, the presence of HS in ECM with abnormal sulfation patterns may trigger the inflammation attracting leukocytes [24,25,26]. Another possible mechanism of inflammation is the stimulation of Toll-like receptors (TLRs) by GAGs. Indeed, in response to tissue injury, the degradation of hyaluronan GAG stimulates TLR4 signaling and mediates a potent inflammatory response activating dendritic cells (DCs) and macrophages [27,28]. In animal models, synovial fibroblasts and fluid showed elevated expression of numerous inflammatory molecules (i.e., tumor necrosis factor), including cytosolic pattern recognition receptors (i.e., TLR) [28]. TLR4 signaling leads to the activation of NF-kB pathway and a subsequent release of cytokines as tumor necrosis factor α (TNF-α) [29]. There are many pieces of evidence to support this theory, such as an increased expression of TLR4 gene in MPS IIIA mouse brain [30] or an increase in TNF-α release in mouse microglia culture added with GAGs from MPS IIIB patients [31]. In addition to TNF-α signaling, IL-1β and IL-6 seem to play a pivotal role in the pathogenesis of MPS [31]. It appears that IL-1β mainly participates in CNS inflammation, while TNF-α is primarily involved in musculoskeletal (MSK) manifestations [32]. IL-1β is not secreted in active form and its activation is mediated by inflammasome proteolytic cleavage [33]. Recently, a two-step model for inflammasome activation has been described. NF-kB pathway promotes NLR family pyrin domain containing 3 (NLRP3) inflammasome transcription (priming step) and other conditions as defective autophagy, lysosomal vacuolation, mitochondrial dysfunction and oxidative stress may mediate the NLRP3 inflammasome assembly and activation (activation step) (Figure 2) [32,34].
ERT is the only approved medical treatment for MPS that aims to reduce GAGs synthesis, however, it has no effect on inflammation. In MPS VI rats, treatment with infliximab prevented the elevation of TNF-α and NF-kB signaling not only in the blood but also in articular chondrocytes and fibroblast-like synoviocytes [35]. In another study on MPS VI rats, TNF-α inhibitor combined with ERT showed an improvement in MSK outcome (particularly in motor activity and mobility) [36].
3. Musculoskeletal Manifestations in MPS
Even with a broad phenotypic spectrum, the involvement of bone, cartilage, ligaments, tendons, joint capsules, and all the soft tissues near the joints represents a common feature among all MPS types [6,37]. The timing and severity of such involvement are unpredictable, as in other organs impaired by GAG infiltration. Table 2 shows the most common MSK manifestations in MPS. Patients with attenuated forms of MPS might first seek medical attention for MSK complaints; thus, pediatric rheumatologists and orthopedic surgeons might be consulted as the first healthcare specialist. Unfortunately, mild MPS subjects might be frequently misdiagnosed as having other diseases such as juvenile idiopathic arthritis, osteochondrodysplasias, or other disorders (Table 3) [3,38,39,40,41].
3.1. Arthropathy and related issues
3.1.1. Joint Stiffness and Contractures
The impaired skeletal remodeling and the GAG infiltration of tissues around and within the joints are the causes of joint stiffness and contracture in all but one type of MPS (hypermobility is characteristic of MPS IV) [1,6,37]. Joint stiffness and contracture in MPS are progressive and present a symmetrical distribution with all joints potentially affected [3]. In MPS patients, the local and systemic signs of inflammation are absent (warmth, swelling, tenderness, fever, increased inflammatory markers). The classical claw hand deformity observed in MPS patients is due to skeletal abnormalities and contracture of interphalangeal (IP) joints with distal IP (DIP) more frequently involved. Furthermore, the bone enlargement near the joints in MPS may result in a swollen appearance mimicking arthritis. In this setting, juvenile idiopathic arthritis (JIA) represents a common differential diagnosis; however, several clues might help clinicians properly differentiate these two conditions (Table 4). Ultrasonography represents a useful tool in joint assessment of these patients since it can detect abnormal intraarticular material with peri-synovial Doppler signal but without effusions and a clear distribution to synovial recesses; flexor and extensor tendons of fingers might show a normal structure, whereas retinacula and flexor tendon pulleys might be thickened [42]. Sometimes the skin over the involved joint might become tight and thick, resembling the skin of scleroderma patients [37]. Alongside congenital arthrogryposis that is thought to result from decreased intrauterine movement [43], two other conditions might mimic the findings of MPS: camptodactyly and cheiroarthropathy. The flexion deformities of the proximal IP (PIP) joints of camptodactyly can be found as isolated features or as part of a syndrome such as camptodactyly-arthropathy-coxa vara-pericarditis (CACP) [43]. The pattern of affected joints (PIP in camptodactyly and DIP in MPS), and the involvement extension (the fifth finger in camptodactyly, whereas all fingers can be progressively affected in MPS) allow for discriminating between MPS and camptodactyly. Furthermore, dysostosis multiplex and other extra-articular features are absent in CACP and other conditions associated with camptodactyly. Cheiroarthropathy is typically associated with type I and type II diabetes; in this condition, PIP joints of the fourth and fifth fingers are involved, even though all the joints of the hand might be affected later on. These changes might be accompanied by skin tightening and thickening resembling scleroderma; indeed, cheiroarthropathy has been previously described as pseudoscleroderma [44]. The simultaneous presence of several MSK abnormalities (pes cavus, metatarsus adductus and equinovarus deformities, hip dysplasia, genu valgum) can affect the patient’s ability to walk [6,45]. Toe-walking can be observed in MPS patients when ankles and Achilles tendons are involved.
3.1.2. Joint Stiffness and Contractures
MPS IV (Morquio syndrome) is the only MPS type characterized by the presence of joint hypermobility due to several abnormalities of periarticular connective tissues and bones (bone hypoplasia and metaphyseal deformity) [46]. Proximal stiffness and distal hypermobility of the joints are the typical findings in MPS IV that may impact daily activity (e.g., dressing and personal care). Furthermore, these patients present a high risk of atlanto-axial subluxation caused by the concomitant presence of joint hypermobility and odontoid hypoplasia [37,47,48].
3.1.3. Trigger Digits and Carpal Tunnel Syndrome (CTS)
The GAG deposition in the flexor tendons and capsular tissues is responsible for the trigger finger. Triggering is usually a clinical diagnosis made upon the usual locking and catching symptoms along with ultrasound findings that help understand the underlying cause [49,50]. Indeed, inflammatory tenosynovitis may also provoke a discrepancy between the flexor tendon sheath’s size and the enclosing fibro-osseous canal, ultimately resulting in a trigger digit [51]. Since CTS is rare in children, the presence of this uncommon sign should raise the suspicion of a possible MPS, especially when associated with a trigger finger [52]. The thickening of the flexor retinaculum and tendon sheaths due to GAG deposition causes median nerve compression [53]. The early diagnosis of CTS in MPS patients might be challenging given the lack of typical complaints (numbness or pain) partially due to intellectual and verbal limitations of these subjects [3,54,55]. Data coming from an international registry on MPS I confirmed CTS as a common manifestation (30%); however, in the attenuated form, the latency between the CTS and MPS diagnosis resulted in more than 7 years [56]. Other experiences confirmed the CTS as common finding in mild diseases. In a case series reporting thirteen patients with attenuated MPS I, ten patients presented CTS, with two of them having concomitant triggering [3]. In another study, the electromyographic or nerve conduction velocity testing was used to diagnose CTS in 22 patients with MPS. Of the 17 subjects with CTS, 8 children (45 fingers) had trigger digits too [55].
3.2. Skeletal Involvement
The skeletal alterations seen in MPS and the related radiographic changes are regrouped under the term result from impairment in endochondral and membranous bone growth [9]. All bones of the body can be affected. Usually, long bones of the extremities show thickened diaphysis and hypoplastic epiphyses. The Madelung’s deformity with the distal radius curved towards a hypoplastic ulna, and the bullet shape metacarpals (short and thin with proximal tapering) are the typical alteration of upper limbs. Other abnormalities include thick calvarium, j-shaped sella turcica, enlargement of the skull, short and thickened clavicles, and oar-shaped ribs [37,45,57,58].
3.2.1. Kyphosis
Kyphosis might occur in all MPS types but is almost always present in severe forms of MPS I [59,60]. The kyphotic deformity is often due to hypoplastic vertebral bodies, especially in the anterior-superior part, and can be associated with scoliosis [45]. Even after hematopoietic stem cell transplant (HSCT), the progression of kyphosis often requires a surgical intervention despite conservative treatments such as strengthening activities and physical therapy [57]. It is reasonable to postpone surgery as long as possible both to allow for the maximum growth of the spine and for the consideration of the high risk of these procedures in MPS. However, if signs of myelopathy are detected, invasive procedures cannot be delayed. Young children who are not eligible for surgery may benefit from bracing [59].
3.2.2. Hip Dysplasia
Hip dysplasia in MPS comes from the variable combination of the flattened acetabulum, hypoplasia of the proximal epiphysis in its medial portion, and coxa valga. Therefore, children may develop several dysfunctions and limitations due to progressive instability and a higher risk of dislocation. Hip dysplasia affects several types of MPS (MPS I, MPS II, MPS III, MPS IV, and MPS VI), occurring in almost all subjects with Hurler syndrome (severe MPS I) [6]. Hip pain has been observed as presenting manifestation of MPS, and it can be easily misinterpreted [40,61]. In this regard, ultrasound may help in the diagnosis along with the whole clinical phenotype [62]. Hip surgery is deemed necessary in Hurler syndrome patients given its severity; on the contrary, it is not recommended for MPS III and IV due to the risk of osteonecrosis of the femur head [63,64,65].
3.2.3. Genu Valgum
Knee deformity is usually severe in MPS I, MPS IV, and MPS VI requiring surgery [46,66,67]. Total knee arthroplasty is necessary for adult MPS patients with severe arthrosis due to impairment of articular cartilage [68]. Eight-plate hemiepiphysiodeses at the distal femoral and/or proximal tibial physis were performed in 23 patients with MPS IV progressive or severe genu valgum with a mean follow-up of 44 months. The mean age at surgery was 8.3 years. Outcome measures documented a significative intermalleolar distance reduction without major complications (infections, implant failures, or loosening); three patients required a repeated procedure after a mean 22 months after plate removal [67]. Compared to osteotomy, hemiepiphysiodesis is less invasive and has a lower mobility impact.
3.2.4. Spinal Cord Compression
Hypoplasia of the odontoid process is frequently observed in MPS, especially in Hurler syndrome and Morquio syndrome. Odontoid hypoplasia is the leading cause of atlanto-axial instability predisposing to subluxation that may result in spinal cord compression in combination with GAG infiltration of the dura [69,70,71]. The resulting development of spastic tetraparesis is the most common finding; however, hemiparesis and paraparesis have also been documented [70,71]. HSCT and enzyme replacement therapy (ERT) seem to have a positive impact on spinal cord compression; however, larger studies are needed to verify this opportunity [72,73].
3.2.5. Consideration on Surgery in MPS
The treatment approach for skeletal abnormalities in MPS is mainly based on individual cases. However, severe and progressive deformities that may have serious consequences (e.g., mobility limitation, spinal cord compression) often require invasive procedures [69,74]. Patients with MPS have high risk of surgery for many reasons. Before surgical interventions, physicians should carefully assess spinal alterations such as neck stiffness, atlanto-axial instability, and cervical/occipital stenosis. Furthermore, endotracheal intubation can be challenging for these patients for several reasons: macroglossia, hypertrophy of the adenoids and tonsils, reduced mouth opening, and increased secretion. Fiberoptic intubation through a supraglottic airway seems associated with the lowest risk of perioperative adverse events and the lowest need for the transition to a rescue airway technique [75]. Other risks might lie in restrictive pulmonary disease, hypoplastic cartilages of the trachea, and cardiac involvement (mild to severe valvulitis and cardiomyopathy). Therefore, such patients need an accurate pre-operative assessment as well as a close post-operative observation also for minor procedures [45,57].
3.2.6. Short Stature
Growth impairment is a frequent manifestation of all MPS types and is caused by the alteration of the scheduled chondrocyte maturation in the growth plate [76]. The result is a non-harmonic short stature with axial development more compromised than appendicular growth [77]. The efficacy of recombinant growth hormone in these patients is variable, with a high rate of unresponsiveness. Final height can be severely impaired even though attenuated types might have a subtle decrease in growth velocity or even a normal maturation [78]. The slowing down of growth in untreated MPS I patients starts near 2 years of age and usually becomes evident by the age of 9 (below the third percentile) [79,80]. Data coming from the MPS I Registry documented a slower decline in height z-scores during laronidase ERT period compared to the natural history interval; however, the median height remained below normal standards overall [80,81]. An algorithm for early diagnosis of attenuated MPS I based on the primary observation of growth impairment in children above 5 years of age has been proposed [82].
3.3. Musculoskeletal Biomarkers
Nowadays, there are not validated biomarkers able to predict the progression of the disease or the response to therapy. TNF-α levels, regardless of ERT therapy, are higher in patients suffering from MPS when compared with healthy controls, and they are associated with MSK symptoms and limitations. However, elevated levels of TNF-α appear not to correlate with general health score [29]. In MPS, inflammation and direct chondrocyte apoptosis mediated by KS lead to osteoclasts, osteoblasts and chondrocytes dysfunction resulting in abnormal bone matrix deposition and failure of endochondral ossification [83]. In MPS IVA, KS accumulates in cartilage, leading to specific skeletal abnormalities. In these patients KS, KS sulfation levels, chondroitin-6-sulfate levels, and collagen type II are potential biomarkers associated with bone and cartilage disease [84]. Increased osteoblast activity may be suggested by elevated levels of osteocalcin in MPS patients [85]. In a study on MPS VII dogs, bone formation markers, such as alkaline phosphatase, osteopontin, and osteocalcin were lower in MPS than in controls suggesting a delay in bone formation [86].
On the other hand, elevated levels of IL-6 seem to predict progression in joint contracture, short stature, and hip dysplasia [87]. Simonaro et al. reported chondrocyte apoptosis in the articular cartilage of rats and cats with MPS VI. They found increased levels of IL-1, TNF- α, and nitric oxide in chondrocyte cultures of MPS animal model compared with normal cells [88]. A recent study on MPS I patients revealed elevated levels of IL-1β, TNF-α, osteocalcin, pyridinolines (PYD), and deoxypyridinolines in MPS patients compared to healthy controls. In addition, IL-6 and PYD levels appear to be associated with progression in joint contracture, short stature, and hip dysplasia [89]. Although the wide heterogeneity of MPS makes finding reliable markers challenging, cytokines and molecules related to the degradation of the ECM might be candidates of MSK biomarkers. Further studies are needed to validate specific and sensitive markers that can help in clinical practice.
3.4. Bone Status in MPS
Patients with MPS are theoretically at risk of low bone mass density (BMD) due to limited mobility secondary to bone dysplasia and contractures. The development of bone mass in children is a dynamic process of adapting to mechanical stimuli. Physical exercise is necessary to stimulate bone cells in order to reinforce bone structure and promote bone remodeling. Fractures after low energy trauma have been described in MPS patients but their incidence is low [90,91,92,93]. In a study on 40 MPS subjects, the prevalence of low BMD was 48% for lumbar vertebra, but the rate of DXA z-score < −2 reduced to 6% after correction for height-for-age z-score (HAZ). Indeed, a high percentage of patients with MPS present with short stature; therefore, DXA should be evaluated by adjusting for height (HAZ) [94]. A recent study on 126 MPS patients of various types found a short stature in 67.5%. Furthermore, 13.5% of patients were immobile, and 28.6% had 25(OH)D3 deficiency. In this cohort, BMD z score was < −2 in 40% of patients. However, after HAZ adjustment, only 2.2% had a BMD z score < −2 [95]. These data suggest the prevalence of low BMD in patients with MPS is probably overestimated and correction for HAZ is necessary due to abnormal bone development in MPS.
4. Other Manifestations of Attenuated MPS
The correlation between underlying mutations and residual enzymatic activity causes the broad phenotypic spectrum, impacting the timing of the manifestations’ development [1,3]. The inter- and intra-phenotypic variability of MPS makes the diagnosis of MPS challenging, especially for attenuated forms. The lack of neurological involvement along with the absence of manifest dysmorphisms such as coarse face and evident skeletal abnormalities may partially explain the frequent diagnosis delay [3]. Subtle disease progression and unspecific early symptoms might be unnoticed by healthcare providers unfamiliar with MPS. Alongside MSK involvement, late-onset MPS patients more frequently have eye- and heart-related issues [39]. Non MKS features are summarized in Table 5.
The most common ocular finding in MPS is corneal clouding which may severely impact visual acuity. It is due to the deposition of GAG granules with a yellowish-grey color in all corneal layers. MPS I and MPS VI have a more severe manifestation than MPS IV and MPS VII; on the other hand, patients with MPS II and III might present very mild corneal involvement [96]. Corneal transplantation is often needed in early adult patients; however, the concomitant presence of retinopathy represents a poor prognostic factor [97,98]. Glaucoma, increased intraocular pressure, optical nerve abnormalities, and retinal degeneration might be associated with corneal clouding, which usually begins around 10 years of age in attenuated MPS I [96,97,99,100]. Patients with MPS I, II, and III are more prone to develop retinal degeneration in adulthood, which is rarely seen in MPS IV [96].
Heart involvement is a common feature across all MPS types representing a significant cause of morbidity and mortality [101,102]. Patients with mild form frequently show valvular disease (thickening, regurgitation and/or stenosis) with mitral and aortic valves commonly affected [103]. The rapidity of the valvular disease progression is influenced by the accumulated GAG; indeed, those diseases characterized by dermatan and heparan sulfate deposition (MPS I, MPS II, and MPS VI) show a severe course of valvulopathy compared to types in which there is no such infiltration (MPS III and MPS IV) [103,104]. GAG storage may also cause left ventricular hypertrophy, coronary artery disease, abnormal diastolic function, arrhythmia, and complete heart block [104,105]. Furthermore, airways obstruction, rib deformity, and involvement of pulmonary vessels may result in pulmonary hypertension. ERT might slower down the valvular disease and ameliorate the ventricular hypertrophy [105,106].
Except for MPS III, cognitive impairment is uncommonly seen in the early stage of attenuated forms. Nevertheless, MPS I and II patients may develop variable degrees of intellectual disabilities during the disease course [39,107].
Among the other possible manifestations, the respiratory system is frequently involved (rhinosinusitis, otitis media, restrictive lung disease, sleep apnea, snoring) due to the GAG submucosal deposition causing airway obstruction, potentially, in every part of the respiratory tract [108]. Tracheomalacia, resulting in tracheal stenosis, might be life-threatening, especially in MPS II and IV, where this complication is more frequent. The first signs may arise during adolescence with further adult progression [109]. The GAG storage in the ear system causes conductive or sensorineural hearing loss, typically in the high-frequency range. Inguinal and umbilical hernias are quite common, especially in attenuated MPS I (till 65% of cases), and often may precede the onset of MSK manifestations [99,110]. Several degrees of hepatosplenomegaly and diarrhea are frequently observed in MPS [100,111].
5. Diagnosis
The accumulation of undegraded GAGs increases these substrates in urine, blood [112,113], and cerebral spinal fluid [114,115]. In children, the first best step in management is measuring the levels of urinary GAGs, as it is an excellent test in terms of sensitivity and specificity [116]. Quantitative analysis allows for the identification of an abnormal accumulation of urinary GAGs, while qualitative (electrophoresis or chromatography) analysis determines the type of accumulated GAGs. It is recommended that the first morning urine is collected because they are concentrated in order to avoid false negative results [117]. An elevated GAG concentration in urine is highly suggestive of MPS, but a negative test does not rule out the diagnosis [118]; in fact, a false negative test may occur in MPS III and IV [119,120] due to GAGs concentration declining with age. Definitive diagnosis requires enzyme activity assays, usually tested on peripheral blood leukocytes, and gene sequencing is necessary for identifying the underling mutation.
Prenatal diagnosis is based on measuring the enzyme activity in cultivated chorionic villi or amniocytes in case of a family history of MPS or hydrops fetalis. Testing for MPS I has been included in the United States newborn screening panel since 2016, and it is currently underway in other countries [121,122].
6. Conclusions
The suspicion of a possible diagnosis of MPS should rise based on the concomitant presence of some of the abovementioned manifestations. However, physicians may focus their attention to symptoms related to each own expertise and overlook other possible hints. In this regard the anamnesis plays a crucial role; a careful medical history of the patient and the family may reveal unexpected clues. A semi-structured medical history form has been proposed to help with medical interviews of suspected MPS patients [109]. Furthermore, the Cimaz algorithm, despite its countless variations, remains a milestone for the diagnosis of attenuated MPS with joint involvement [123]. MPS manifestations might be overlooked, carrying the risk of misdiagnosis. Therefore, awareness of MPS phenotypes should be raised, particularly among pediatric rheumatologists and orthopedic surgeons, since MSK involvement is common in attenuated forms. Alongside progressive joint contracture without inflammation, looking for other features in the medical history might avoid mistakes and delays in diagnosis. Once the suspicion of MPS is raised, urinary GAG is recommended, with enzyme assay and genetic testing as the following tests if an abnormal pattern is found.
Author Contributions
Conceptualization, A.M. and S.C.; validation, A.M., R.F.C.; writing—original draft preparation, A.M., S.C.; writing—review and editing, A.M., R.F.C.; supervision, A.M., R.F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
This manuscript is dedicated to the memory of Rolando Cimaz, an inspiring mentor and beloved friend.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muenzer, J. Overview of the Mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. 5), v4–v12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, K.L.; Flanigan, K.M. Update in the Mucopolysaccharidoses. Semin. Pediatr. Neurol. 2021, 37, 100874. [Google Scholar] [CrossRef] [PubMed]
- Cimaz, R.; Vijay, S.; Haase, C.; Coppa, G.V.; Bruni, S.; Wraith, E.; Guffon, N. Attenuated Type I Mucopolysaccharidosis in the Differential Diagnosis of Juvenile Idiopathic Arthritis: A Series of 13 Patients with Scheie Syndrome. Clin. Exp. Rheumatol. 2006, 24, 196–202. [Google Scholar] [PubMed]
- Michaud, M.; Belmatoug, N.; Catros, F.; Ancellin, S.; Touati, G.; Levade, T.; Gaches, F. Mucopolysaccharidosis: A review. Rev. Med. Interne 2020, 41, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Baehner, F.; Schmiedeskamp, C.; Krummenauer, F.; Miebach, E.; Bajbouj, M.; Whybra, C.; Kohlschütter, A.; Kampmann, C.; Beck, M. Cumulative Incidence Rates of the Mucopolysaccharidoses in Germany. J. Inherit. Metab. Dis. 2005, 28, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Cimaz, R.; La Torre, F. Mucopolysaccharidoses. Curr. Rheumatol. Rep. 2014, 16, 389. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.; Ellaway, C.; Foster, H.E.; Giugliani, R.; Goizet, C.; Goring, S.; Hawley, S.; Jurecki, E.; Khan, Z.; Lampe, C.; et al. Understanding the Early Presentation of Mucopolysaccharidoses Disorders. J. Inborn Errors Metab. Screen. 2018, 6, 2326409818800346. [Google Scholar] [CrossRef]
- Schulze-Frenking, G.; Jones, S.A.; Roberts, J.; Beck, M.; Wraith, J.E. Effects of Enzyme Replacement Therapy on Growth in Patients with Mucopolysaccharidosis Type II. J. Inherit. Metab. Dis. 2011, 34, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Clarke, L.A. Pathogenesis of Skeletal and Connective Tissue Involvement in the Mucopolysaccharidoses: Glycosaminoglycan Storage Is Merely the Instigator. Rheumatology 2011, 50 (Suppl. 5), v13–v18. [Google Scholar] [CrossRef] [Green Version]
- Opoka-Winiarska, V.; Jurecka, A.; Emeryk, A.; Tylki-Szymańska, A. Osteoimmunology in Mucopolysaccharidoses Type I, II, VI and VII. Immunological Regulation of the Osteoarticular System in the Course of Metabolic Inflammation. Osteoarthr. Cartil. 2013, 21, 1813–1823. [Google Scholar] [CrossRef]
- Mitrovic, S.; Gouze, H.; Gossec, L.; Schaeverbeke, T.; Fautrel, B. Mucopolysaccharidoses Seen in Adults in Rheumatology. Jt. Bone Spine 2017, 84, 663–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.-J.; Esko, J.D.; Crawford, B.E. Disease-Specific Non-Reducing End Carbohydrate Biomarkers for Mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan Sulfate Inhibits Hematopoietic Stem and Progenitor Cell Migration and Engraftment in Mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwyer, C.A.; Scudder, S.L.; Lin, Y.; Dozier, L.E.; Phan, D.; Allen, N.J.; Patrick, G.N.; Esko, J.D. Neurodevelopmental Changes in Excitatory Synaptic Structure and Function in the Cerebral Cortex of Sanfilippo Syndrome IIIA Mice. Sci. Rep. 2017, 7, 46576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and Their Interactions with Proteins. Chem. Biol. Drug. Des. 2008, 72, 455–482. [Google Scholar] [CrossRef]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñonuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis Type I, Unique Structure of Accumulated Heparan Sulfate and Increased N-Sulfotransferase Activity in Mice Lacking α-l-Iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef] [Green Version]
- Futerman, A.H.; van Meer, G. The Cell Biology of Lysosomal Storage Disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef]
- Pereira, V.G.; Gazarini, M.L.; Rodrigues, L.C.; da Silva, F.H.; Han, S.W.; Martins, A.M.; Tersariol, I.L.S.; D’Almeida, V. Evidence of Lysosomal Membrane Permeabilization in Mucopolysaccharidosis Type I: Rupture of Calcium and Proton Homeostasis. J. Cell Physiol. 2010, 223, 335–342. [Google Scholar] [CrossRef]
- Pereira, V.G.; Martins, A.M.; Micheletti, C.; D’Almeida, V. Mutational and Oxidative Stress Analysis in Patients with Mucopolysaccharidosis Type I Undergoing Enzyme Replacement Therapy. Clin. Chim. Acta 2008, 387, 75–79. [Google Scholar] [CrossRef]
- Filippon, L.; Vanzin, C.S.; Biancini, G.B.; Pereira, I.N.; Manfredini, V.; Sitta, A.; do Carmo R. Peralba, M.; Schwartz, I.V.D.; Giugliani, R.; Vargas, C.R. Oxidative Stress in Patients with Mucopolysaccharidosis Type II before and during Enzyme Replacement Therapy. Mol. Genet. Metab. 2011, 103, 121–127. [Google Scholar] [CrossRef]
- Arfi, A.; Richard, M.; Gandolphe, C.; Bonnefont-Rousselot, D.; Thérond, P.; Scherman, D. Neuroinflammatory and Oxidative Stress Phenomena in MPS IIIA Mouse Model: The Positive Effect of Long-Term Aspirin Treatment. Mol. Genet. Metab. 2011, 103, 18–25. [Google Scholar] [CrossRef]
- Zalfa, C.; Verpelli, C.; D’Avanzo, F.; Tomanin, R.; Vicidomini, C.; Cajola, L.; Manara, R.; Sala, C.; Scarpa, M.; Vescovi, A.L.; et al. Glial Degeneration with Oxidative Damage Drives Neuronal Demise in MPSII Disease. Cell Death Dis. 2016, 7, e2331. [Google Scholar] [CrossRef] [PubMed]
- Reolon, G.K.; Reinke, A.; de Oliveira, M.R.; Braga, L.M.; Camassola, M.; Andrades, M.E.; Moreira, J.C.F.; Nardi, N.B.; Roesler, R.; Dal-Pizzol, F. Alterations in Oxidative Markers in the Cerebellum and Peripheral Organs in MPS I Mice. Cell Mol. Neurobiol. 2009, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lever, R.; Page, C. Glycosaminoglycans, Airways Inflammation and Bronchial Hyperresponsiveness. Pulm. Pharmacol. Ther. 2001, 14, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Matsumoto, M.; Patel, S.; Lee, L.; Guan, J.-L.; Li, S. Role of Cell Surface Heparan Sulfate Proteoglycans in Endothelial Cell Migration and Mechanotransduction. J. Cell Physiol. 2005, 203, 166–176. [Google Scholar] [CrossRef]
- Hampe, C.S.; Eisengart, J.B.; Lund, T.C.; Orchard, P.J.; Swietlicka, M.; Wesley, J.; McIvor, R.S. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1838. [Google Scholar] [CrossRef]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan Activate Dendritic Cells via Toll-like Receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; D’Angelo, M.; He, X.; Eliyahu, E.; Shtraizent, N.; Haskins, M.E.; Schuchman, E.H. Mechanism of Glycosaminoglycan-Mediated Bone and Joint Disease: Implications for the Mucopolysaccharidoses and Other Connective Tissue Diseases. Am. J. Pathol. 2008, 172, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.E.; Vehe, R.K.; Rudser, K.; Kunin-Batson, A.; Utz, J.J.; Dickson, P.; Shapiro, E.; Whitley, C.B. Elevated TNF-α Is Associated with Pain and Physical Disability in Mucopolysaccharidosis Types I, II, and VI. Mol. Genet. Metab. 2016, 117, 427–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, H.; Ellison, S.M.; Holley, R.J.; O’Leary, C.; Liao, A.; Asadi, J.; Glover, E.; Ghosh, A.; Jones, S.; Wilkinson, F.L.; et al. Haematopoietic Stem Cell Gene Therapy with IL-1Ra Rescues Cognitive Loss in Mucopolysaccharidosis IIIA. EMBO Mol. Med. 2020, 12, e11185. [Google Scholar] [CrossRef]
- Ausseil, J.; Desmaris, N.; Bigou, S.; Attali, R.; Corbineau, S.; Vitry, S.; Parent, M.; Cheillan, D.; Fuller, M.; Maire, I.; et al. Early Neurodegeneration Progresses Independently of Microglial Activation by Heparan Sulfate in the Brain of Mucopolysaccharidosis IIIB Mice. PLoS ONE 2008, 3, e2296. [Google Scholar] [CrossRef] [PubMed]
- Mandolfo, O.; Parker, H.; Bigger, B. Innate Immunity in Mucopolysaccharide Diseases. Int. J. Mol. Sci. 2022, 23, 1999. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like Receptor 4 Pathway and Use of TNF-Alpha Antagonists for Treatment of the Mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Eliyahu, E.; Wolfson, T.; Ge, Y.; Jepsen, K.J.; Schuchman, E.H.; Simonaro, C.M. Anti-TNF-Alpha Therapy Enhances the Effects of Enzyme Replacement Therapy in Rats with Mucopolysaccharidosis Type VI. PLoS ONE 2011, 6, e22447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morishita, K.; Petty, R.E. Musculoskeletal Manifestations of Mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl 5), v19–v25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghadam, S.H.; Ghahvechi, M.; Mozafari, F.; Sayarifard, F.; Mousavi, M.-S.; Rostami, R.; Ziaee, V. Mucopolysaccharidosis Type I in Children, a Forgotten Diagnosis Responsible for Undiagnosed Musculoskeletal Complaints: Report of Two Cases. Acta Med. 2019, 62, 161–165. [Google Scholar] [CrossRef] [Green Version]
- Vijay, S.; Wraith, J.E. Clinical Presentation and Follow-up of Patients with the Attenuated Phenotype of Mucopolysaccharidosis Type I. Acta Paediatr. 2005, 94, 872–877. [Google Scholar] [CrossRef]
- Wiśniewska, K.; Wolski, J.; Gaffke, L.; Cyske, Z.; Pierzynowska, K.; Węgrzyn, G. Misdiagnosis in Mucopolysaccharidoses. J. Appl. Genet. 2022, 63, 475–495. [Google Scholar] [CrossRef]
- Chakraborty, P.P.; Biswas, S.N.; Ray, S.; Dey, S.K. Mucopolysaccharidosis Type I Disguised as Rickets. BMJ Case Rep. 2016, 2016, bcr2016215416. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Inbar-Feigenberg, M.; Raiman, J.; Bisch, M.; Chakraborty, P.; Mitchell, J.; Di Geso, L. Ultrasound Findings of Finger, Wrist and Knee Joints in Mucopolysaccharidosis Type I. Mol. Genet. Metab. 2021, 133, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Ty, J.M.; James, M.A. Failure of Differentiation: Part II (Arthrogryposis, Camptodactyly, Clinodactyly, Madelung Deformity, Trigger Finger, and Trigger Thumb). Hand Clin. 2009, 25, 195–213. [Google Scholar] [CrossRef]
- Del Rosso, A.; Cerinic, M.M.; De Giorgio, F.; Minari, C.; Rotella, C.M.; Seghieri, G. Rheumatological Manifestations in Diabetes Mellitus. Curr. Diabetes Rev. 2006, 2, 455–466. [Google Scholar] [CrossRef] [PubMed]
- White, K.K. Orthopaedic Aspects of Mucopolysaccharidoses. Rheumatology 2011, 50 (Suppl. 5), v26–v33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaño, A.M.; Tomatsu, S.; Gottesman, G.S.; Smith, M.; Orii, T. International Morquio a Registry: Clinical Manifestation and Natural Course of Morquio a Disease. J. Inherit. Metab. Dis. 2007, 30, 165–174. [Google Scholar] [CrossRef]
- Moon, E.; Lee, S.; Chong, S.; Park, J.H. Atlantoaxial Instability Treated with Free-Hand C1-C2 Fusion in a Child with Morquio Syndrome. Childs Nerv. Syst. 2020, 36, 1785–1789. [Google Scholar] [CrossRef]
- Northover, H.; Cowie, R.A.; Wraith, J.E. Mucopolysaccharidosis Type IVA (Morquio Syndrome): A Clinical Review. J. Inherit. Metab. Dis. 1996, 19, 357–365. [Google Scholar] [CrossRef]
- Welman, T.; Young, K.; Larkin, J.; Horwitz, M.D. Trigger Finger from Ocean Rowing: An Observational Study. Hand 2022, 17, 254–260. [Google Scholar] [CrossRef]
- Kim, H.-R.; Lee, S.-H. Ultrasonographic Assessment of Clinically Diagnosed Trigger Fingers. Rheumatol. Int. 2010, 30, 1455–1458. [Google Scholar] [CrossRef]
- Ryzewicz, M.; Wolf, J.M. Trigger Digits: Principles, Management, and Complications. J. Hand Surg. Am. 2006, 31, 135–146. [Google Scholar] [CrossRef]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I Mucopolysaccharidosis I: Management and Treatment Guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- al-Qattan, M.M.; Thomson, H.G.; Clarke, H.M. Carpal Tunnel Syndrome in Children and Adolescents with No History of Trauma. J. Hand Surg. Br. 1996, 21, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Yuen, A.; Dowling, G.; Johnstone, B.; Kornberg, A.; Coombs, C. Carpal Tunnel Syndrome in Children with Mucopolysaccaridoses. J. Child Neurol. 2007, 22, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Van Heest, A.E.; House, J.; Krivit, W.; Walker, K. Surgical Treatment of Carpal Tunnel Syndrome and Trigger Digits in Children with Mucopolysaccharide Storage Disorders. J. Hand Surg. Am. 1998, 23, 236–243. [Google Scholar] [CrossRef]
- Viskochil, D.; Muenzer, J.; Guffon, N.; Garin, C.; Munoz-Rojas, M.V.; Moy, K.A.; Hutchinson, D.T. Carpal Tunnel Syndrome in Mucopolysaccharidosis I: A Registry-Based Cohort Study. Dev. Med. Child Neurol. 2017, 59, 1269–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ponti, G.; Donsante, S.; Frigeni, M.; Pievani, A.; Corsi, A.; Bernardo, M.E.; Riminucci, M.; Serafini, M. MPSI Manifestations and Treatment Outcome: Skeletal Focus. Int. J. Mol. Sci. 2022, 23, 11168. [Google Scholar] [CrossRef]
- Chen, S.J.; Li, Y.W.; Wang, T.R.; Hsu, J.C. Bony Changes in Common Mucopolysaccharidoses. Acta Paediatr. Sin. 1996, 37, 178–184. [Google Scholar]
- Tandon, V.; Williamson, J.B.; Cowie, R.A.; Wraith, J.E. Spinal Problems in Mucopolysaccharidosis I (Hurler Syndrome). J. Bone Jt. Surg. Br. 1996, 78, 938–944. [Google Scholar] [CrossRef]
- Weisstein, J.S.; Delgado, E.; Steinbach, L.S.; Hart, K.; Packman, S. Musculoskeletal Manifestations of Hurler Syndrome: Long-Term Follow-up after Bone Marrow Transplantation. J. Pediatr. Orthop. 2004, 24, 97–101. [Google Scholar] [CrossRef]
- Chakraborty, P.P.; Patra, S.; Biswas, S.N.; Barman, H. Attenuated Form of Type II Mucopolysaccharidoses (Hunter Syndrome): Pitfalls and Potential Clues in Diagnosis. BMJ Case. Rep. 2018, 2018, bcr2018224392. [Google Scholar] [CrossRef] [PubMed]
- Żuber, Z.; Jurecka, A.; Różdżyńska-Świątkowska, A.; Migas-Majoch, A.; Lembas, A.; Kieć-Wilk, B.; Tylki-Szymańska, A. Ultrasonographic Features of Hip Joints in Mucopolysaccharidoses Type I and II. PLoS ONE 2015, 10, e0123792. [Google Scholar] [CrossRef] [PubMed]
- White, K.K.; Steinman, S.; Mubarak, S.J. Cervical Stenosis and Spastic Quadriparesis in Morquio Disease (MPS IV). A Case Report with Twenty-Six-Year Follow-Up. J. Bone Jt. Surg. Am. 2009, 91, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Yasunaga, Y.; Ikuta, Y.; Harada, A.; Kusaka, O.; Sukegawa, K. Femoral Head Dysplasia in Morquio Disease Type A: Bilateral Varus Osteotomy of the Femur. Acta Orthop. Scand. 2001, 72, 18–21. [Google Scholar] [CrossRef]
- Taylor, C.; Brady, P.; O’Meara, A.; Moore, D.; Dowling, F.; Fogarty, E. Mobility in Hurler Syndrome. J. Pediatr. Orthop. 2008, 28, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Odunusi, E.; Peters, C.; Krivit, W.; Ogilvie, J. Genu Valgum Deformity in Hurler Syndrome after Hematopoietic Stem Cell Transplantation: Correction by Surgical Intervention. J. Pediatr. Orthop. 1999, 19, 270–274. [Google Scholar] [CrossRef]
- Cooper, G.A.; Southorn, T.; Eastwood, D.M.; Bache, C.E. Lower Extremity Deformity Management in MPS IVA, Morquio-Brailsford Syndrome: Preliminary Report of Hemiepiphysiodesis Correction of Genu Valgum. J. Pediatr. Orthop. 2016, 36, 376–381. [Google Scholar] [CrossRef]
- De Waal Malefijt, M.C.; van Kampen, A.; van Gemund, J.J. Total Knee Arthroplasty in Patients with Inherited Dwarfism—A Report of Five Knee Replacements in Two Patients with Morquio’s Disease Type A and One with Spondylo-Epiphyseal Dysplasia. Arch. Orthop. Trauma Surg. 2000, 120, 179–182. [Google Scholar] [CrossRef]
- Pauchard, N.; Garin, C.; Jouve, J.L.; Lascombes, P.; Journeau, P. Perioperative Medullary Complications in Spinal and Extra-Spinal Surgery in Mucopolysaccharidosis: A Case Series of Three Patients. JIMD Rep. 2014, 16, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Kachur, E.; Del Maestro, R. Mucopolysaccharidoses and Spinal Cord Compression: Case Report and Review of the Literature with Implications of Bone Marrow Transplantation. Neurosurgery 2000, 47, 223–228. [Google Scholar] [CrossRef]
- Paulson, G.W.; Meagher, J.N.; Burkhart, J. Spinal Pachymeningitis Secondary to Mucopolysaccharidosis. Case Report. J. Neurosurg. 1974, 41, 618–621. [Google Scholar] [CrossRef] [PubMed]
- Peck, S.H.; Casal, M.L.; Malhotra, N.R.; Ficicioglu, C.; Smith, L.J. Pathogenesis and Treatment of Spine Disease in the Mucopolysaccharidoses. Mol. Genet Metab. 2016, 118, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, G.; Maximova, N.; Zennaro, F.; Gregori, M.; Tamaro, P. Hematopoietic Stem Cell Transplantation Effects on Spinal Cord Compression in Hurler. Pediatr. Transplant. 2014, 18, E96–E99. [Google Scholar] [CrossRef]
- Hofmann, A.; Heyde, C.-E.; Völker, A.; Schumann, E.; Heinz von der Höh, N. Treatment of Severe Kyphoscoliosis in Children with Mucopolysaccharidosis Type I (Pfaundler-Hurler Syndrome) Using the Growing Rod Technique: A Case Series with Mid-Term Results. World Neurosurg. 2020, 139, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, T.; Muschol, N.M.; Sehner, S.; Punke, M.A.; Haas, S.A.; Roeher, K.; Breyer, S.; Koehn, A.F.; Ullrich, K.; Zöllner, C.; et al. Airway Management and Perioperative Adverse Events in Children with Mucopolysaccharidoses and Mucolipidoses: A Retrospective Cohort Study. Paediatr. Anaesth. 2020, 30, 181–190. [Google Scholar] [CrossRef]
- Silveri, C.P.; Kaplan, F.S.; Fallon, M.D.; Bayever, E.; August, C.S. Hurler Syndrome with Special Reference to Histologic Abnormalities of the Growth Plate. Clin. Orthop. Relat. Res. 1991, 269, 305–311. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Sakkers, R.J.B.; Boelens, J.; de Koning, T.J.; Wulffraat, N.M. Musculoskeletal Manifestations of Lysosomal Storage Disorders. Ann. Rheum. Dis. 2009, 68, 1659–1665. [Google Scholar] [CrossRef]
- Polgreen, L.E.; Miller, B.S. Growth Patterns and the Use of Growth Hormone in the Mucopolysaccharidoses. J. Pediatr. Rehabil. Med. 2010, 3, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Guffon, N.; Journeau, P.; Brassier, A.; Leger, J.; Chevallier, B. Growth Impairment and Limited Range of Joint Motion in Children Should Raise Suspicion of an Attenuated Form of Mucopolysaccharidosis: Expert Opinion. Eur. J. Pediatr. 2019, 178, 593–603. [Google Scholar] [CrossRef] [Green Version]
- Viskochil, D.; Clarke, L.A.; Bay, L.; Keenan, H.; Muenzer, J.; Guffon, N. Growth Patterns for Untreated Individuals with MPS I: Report from the International MPS I Registry. Am. J. Med. Genet. A 2019, 179, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Polgreen, L.E.; Bay, L.; Clarke, L.A.; Guffon, N.; Jones, S.A.; Muenzer, J.; Flores, A.L.; Wilson, K.; Viskochil, D. Growth in Individuals with Attenuated Mucopolysaccharidosis Type I during Untreated and Treated Periods: Data from the MPS I Registry. Am. J. Med. Genet. A 2022, 188, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Baronio, F.; Zucchini, S.; Zulian, F.; Salerno, M.; Parini, R.; Cattoni, A.; Deodato, F.; Gaeta, A.; Bizzarri, C.; Gasperini, S.; et al. Proposal of an Algorithm to Early Detect Attenuated Type I Mucopolysaccharidosis (MPS Ia) among Children with Growth Abnormalities. Medicina 2022, 58, 97. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Utsunomiya, A. Bone Biomarkers in Mucopolysaccharidoses. Int. J. Mol. Sci. 2021, 22, 12651. [Google Scholar] [CrossRef]
- De Franceschi, L.; Roseti, L.; Desando, G.; Facchini, A.; Grigolo, B. A Molecular and Histological Characterization of Cartilage from Patients with Morquio Syndrome. Osteoarthr. Cartil. 2007, 15, 1311–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Byers, S.; Casal, M.L.; Smith, L.J. Failures of Endochondral Ossification in the Mucopolysaccharidoses. Curr. Osteoporos. Rep. 2020, 18, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Peck, S.H.; O’Donnell, P.J.M.; Kang, J.L.; Malhotra, N.R.; Dodge, G.R.; Pacifici, M.; Shore, E.M.; Haskins, M.E.; Smith, L.J. Delayed Hypertrophic Differentiation of Epiphyseal Chondrocytes Contributes to Failed Secondary Ossification in Mucopolysaccharidosis VII Dogs. Mol. Genet. Metab. 2015, 116, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Mills, P.; Davison, J.; Cleary, M.; Gissen, P.; Banushi, B.; Doykov, I.; Dorman, M.; Mills, K.; Heywood, W.E. Free Urinary Glycosylated Hydroxylysine as an Indicator of Altered Collagen Degradation in the Mucopolysaccharidoses. J. Inherit. Metab. Dis. 2020, 43, 309–317. [Google Scholar] [CrossRef]
- Simonaro, C.M.; D’Angelo, M.; Haskins, M.E.; Schuchman, E.H. Joint and Bone Disease in Mucopolysaccharidoses VI and VII: Identification of New Therapeutic Targets and Biomarkers Using Animal Models. Pediatr. Res 2005, 57, 701–707. [Google Scholar] [CrossRef] [Green Version]
- Lund, T.C.; Doherty, T.M.; Eisengart, J.B.; Freese, R.L.; Rudser, K.D.; Fung, E.B.; Miller, B.S.; White, K.K.; Orchard, P.J.; Whitley, C.B.; et al. Biomarkers for Prediction of Skeletal Disease Progression in Mucopolysaccharidosis Type I. JIMD Rep. 2021, 58, 89–99. [Google Scholar] [CrossRef]
- Fung, E.B.; Johnson, J.A.; Madden, J.; Kim, T.; Harmatz, P. Bone Density Assessment in Patients with Mucopolysaccharidosis: A Preliminary Report from Patients with MPS II and VI. J. Pediatr. Rehabil. Med. 2010, 3, 13–23. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Shih, S.-C.; Chuang, C.-K.; Chen, M.-R.; Niu, D.-M.; Lin, S.-P. Assessment of Bone Mineral Density by Dual Energy X-Ray Absorptiometry in Patients with Mucopolysaccharidoses. Orphanet J. Rare. Dis. 2013, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Guiral, J.; Sanchez, J.M.; Gonzalez, M.A. Stress Fracture of the Femoral Neck in a Young Adult with Maroteaux-Lamy Syndrome. Acta Orthop. Belg. 1992, 58, 91–92. [Google Scholar] [PubMed]
- Ichikawa, T.; Nishimura, G.; Tsukune, Y.; Dezawa, A.; Miki, H. Progressive Bone Resorption after Pathological Fracture of the Femoral Neck in Hunter’s Syndrome. Pediatr. Radiol. 1999, 29, 914–916. [Google Scholar] [CrossRef] [PubMed]
- Polgreen, L.E.; Thomas, W.; Fung, E.; Viskochil, D.; Stevenson, D.A.; Steinberger, J.; Orchard, P.; Whitley, C.B.; Ensrud, K.E. Low Bone Mineral Content and Challenges in Interpretation of Dual-Energy X-ray Absorptiometry in Children with Mucopolysaccharidosis Types I, II, and VI. J. Clin. Densitom. 2014, 17, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kor, D.; Bulut, F.D.; Kılavuz, S.; Şeker Yılmaz, B.; Köşeci, B.; Kara, E.; Kaya, Ö.; Başaran, S.; Seydaoğlu, G.; Önenli Mungan, N. Evaluation of Bone Health in Patients with Mucopolysaccharidosis. J. Bone. Miner. Metab. 2022, 40, 498–507. [Google Scholar] [CrossRef]
- Del Longo, A.; Piozzi, E.; Schweizer, F. Ocular Features in Mucopolysaccharidosis: Diagnosis and Treatment. Ital. J. Pediatr. 2018, 44, 125. [Google Scholar] [CrossRef] [Green Version]
- Fenzl, C.R.; Teramoto, K.; Moshirfar, M. Ocular Manifestations and Management Recommendations of Lysosomal Storage Disorders I: Mucopolysaccharidoses. Clin. Ophthalmol. 2015, 9, 1633–1644. [Google Scholar] [CrossRef] [Green Version]
- Ashworth, J.L.; Biswas, S.; Wraith, E.; Lloyd, I.C. Mucopolysaccharidoses and the Eye. Surv. Ophthalmol. 2006, 51, 1–17. [Google Scholar] [CrossRef]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The Natural History of MPS I: Global Perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.A.; Beck, M.; Clarke, J.T.R.; Cox, G.F. Childhood Onset of Scheie Syndrome, the Attenuated Form of Mucopolysaccharidosis I. J. Inherit. Metab. Dis. 2010, 33, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Braunlin, E.; Wang, R. Cardiac Issues in Adults with the Mucopolysaccharidoses: Current Knowledge and Emerging Needs. Heart 2016, 102, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Boffi, L.; Russo, P.; Limongelli, G. Early Diagnosis and Management of Cardiac Manifestations in Mucopolysaccharidoses: A Practical Guide for Paediatric and Adult Cardiologists. Ital. J. Pediatr. 2018, 44, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The Natural Course and the Impact of Therapies of Cardiac Involvement in the Mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Moretto, A.; Bosatra, M.G.; Marchesini, L.; Tesoro, S. Anesthesiological Risks in Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 116. [Google Scholar] [CrossRef] [PubMed]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac Disease in Patients with Mucopolysaccharidosis: Presentation, Diagnosis and Management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [Green Version]
- Kampmann, C.; Lampe, C.; Whybra-Trümpler, C.; Wiethoff, C.M.; Mengel, E.; Arash, L.; Beck, M.; Miebach, E. Mucopolysaccharidosis VI: Cardiac Involvement and the Impact of Enzyme Replacement Therapy. J. Inherit. Metab. Dis. 2014, 37, 269–276. [Google Scholar] [CrossRef]
- Yund, B.; Rudser, K.; Ahmed, A.; Kovac, V.; Nestrasil, I.; Raiman, J.; Mamak, E.; Harmatz, P.; Steiner, R.; Lau, H.; et al. Cognitive, Medical, and Neuroimaging Characteristics of Attenuated Mucopolysaccharidosis Type II. Mol. Genet. Metab. 2015, 114, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Scarpa, M.; Tobin, M.J.; Ward, S.A.; Rapoport, D.M. Respiratory and Sleep Disorders in Mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef]
- Rigoldi, M.; Verrecchia, E.; Manna, R.; Mascia, M.T. Clinical Hints to Diagnosis of Attenuated Forms of Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 132. [Google Scholar] [CrossRef] [Green Version]
- Bruni, S.; Lavery, C.; Broomfield, A. The Diagnostic Journey of Patients with Mucopolysaccharidosis I: A Real-World Survey of Patient and Physician Experiences. Mol. Genet. Metab. Rep. 2016, 8, 67–73. [Google Scholar] [CrossRef]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early Diagnostic Signs in Infants and Children. Ital. J. Pediatr. 2018, 44, 133. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Okamura, K.; Maeda, H.; Taketani, T.; Castrillon, S.V.; Gutierrez, M.A.; Nishioka, T.; Fachel, A.A.; Orii, K.O.; Grubb, J.H.; et al. Keratan Sulphate Levels in Mucopolysaccharidoses and Mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 187–202. [Google Scholar] [CrossRef]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Peña, O.M.; Montaño, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan Sulfate Levels in Mucopolysaccharidoses and Mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Capece, G.; Cecio, A.; D’Auria, N.; Di Iorio, G.; Ronsisvalle, L.; Di Natale, P. Sanfilippo B Syndrome (MPS III B): Case Report with Analysis of CSF Mucopolysaccharides and Conjunctival Biopsy. J. Neurol. 1981, 225, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Rojas, M.-V.; Vieira, T.; Costa, R.; Fagondes, S.; John, A.; Jardim, L.B.; Vedolin, L.M.; Raymundo, M.; Dickson, P.I.; Kakkis, E.; et al. Intrathecal Enzyme Replacement Therapy in a Patient with Mucopolysaccharidosis Type I and Symptomatic Spinal Cord Compression. Am. J. Med. Genet. A 2008, 146A, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans Analysis in Blood and Urine of Patients with Mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E. Urine Analysis in the Diagnosis of Mucopolysaccharide Disorders. Ann. Clin. Biochem. 1998, 35, 207–225. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, K.; Janani, S.; Priya, S.; Elango, E.M.; Sundari, R.M. Diagnosis of Mucopolysaccharidoses: How to Avoid False Positives and False Negatives. Indian. J. Pediatr. 2004, 71, 29–32. [Google Scholar] [CrossRef]
- Longdon, K.; Pennock, C.A. Abnormal Keratan Sulphate Excretion. Ann. Clin. Biochem. 1979, 16, 152–154. [Google Scholar] [CrossRef]
- Linker, A.; Evans, L.R.; Langer, L.O. Morquio’s Disease and Mucopolysaccharide Excretion. J. Pediatr. 1970, 77, 1039–1047. [Google Scholar] [CrossRef]
- Clarke, L.A.; Atherton, A.M.; Burton, B.K.; Day-Salvatore, D.L.; Kaplan, P.; Leslie, N.D.; Scott, C.R.; Stockton, D.W.; Thomas, J.A.; Muenzer, J. Mucopolysaccharidosis Type I Newborn Screening: Best Practices for Diagnosis and Management. J. Pediatr. 2017, 182, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn Screening in Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 126. [Google Scholar] [CrossRef] [PubMed]
- Cimaz, R.; Coppa, G.V.; Koné-Paut, I.; Link, B.; Pastores, G.M.; Elorduy, M.R.; Spencer, C.; Thorne, C.; Wulffraat, N.; Manger, B. Joint Contractures in the Absence of Inflammation May Indicate Mucopolysaccharidosis. Pediatr. Rheumatol. Online J. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
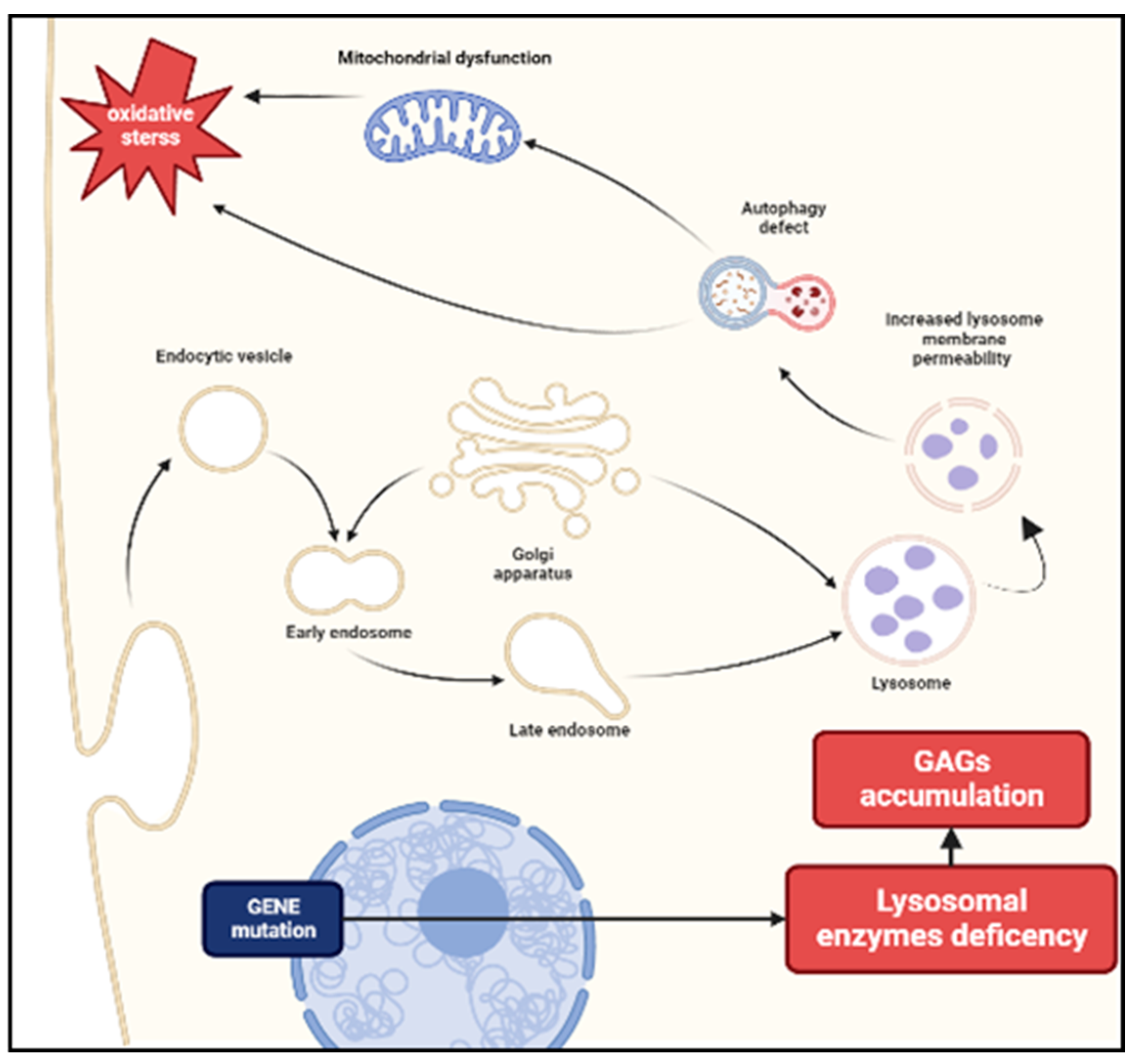
Figure 1.
Pathogenesis of MPS. The deficiency of enzymes necessary for the degradation of glycosaminoglycans (GAGs) leads to an accumulation of these substrates in lysosomes. Increased lysosomal membrane permeability influences cellular homeostasis and causes impaired vesicular fusion and autophagy. A secondary mitochondrial dysfunction may cause a release of reactive oxygen species resulting in oxidative stress.
Figure 1.
Pathogenesis of MPS. The deficiency of enzymes necessary for the degradation of glycosaminoglycans (GAGs) leads to an accumulation of these substrates in lysosomes. Increased lysosomal membrane permeability influences cellular homeostasis and causes impaired vesicular fusion and autophagy. A secondary mitochondrial dysfunction may cause a release of reactive oxygen species resulting in oxidative stress.

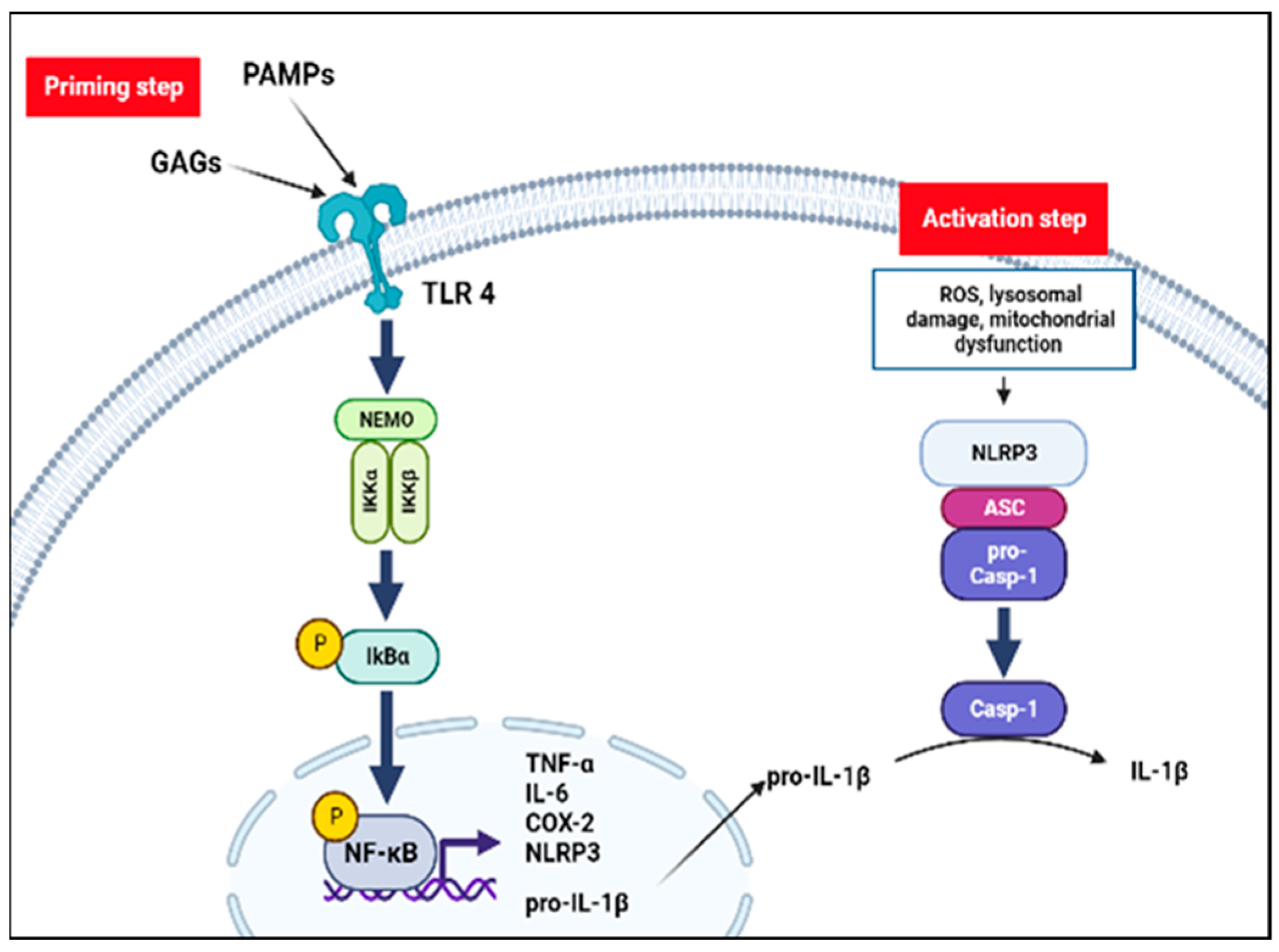
Figure 2.
The inflammatory hypothesis in MPS. Inflammasome activation requires two-step. Pathogen-associated molecular-patterns (PAMPs) and unfractionated glycosaminoglycans (GAGs) stimulate inflammatory processes through a Toll-like receptor 4 (TLR4) pathway: the inhibitory subunit of NF-kB alpha (IκBα) is phosphorylated by the NF-kappa-B essential modulator (NEMO) complex liberating NF-κB dimer. Translocation of NF-kB dimer to the nucleus promotes NOD-like receptors family pyrin domain containing 3 (NLRP3) and proinflammatory cytokines transcription (priming step). Lysosomal damage, mitochondrial dysfunction and oxidative stress may mediate the NLRP3 inflammasome assembly and activation (activation step). NLRP3 together with the adaptor apoptosis-associated speck-like protein containing a CARD (ASC) protein promotes the activation of caspase-1 and forms NLRP3 inflammasome complex. Finally, caspase-1 mediates conversion to the active form of IL-1β.
Figure 2.
The inflammatory hypothesis in MPS. Inflammasome activation requires two-step. Pathogen-associated molecular-patterns (PAMPs) and unfractionated glycosaminoglycans (GAGs) stimulate inflammatory processes through a Toll-like receptor 4 (TLR4) pathway: the inhibitory subunit of NF-kB alpha (IκBα) is phosphorylated by the NF-kappa-B essential modulator (NEMO) complex liberating NF-κB dimer. Translocation of NF-kB dimer to the nucleus promotes NOD-like receptors family pyrin domain containing 3 (NLRP3) and proinflammatory cytokines transcription (priming step). Lysosomal damage, mitochondrial dysfunction and oxidative stress may mediate the NLRP3 inflammasome assembly and activation (activation step). NLRP3 together with the adaptor apoptosis-associated speck-like protein containing a CARD (ASC) protein promotes the activation of caspase-1 and forms NLRP3 inflammasome complex. Finally, caspase-1 mediates conversion to the active form of IL-1β.


{kind=link}
{kind=link}
Table 1.
Genetical and demographical features of MPS.
| Disease | OMIM | Enzyme Deficiency | Gene (Locus) | Inheritance | Incidence (1/100,000 Live Births) | GAG | CNS | Approved Therapy |
|---|---|---|---|---|---|---|---|---|
| MPS I | α-L-Iduronidase | IDUA (4p16.3) | AR | 0.69–1.66 | Dermatan sulfate, heparan sulfate | + | Laronidase | |
| Type Hurler | 607,014 | +++ | ||||||
| Type Scheie | 607,016 | − | ||||||
| Type Hurler/Scheie | 607,016 | +/− | ||||||
| MPS II (Hunter syndrome) | 309,900 | Iduronate sulphate sulphatase | IDS (Xq28) | X-Linked recessive | 0.3–0.71 | Dermatan sulfate, heparan sulfate | + | Idrosulfase |
| MPS III (Sanfilippo syndrome) | AR | Heparan sulfate | +++ | NA | ||||
| Type III-A | 252,900 | Heparan-S-sulphate-sulphaminidase | SGSH (17q25.3) | 0.29–1.89 | ||||
| Type III-B | 252,920 | N-Acetyl-D-glucosaminidase | NAGLU (17q21.2) | 0.42–0.72 | ||||
| Type III-C | 252,930 | Acetyl-CoA-glucosaminide-N-acetyltransferase | HGSNAT (8p11.21) | 0.07–0.21 | ||||
| Type III-D | 252,940 | N-Acetylglucosaminine-6-sulphate-sulphatase | GNS (12q14.3) | 0.1 | ||||
| MPS IV (Morquio syndrome) | AR | 0.2–1.3 | − | |||||
| Type IV-A | 253,000 | Galactosamine-6-sulphate-sulphatase | GALNS (16q24.3) | 0.2–1.3 | A: Keratan sulfate, chondroitin sulfate | Elosulfase alpha | ||
| Type IV-B | 253,010 | β-galactosidase | GALNS (16q24.3) | 0.02–0.14 | B: Keratan sulfate | |||
| MPS VI (Maroteaux–Lamy syndrome) | 253,200 | Arylsulfatase B | ARSB (5q14.1) | AR | 0.36–1.3 | Dermatan sulfate, heparan sulfate | − | Galsulfase |
| MPS VII (Sly syndrome) | 253,220 | β-Glucuronidase | GUSB (7q11.21) | Automosal recessive | 0.05–0.29 | Dermatan sulfate, heparan sulfate, chondroitin sulfate | +/− | Vestronidase alpha |
| MPS IX (Hyaluronidase deficiency) | 601,492 | Hyaluronidase | Automosal recessive | <0.01 | Hyaluronan | − | NA | |
MPS = Mucopolysaccharidosis; OMIM = Online Mendelian Inheritance in Man; CNS = central nervous system; GAG = glycosaminoglycans; AR = autosomal recessive; NA = not available.
Table 2.
Musculoskeletal features of MPS.
| MPS Type | Musculoskeletal Features |
|---|---|
| MPS I (Hurler, Hruler-Scheie, Scheie) | Dysostosis multiplex, short stature (disproportionate), joint contractures, carpal tunnel syndrome, trigger digits, odontoid hypoplasia, atlanto-axial instability, acetabular dysplasia, coxa valga, genu valgum |
| MPS II (Hunter) | Dysostosis multiplex, short stature (disproportionate), joint contractures, carpal tunnel syndrome, trigger digits, odontoid hypoplasia, atlanto-axial instability, acetabular dysplasia, coxa valga, genu valgum |
| MPS III (Sanfilippo) | Mild short stature and contractures (mainly elbow joint) |
| MPS IV (Morquio) | Severe skeletal dysplasia, dysostosis multiplex, short stature (disproportionate), joint hypermobility, odontoid hypoplasia, atlanto-axial instability, acetabular dysplasia, hip dislocations, coxa valga, genu valgum, pes planus, pectus carinatum |
| MPS VI (Maroteaux-Lamy) | Dysostosis multiplex, short stature (disproportionate), joint contractures, carpal tunnel syndrome, odontoid hypoplasia, atlanto-axial instability, acetabular dysplasia, coxa valga, genu valgum, trigger digits, pectus carinatum |
| MPS VII (Sly) | Dysostosis multiplex, short stature (disproportionate), joint contractures, odontoid hypoplasia, atlanto-axial instability, acetabular dysplasia, pectus carinatum |
| MPS IX (Hyaluronidase deficiency) | Short stature, periarticular soft tissue masses, nodular synovial masses, joint effusions, acetabular erosions |
Table 3.
Differential diagnosis of musculoskeletal manifestations in MPS.
| Inflammatory diseases | Inflammatory arthritis Scleroderma Dermatomyositis and polymyositis |
| Distal extremity conditions | Arthrogryposis Camptodactyly Clinodactyly Trigger finger (isolated) Carpal tunnel syndrome (isolated) |
| Osteochondrodysplasias | Epiphyseal dysplasia Spondyloepiphyseal dysplasia congenital (including the X-linked form) Spondylometaphyseal dysplasia Dystrophic dysplasias Osteogenesis imperfecta Other dysplasias |
| Other metabolic diseases | Gaucher’s disease Fabry’s disease Pompe’s disease Rickets Hypophosphatasia Diabetic cheiroarthropathy |
| Miscellaneous | Legg-Perthes-Calvé disease Growing pains Amplified musculoskeletal pain syndrome (AMPS) Muscular dystrophy Polyneuropathy Ehler-Danlos syndrome |
Table 4.
MPS arthropathy vs. Juvenile idiopathic arthritis (JIA).
| Features of Joint Involvement | MPS | JIA |
|---|---|---|
| Involved joints | DIP | PIP and MCP |
| Stiffness temporal pattern | Continuous | Morning 1 |
| Clinical signs | Stiffness and contracture 2 | Joint swelling, warmth, and tenderness |
| Inflammatory markers | Normal | Normal/raised |
| Response to anti-inflammatory drugs 3 | No | Yes |
DIP: distal interphalangeal joints; PIP: proximal interphalangeal joints, MCP: metacarpophalangeal joints; 1 usually worse in the morning, exacerbated by rest, and relieved by activity; 2 some joints may have a swollen appearance, caused by underlying bony enlargement rather than synovial effusion; 3 steroidal and non-steroidal anti-inflammatory drugs.
Table 5.
Non musculoskeletal features of different type of MPS.
| Neurological | ENT | GI | Cardiological | Ophthalmological | Hepato-Splenomegaly | Skin Involvement | |
|---|---|---|---|---|---|---|---|
| MPS I H | Hydrocephalus Psychomotor retardation Behavior trouble Peripheral compression Atlanto-axial instability | Deafness (+++ H) Recurrent sinopulmonary infections Chronic rhinitis | Umbilical or inguinal hernias | Valve disease Cardiomyopathy Endocardial fibroelastosis Coronary heart disease | Corneal clouding Retinopathy Optical nerve compression | ++ | Thickened and rough skin texture Pebbly papules (rare) |
| MPS I S | - | - | ++ | ||||
| MPS I HS | Pachymeningitis cervicalis Typically normal intelligence | Umbilical or inguinal hernias | ++ | ||||
| MPS II | Neurocognitive decline Behavior trouble Some patients have normal intelligence | Deafness | - | Valve disease Cardiomyopathy Endocardial fibroelastosis Coronary heart disease | - | ++ | Pebbly papules |
| MPS III | Neurocognitive decline Behavior trouble Intellectual disability | Recurrent sinopulmonary infections Deafness | Umbilical or inguinal hernias | Milder forms of valve disease and Cardiomyopathy | - | + | Thickened and rough skin texture |
| MPS IV | - | Recurrent sinopulmonary infections Deafness | - | Milder forms of valve disease and Cardiomyopathy | Mild corneal opacities | + | Thickened and rough skin texture |
| MPS VI | Pachymeningitis cervicalis with normal intelligence | Recurrent sinopulmonary infections OSAS Pulmonary hypertension | - | Valve disease Cardiomyopathy Endocardial fibroelastosis Coronary heart disease | Corneal clouding | ++ | Thickened and rough skin texture |
| MPS VII | Hydrops fetalis Intellectual disability (mild or absent) | ++ | - | Milder forms of valve disease and Cardiomyopathy | Corneal clouding Retinopathy Optical nerve compression | + | Thickened and rough skin texture |
MPS = mucopolysaccharidoses; H = Hurler; S = Scheie; HS = Hurler-Scheie; GI = gastrointestinal; ENT = ear nose throat; OSAS = obstructive sleep apnea syndrome.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Costi, S.; Caporali, R.F.; Marino, A. Mucopolysaccharidosis: What Pediatric Rheumatologists and Orthopedics Need to Know. Diagnostics 2023, 13, 75. https://doi.org/10.3390/diagnostics13010075
AMA Style
Costi S, Caporali RF, Marino A. Mucopolysaccharidosis: What Pediatric Rheumatologists and Orthopedics Need to Know. Diagnostics. 2023; 13(1):75. https://doi.org/10.3390/diagnostics13010075
Chicago/Turabian StyleCosti, Stefania, Roberto Felice Caporali, and Achille Marino. 2023. "Mucopolysaccharidosis: What Pediatric Rheumatologists and Orthopedics Need to Know" Diagnostics 13, no. 1: 75. https://doi.org/10.3390/diagnostics13010075
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.