
Biomarker Candidates for Alzheimer’s Disease Unraveled through In Silico Differential Gene Expression Analysis

 ,
,  ,
,  ,
, 

 , and
, and 
Abstract
:1. Introduction
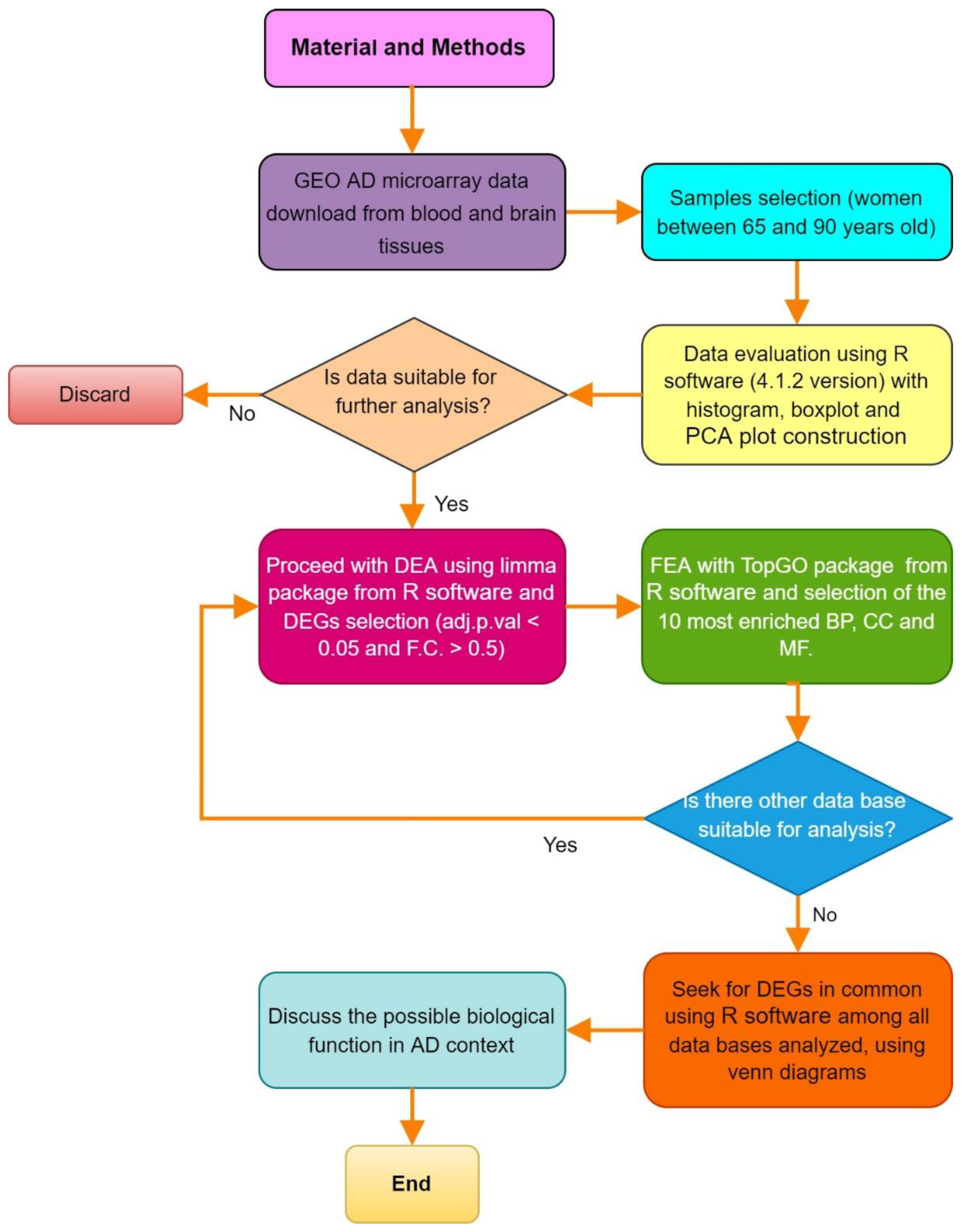
2. Materials and Methods
2.1. Microarray Processing
2.2. Differential Expression Analysis
2.3. Functional Enrichment Analysis
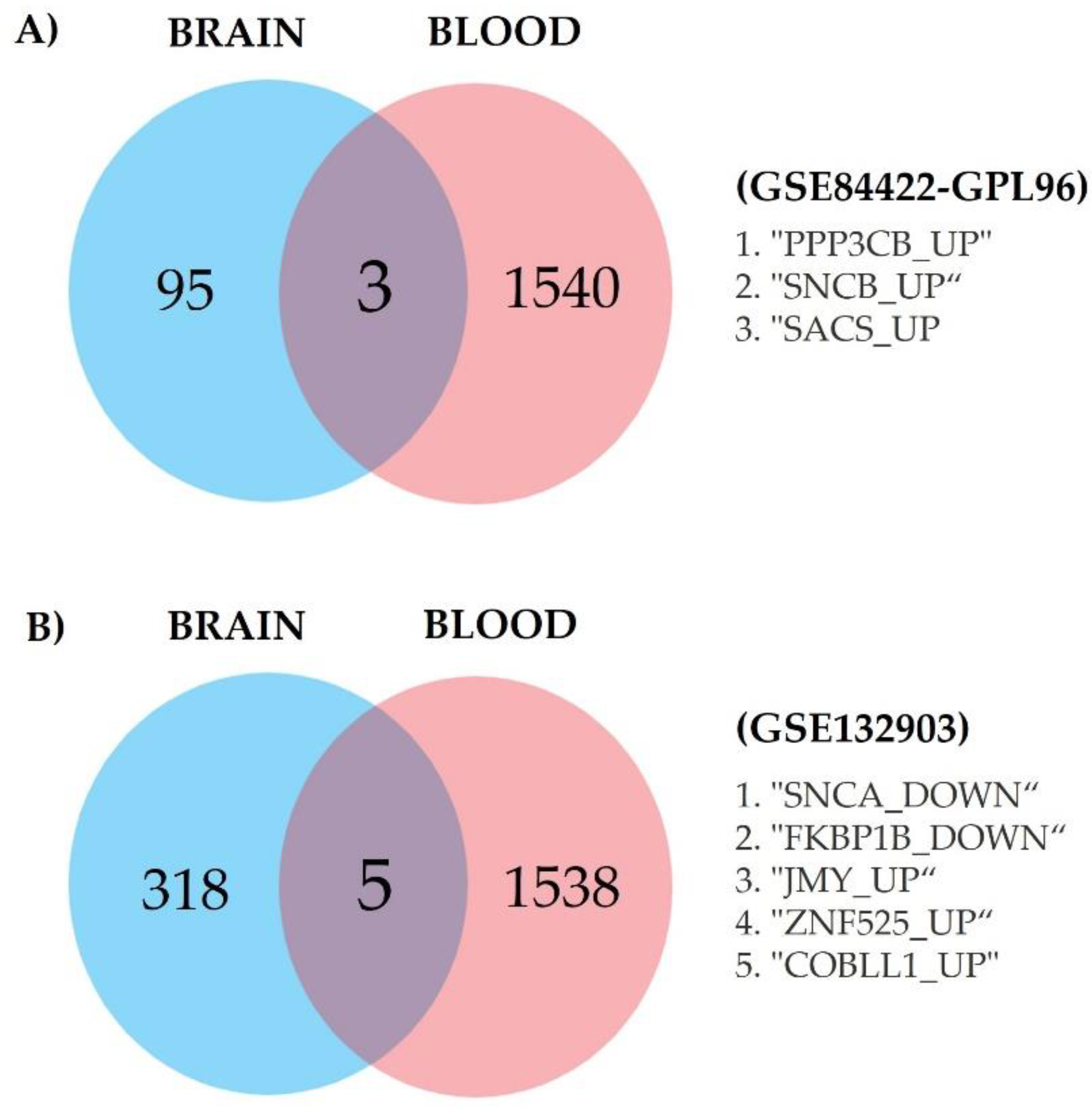
2.4. Genes in Common and Selection of Genes of Interest
3. Results
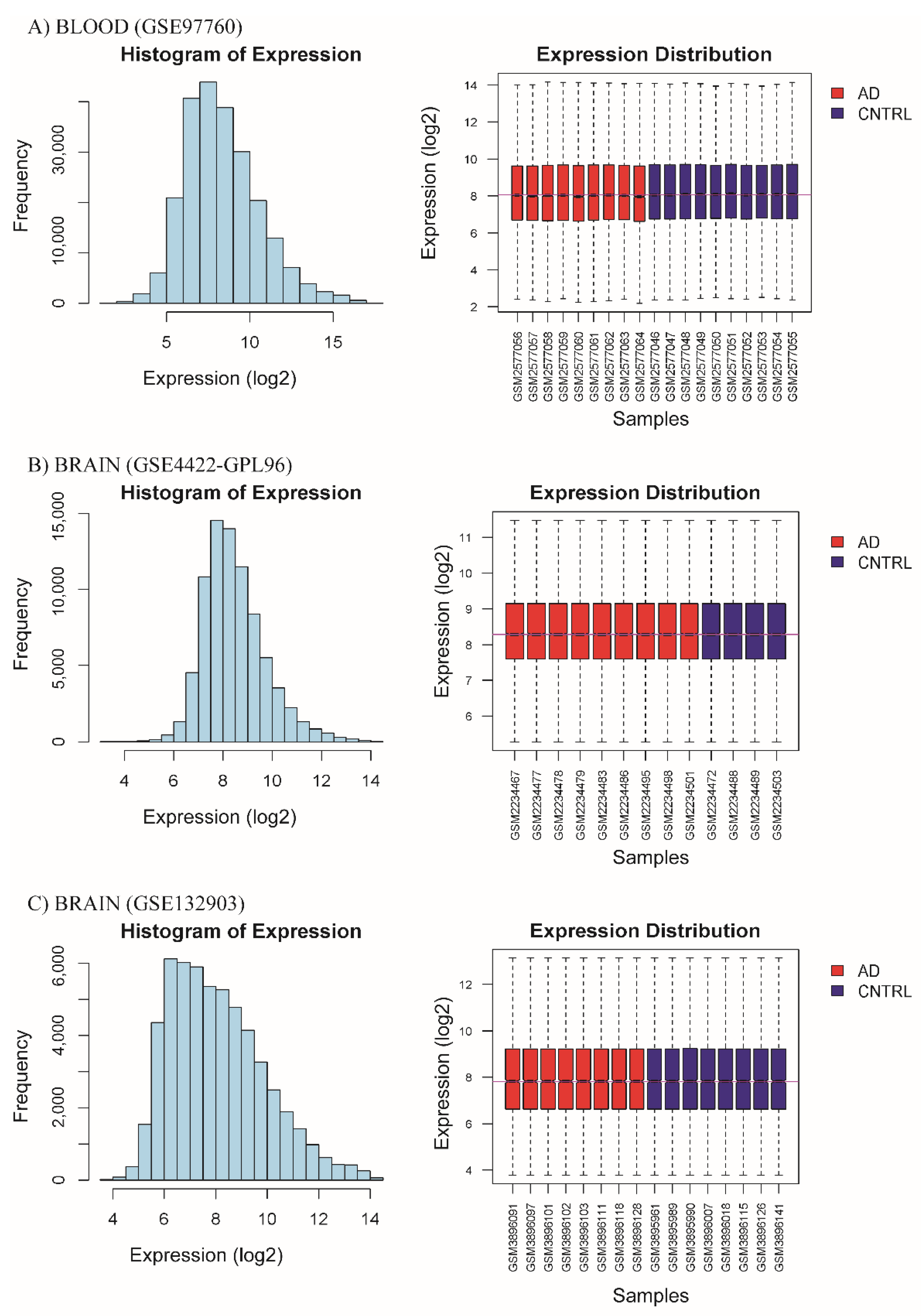
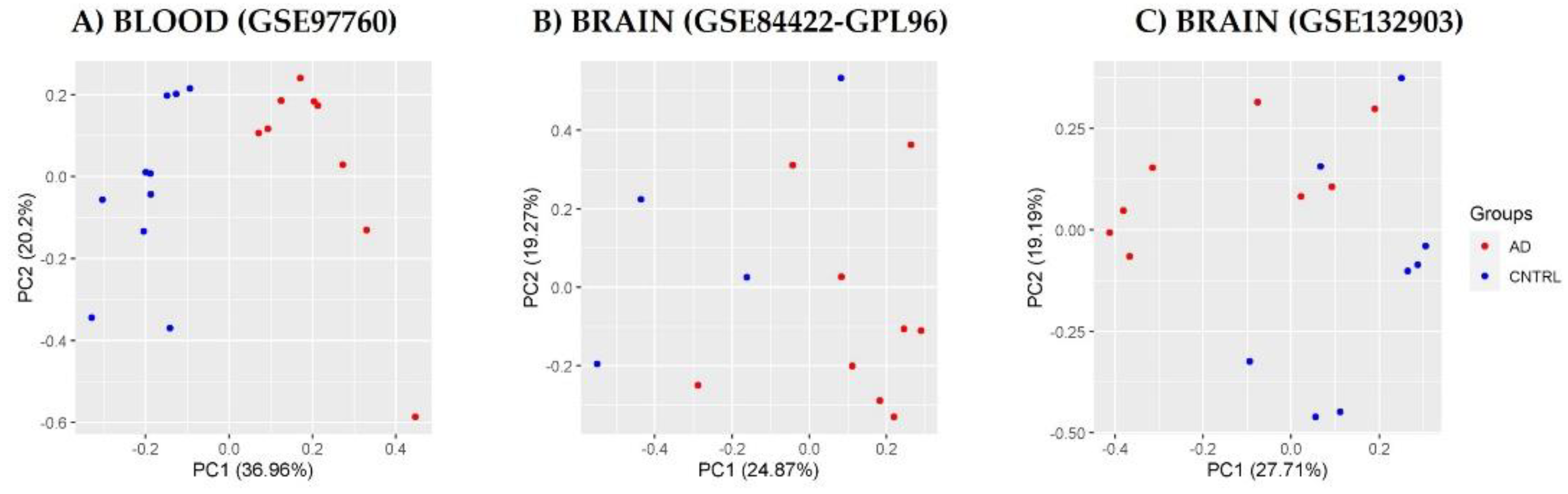
3.1. Homogenization of Raw Database Data
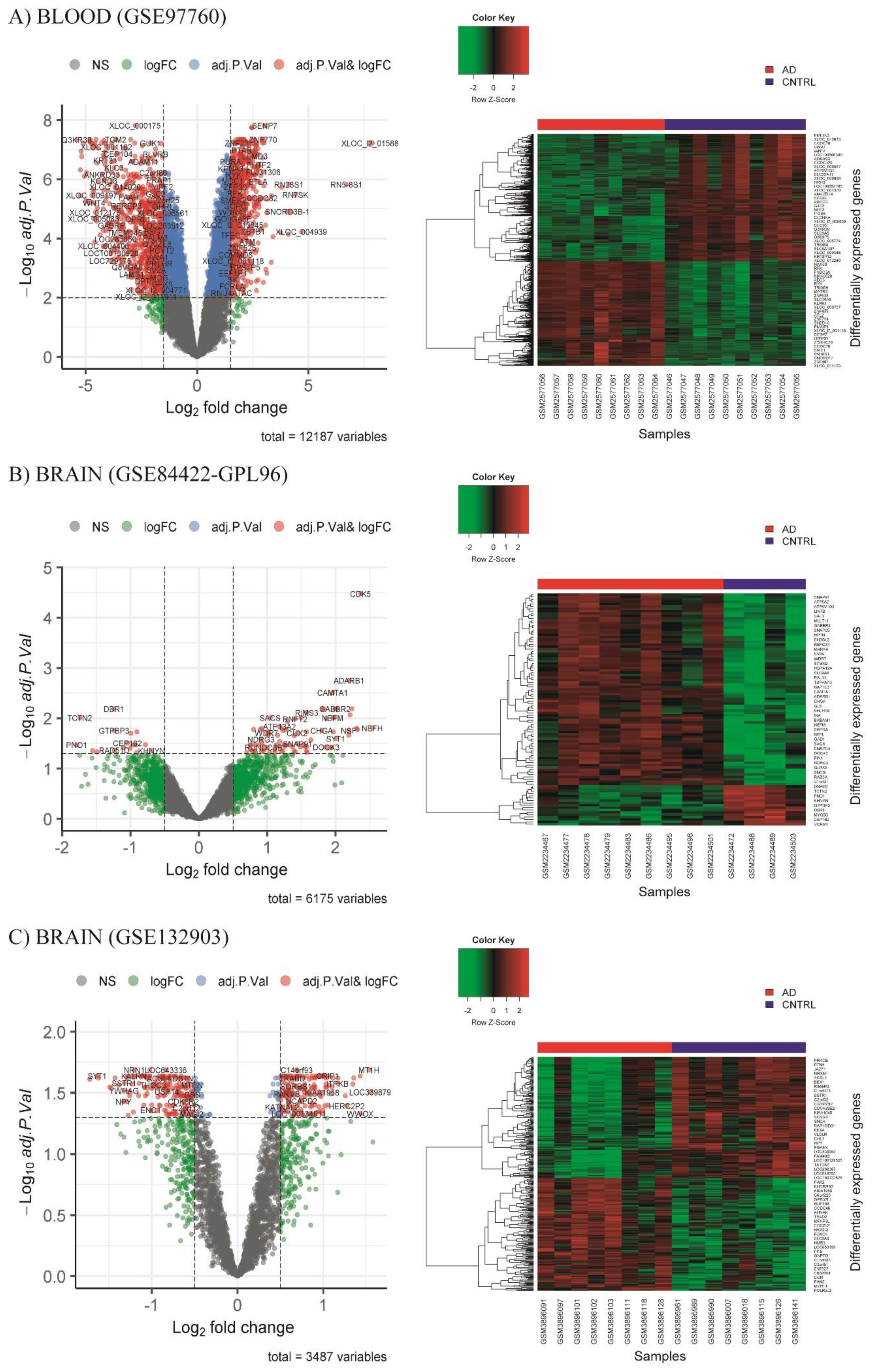
3.2. Differential Expression Analysis (DEA)
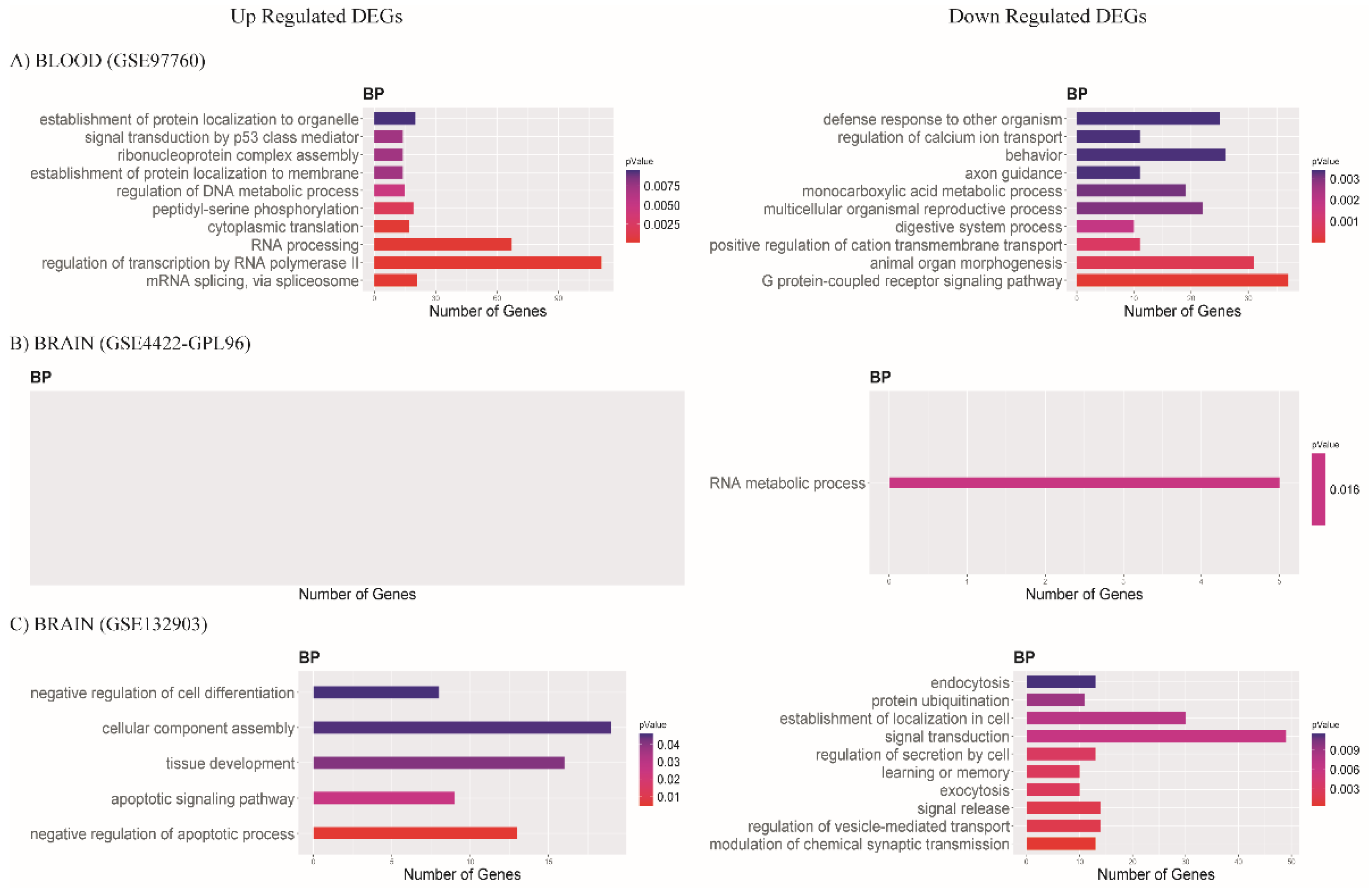
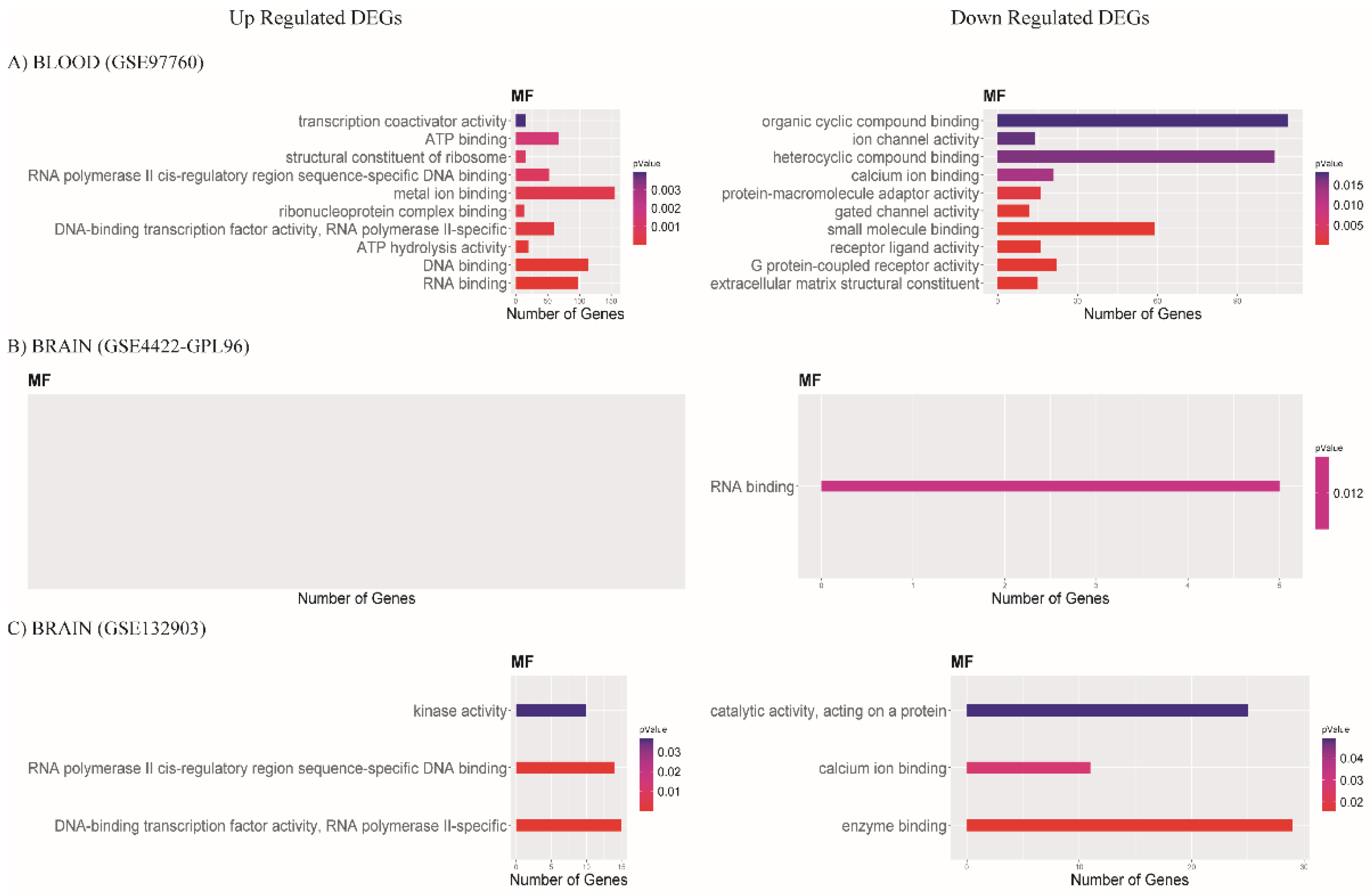
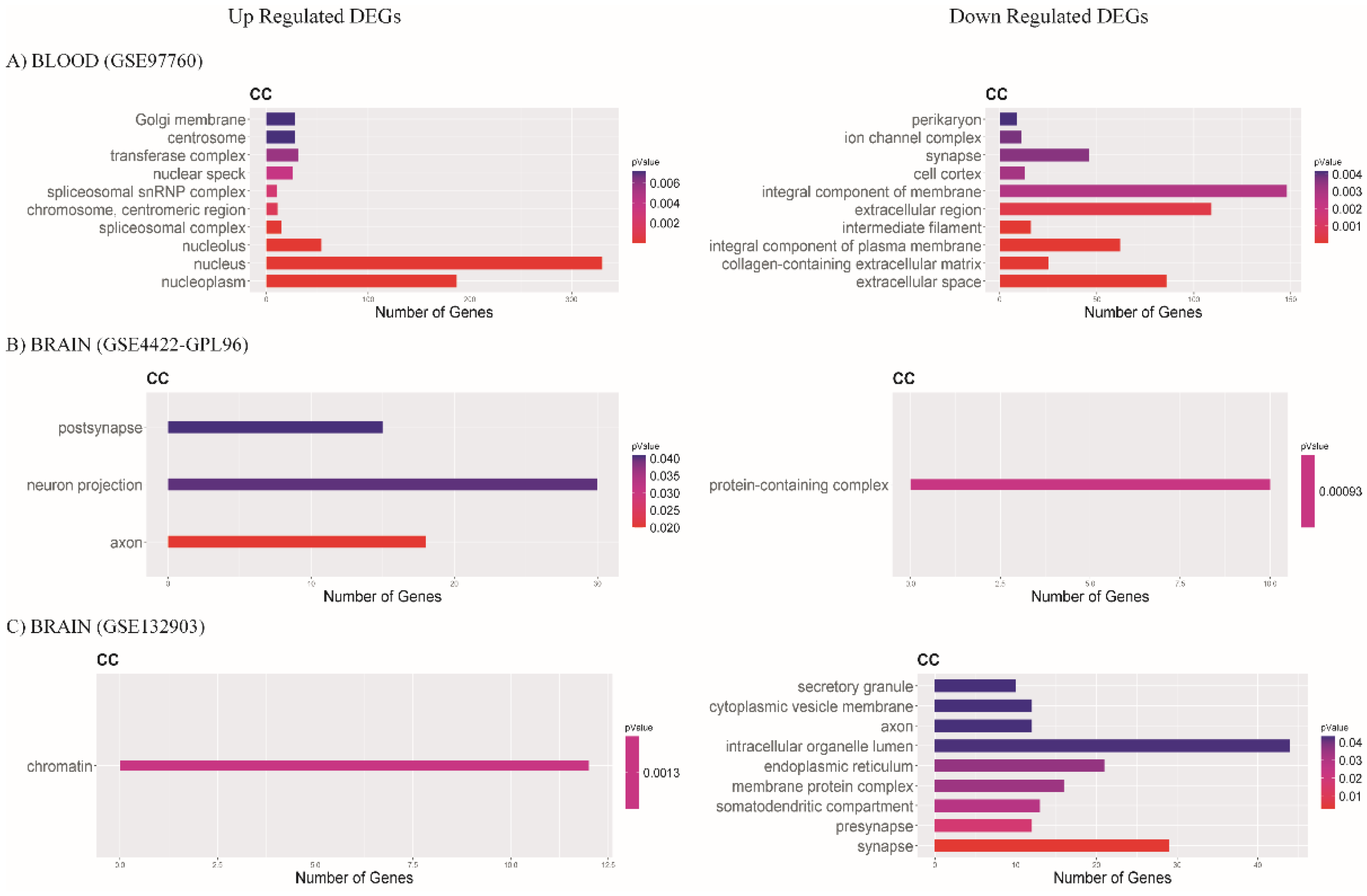
3.3. Functional Enrichment Analysis of DEGs
3.3.1. Biological Process
3.3.2. Molecular Function
3.3.3. Cellular Component
3.4. Genes of Great Interest as Potential AD Biomarkers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez, O.L.; Kuller, L.H. Epidemiology of aging and associated cognitive disorders: Prevalence and incidence of Alzheimer’s disease and other dementias. Handb. Clin. Neurol. 2019, 167, 139–148. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-T.; Beiser, A.S.; Breteler, M.M.B.; Fratiglioni, L.; Helmer, C.; Hendrie, H.C.; Honda, H.; Ikram, M.A.; Langa, K.M.; Lobo, A.; et al. The changing prevalence and incidence of dementia over time—Current evidence. Nat. Rev. Neurol. 2017, 13, 327–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, S.; Peetoom, K.; Bakker, C.; Van Der Flier, W.M.; Papma, J.M.; Koopmans, R.; Verhey, F.R.J.; De Vugt, M.; Köhler, S.; Withall, A.; et al. Global Prevalence of Young-Onset Dementia. JAMA Neurol. 2021, 78, 1080. [Google Scholar] [CrossRef] [PubMed]
- 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022, 18, 700–789. [CrossRef]
- Gauthier, S.R.-N.P.; Morais, J.A.; Webster, C. World Alzheimer Report 2021: Journey through the Diagnosis of Dementia. Alzheimer´s Disease International (ADI): London, UK. 2021, pp. 1–314. Available online: https://www.alzint.org/what-we-do/research/world-alzheimer-report/ (accessed on 22 March 2022).
- Patterson, C. World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. Alzheimer Disease International (ADI), The Global Voice on Dementia. London, UK. 2018, pp. 1–48. Available online: https://www.alzint.org/what-we-do/research/world-alzheimer-report/ (accessed on 22 March 2022).
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Rajan, K.B.; Weuve, J.; Barnes, L.L.; McAninch, E.A.; Wilson, R.S.; Evans, D.A. Population estimate of people with clinical Alzheimer’s disease and mild cognitive impairment in the United States (2020–2060). Alzheimer’s Dement. 2021, 17, 1966–1975. [Google Scholar] [CrossRef]
- Barnes, L.L.; Wilson, R.S.; Schneider, J.A.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Gender, cognitive decline, and risk of AD in older persons. Neurology 2003, 60, 1777–1781. [Google Scholar] [CrossRef]
- Wilson, R.S.; Mendes De Leon, C.F.; Barnes, L.L.; Schneider, J.A.; Bienias, J.L.; Evans, D.A.; Bennett, D.A. Participation in cognitively stimulating activities and risk of incident Alzheimer disease. JAMA 2002, 287, 742–748. [Google Scholar] [CrossRef]
- Živančević, K.; Baralić, K.; Bozic, D.; Miljaković, E.A.; Djordjevic, A.B.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Bulat, P.; Đukić-Ćosić, D. Involvement of environmentally relevant toxic metal mixture in Alzheimer’s disease pathway alteration and protective role of berberine: Bioinformatics analysis and toxicogenomic screening. Food Chem. Toxicol. 2022, 161, 112839. [Google Scholar] [CrossRef]
- Lafta, H.; AlMayaly, I. Potential effect of several heavy metals with Alzheimer’s disease infection. Ann. Rom. Soc. Cell Biol. 2021, 25, 3476–3484. [Google Scholar]
- Živančević, K.; Baralić, K.; Jorgovanović, D.; Buha Djordjević, A.; Ćurčić, M.; Antonijević Miljaković, E.; Antonijević, B.; Bulat, Z.; Đukić-Ćosić, D. Elucidating the influence of environmentally relevant toxic metal mixture on molecular mechanisms involved in the development of neurodegenerative diseases: In silico toxicogenomic data-mining. Environ. Res. 2021, 194, 110727. [Google Scholar] [CrossRef] [PubMed]
- Medehouenou, T.C.M.; Ayotte, P.; Carmichael, P.-H.; Kröger, E.; Verreault, R.; Lindsay, J.; Dewailly, É.; Tyas, S.L.; Bureau, A.; Laurin, D. Exposure to polychlorinated biphenyls and organochlorine pesticides and risk of dementia, Alzheimer’s disease and cognitive decline in an older population: A prospective analysis from the Canadian Study of Health and Aging. Environ. Health 2019, 18, 57. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Zhang, Y.; Liu, L.; Yan, H. Pesticide exposure and risk of Alzheimer’s disease: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 32222. [Google Scholar] [CrossRef] [Green Version]
- Bagyinszky, E.; Youn, Y.C.; An, S.; Kim, S. The genetics of Alzheimer’s disease. Clin. Interv. Aging 2014, 9, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J. Clin. Investig. 2005, 115, 1449–1457. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Barthélemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e1287. [Google Scholar] [CrossRef] [Green Version]
- Hanseeuw, B.J.; Betensky, R.A.; Jacobs, H.I.L.; Schultz, A.P.; Sepulcre, J.; Becker, J.A.; Cosio, D.M.O.; Farrell, M.; Quiroz, Y.T.; Mormino, E.C.; et al. Association of Amyloid and Tau With Cognition in Preclinical Alzheimer Disease. JAMA Neurol. 2019, 76, 915. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cai, L.; Zhou, K.; Cui, M.; Yao, S. Biodistribution and Dosimetry Evaluation for a Novel Tau Tracer [(18)F]-S16 in Healthy Volunteers and Its Application in Assessment of Tau Pathology in Alzheimer’s Disease. Front. Bioeng. Biotechnol. 2021, 9, 812818. [Google Scholar] [CrossRef]
- Gomar, J.J.; Tan, G.; Halpern, J.; Gordon, M.L.; Greenwald, B.; Koppel, J. Increased retention of tau PET ligand [(18)F]-AV1451 in Alzheimer’s Disease Psychosis. Transl. Psychiatry 2022, 12, 82. [Google Scholar] [CrossRef] [PubMed]
- Binder, J.; Ursu, O.; Bologa, C.; Jiang, S.; Maphis, N.; Dadras, S.; Chisholm, D.; Weick, J.; Myers, O.; Kumar, P.; et al. Machine learning prediction and tau-based screening identifies potential Alzheimer’s disease genes relevant to immunity. Commun. Biol. 2022, 5, 125. [Google Scholar] [CrossRef] [PubMed]
- Malek-Ahmadi, M.; Perez, S.E.; Chen, K.; Mufson, E.J. Braak Stage, Cerebral Amyloid Angiopathy, and Cognitive Decline in Early Alzheimer’s Disease. J. Alzheimers Dis. 2020, 74, 189–197. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, A.B. Alzheimer’s disease Braak Stage progressions: Reexamined and redefined as Borrelia infection transmission through neural circuits. Med. Hypotheses 2007, 68, 1059–1064. [Google Scholar] [CrossRef]
- Reitz, C. Genetic diagnosis and prognosis of Alzheimer’s disease: Challenges and opportunities. Expert Rev. Mol. Diagn. 2015, 15, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Albert, M.S.; Dekosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Hane, F.T.; Robinson, M.; Lee, B.Y.; Bai, O.; Leonenko, Z.; Albert, M.S. Recent Progress in Alzheimer’s Disease Research, Part 3: Diagnosis and Treatment. J. Alzheimer’s Dis. 2017, 57, 645–665. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Craig-Schapiro, R.; Perrin, R.J.; Roe, C.M.; Xiong, C.; Carter, D.; Cairns, N.J.; Mintun, M.A.; Peskind, E.R.; Li, G.; Galasko, D.R.; et al. YKL-40: A Novel Prognostic Fluid Biomarker for Preclinical Alzheimer’s Disease. Biol. Psychiatry 2010, 68, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Talwar, P.; Silla, Y.; Grover, S.; Gupta, M.; Agarwal, R.; Kushwaha, S.; Kukreti, R. Genomic convergence and network analysis approach to identify candidate genes in Alzheimer’s disease. BMC Genom. 2014, 15, 199. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Kim, Y.; Bhalla, M.; Lee, S.; Seo, J. Genomics: New Light on Alzheimer’s Disease Research. Int. J. Mol. Sci. 2018, 19, 3771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef] [PubMed]
- Alves, S.R.; da Cruz, E.S.C.; Martins, I.; Henriques, A.G.; da Cruz, E.S.O.A.B. A Bioinformatics Approach Toward Unravelling the Synaptic Molecular Crosstalk Between Alzheimer’s Disease and Diabetes. J. Alzheimers Dis. 2022, 86, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, S.; Ma, C.; Wang, X.; Shuang, Y.; Lin, H.; Guan, Z.; Yang, L. Bioinformatics analysis indicates that microRNA-628-5p overexpression may alleviate Alzheimer’s disease by targeting TYROBP. Mol. Med. Rep. 2020, 23, 142. [Google Scholar] [CrossRef]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Sharma, R.; Shrivastava, A.; Ambasta, R.K.; Kumar, P. Stress-Induced Synaptic Dysfunction and Neurotransmitter Release in Alzheimer’s Disease: Can Neurotransmitters and Neuromodulators be Potential Therapeutic Targets? J. Alzheimers Dis. 2017, 57, 1017–1039. [Google Scholar] [CrossRef]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prevot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef] [Green Version]
- Ceylan, H. Integrated Bioinformatics Analysis to Identify Alternative Therapeutic Targets for Alzheimer’s Disease: Insights from a Synaptic Machinery Perspective. J. Mol. Neurosci. 2022, 72, 273–286. [Google Scholar] [CrossRef]
- Kawalia, S.B.; Raschka, T.; Naz, M.; de Matos Simoes, R.; Senger, P.; Hofmann-Apitius, M. Analytical Strategy to Prioritize Alzheimer’s Disease Candidate Genes in Gene Regulatory Networks Using Public Expression Data. J. Alzheimers Dis. 2017, 59, 1237–1254. [Google Scholar] [CrossRef] [Green Version]
- Hemenway, C.S.; Heitman, J. Calcineurin. Structure, function, and inhibition. Cell Biochem. Biophys. 1999, 30, 115–151. [Google Scholar] [CrossRef]
- Hemmings, B.A.; Adams-Pearson, C.; Maurer, F.; Muller, P.; Goris, J.; Merlevede, W.; Hofsteenge, J.; Stone, S.R. alpha- and beta-forms of the 65-kDa subunit of protein phosphatase 2A have a similar 39 amino acid repeating structure. Biochemistry 1990, 29, 3166–3173. [Google Scholar] [CrossRef] [PubMed]
- Hata, R.; Masumura, M.; Akatsu, H.; Li, F.; Fujita, H.; Nagai, Y.; Yamamoto, T.; Okada, H.; Kosaka, K.; Sakanaka, M.; et al. Up-regulation of calcineurin Abeta mRNA in the Alzheimer’s disease brain: Assessment by cDNA microarray. Biochem. Biophys. Res. Commun. 2001, 284, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Shaikh, S.; Wang, J.Z.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J. Neurochem. 1995, 65, 732–738. [Google Scholar] [CrossRef]
- Arif, M.; Wei, J.; Zhang, Q.; Liu, F.; Basurto-Islas, G.; Grundke-Iqbal, I.; Iqbal, K. Cytoplasmic retention of protein phosphatase 2A inhibitor 2 (I2PP2A) induces Alzheimer-like abnormal hyperphosphorylation of Tau. J. Biol. Chem. 2014, 289, 27677–27691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibasaki, F.; McKeon, F. Calcineurin functions in Ca(2+)-activated cell death in mammalian cells. J. Cell Biol. 1995, 131, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Asai, A.; Qiu, J.; Narita, Y.; Chi, S.; Saito, N.; Shinoura, N.; Hamada, H.; Kuchino, Y.; Kirino, T. High level calcineurin activity predisposes neuronal cells to apoptosis. J. Biol. Chem. 1999, 274, 34450–34458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Utreja, S.; Miller, S.J.; Saunders, A. Calcineurin overexpression regulates APP metabolism. Adv. Alzheimer’s Dis. 2013, 2, 8. [Google Scholar] [CrossRef]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002, 941, 118–126. [Google Scholar] [CrossRef]
- Oeckl, P.; Halbgebauer, S.; Anderl-Straub, S.; von Arnim, C.A.F.; Diehl-Schmid, J.; Froelich, L.; Grimmer, T.; Hausner, L.; Denk, J.; Jahn, H.; et al. Targeted Mass Spectrometry Suggests Beta-Synuclein as Synaptic Blood Marker in Alzheimer’s Disease. J. Proteome Res. 2020, 19, 1310–1318. [Google Scholar] [CrossRef]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, J.; Carver, J.A. beta-Synuclein: An Enigmatic Protein with Diverse Functionality. Biomolecules 2022, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Halbgebauer, S.; Oeckl, P.; Steinacker, P.; Yilmazer-Hanke, D.; Anderl-Straub, S.; von Arnim, C.; Froelich, L.; Gomes, L.A.; Hausner, L.; Huss, A.; et al. Beta-synuclein in cerebrospinal fluid as an early diagnostic marker of Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2020, 92, 249–356. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, M.; Lariviere, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Maroteaux, L.; Scheller, R.H. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res. Mol. Brain Res. 1991, 11, 335–343. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, I.J.; Pervaiz, N.; Abbasi, A.A. The Parkinson Disease gene SNCA: Evolutionary and structural insights with pathological implication. Sci. Rep. 2016, 6, 24475. [Google Scholar] [CrossRef]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. Alpha-synuclein-positive structures in cases with sporadic Alzheimer’s disease: Morphology and its relationship to tau aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef]
- Wirths, O.; Weickert, S.; Majtenyi, K.; Havas, L.; Kahle, P.J.; Okochi, M.; Haass, C.; Multhaup, G.; Beyreuther, K.; Bayer, T.A. Lewy body variant of Alzheimer’s disease: Alpha-synuclein in dystrophic neurites of A beta plaques. Neuroreport 2000, 11, 3737–3741. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Stewart, T.; Toledo, J.B.; Ginghina, C.; Tang, L.; Atik, A.; Aro, P.; Shaw, L.M.; Trojanowski, J.Q.; Galasko, D.R.; et al. A Longitudinal Study of Total and Phosphorylated alpha-Synuclein with Other Biomarkers in Cerebrospinal Fluid of Alzheimer’s Disease and Mild Cognitive Impairment. J. Alzheimers Dis. 2018, 61, 1541–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldacci, F.; Daniele, S.; Piccarducci, R.; Giampietri, L.; Pietrobono, D.; Giorgi, F.S.; Nicoletti, V.; Frosini, D.; Libertini, P.; Lo Gerfo, A.; et al. Potential Diagnostic Value of Red Blood Cells alpha-Synuclein Heteroaggregates in Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 6451–6459. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.B.; Hong, Y.; Dhe-Paganon, S.; Yoon, H.S. FKBP family proteins: Immunophilins with versatile biological functions. Neurosignals 2008, 16, 318–325. [Google Scholar] [CrossRef]
- Jakob, R.P.; Zoldak, G.; Aumuller, T.; Schmid, F.X. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc. Natl. Acad. Sci. USA 2009, 106, 20282–20287. [Google Scholar] [CrossRef] [Green Version]
- Gant, J.C.; Blalock, E.M.; Chen, K.C.; Kadish, I.; Porter, N.M.; Norris, C.M.; Thibault, O.; Landfield, P.W. FK506-binding protein 1b/12.6: A key to aging-related hippocampal Ca2+ dysregulation? Eur. J. Pharmacol. 2014, 739, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Gant, J.C.; Chen, K.C.; Kadish, I.; Blalock, E.M.; Thibault, O.; Porter, N.M.; Landfield, P.W. Reversal of Aging-Related Neuronal Ca2+ Dysregulation and Cognitive Impairment by Delivery of a Transgene Encoding FK506-Binding Protein 12.6/1b to the hippocampus. J. Neurosci. 2015, 35, 10878–10887. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Mullins, R.D. LC3 and STRAP regulate actin filament assembly by JMY during autophagosome formation. J. Cell Biol. 2019, 218, 251–266. [Google Scholar] [CrossRef] [Green Version]
- Sterner, K.N.; McGowen, M.R.; Chugani, H.T.; Tarca, A.L.; Sherwood, C.C.; Hof, P.R.; Kuzawa, C.W.; Boddy, A.M.; Raaum, R.L.; Weckle, A.; et al. Characterization of human cortical gene expression in relation to glucose utilization. Am. J. Hum. Biol. 2013, 25, 418–430. [Google Scholar] [CrossRef]
- Carroll, E.A.; Gerrelli, D.; Gasca, S.; Berg, E.; Beier, D.R.; Copp, A.J.; Klingensmith, J. Cordon-bleu is a conserved gene involved in neural tube formation. Dev. Biol. 2003, 262, 16–31. [Google Scholar] [CrossRef] [Green Version]
- Ravanelli, A.M.; Klingensmith, J. The actin nucleator Cordon-bleu is required for development of motile cilia in zebrafish. Dev. Biol. 2011, 350, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancina, R.M.; Burza, M.A.; Maglio, C.; Pirazzi, C.; Sentinelli, F.; Incani, M.; Montalcini, T.; Pujia, A.; Congiu, T.; Loche, S.; et al. The COBLL1 C allele is associated with lower serum insulin levels and lower insulin resistance in overweight and obese children. Diabetes Metab. Res. Rev. 2013, 29, 413–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.J.; Lee, S.J.; Lee, E.; Lee, E.Y.; Han, K. Risk of Incident Dementia According to Glycemic Status and Comorbidities of Hyperglycemia: A Nationwide Population-Based Cohort Study. Diabetes Care 2022, 45, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chen, G.; Liu, Z.; Yu, L.; Shang, Y. A bioinformatics analysis to identify novel biomarkers for prognosis of pulmonary tuberculosis. BMC Pulm. Med. 2020, 20, 279. [Google Scholar] [CrossRef]
- Zhao, X.M.; Li, Y.B.; Sun, P.; Pu, Y.D.; Shan, M.J.; Zhang, Y.M. Bioinformatics analysis of key biomarkers for retinoblastoma. J. Int. Med. Res. 2021, 49, 3000605211022210. [Google Scholar] [CrossRef]
- Wei, D.; Li, R.; Si, T.; He, H.; Wu, W. Screening and bioinformatics analysis of key biomarkers in acute myocardial infarction. Pteridines 2021, 32, 79–92. [Google Scholar] [CrossRef]
- Yu, W.; Yu, W.; Yang, Y.; Lu, Y. Exploring the Key Genes and Identification of Potential Diagnosis Biomarkers in Alzheimer’ s Disease Using Bioinformatics Analysis. Front. Aging Neurosci. 2021, 13, 602781. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GEO ID | Tissue | Samples | Condition | Race | Publication and Last Update Date |
|---|---|---|---|---|---|
| GSE97760 | Peripheral blood | CNTRL: 10 AD: 9 | CNTRL: healthy AD: sporadic/advanced | CNTRL: not provided AD: not provided | Published on 14 April 2017/updated on 27 March 2018 |
| GSE84422-GPL96 | Hippocampus | CNTRL: 4 AD: 9 | CNTRL: healthy AD: sporadic/definite | CNTRL: 4 white AD: 8 white/1black | Published on 19 August 2016/updated on 26 June 2019 |
| GSE132903 | Middle temporal gyrus | CNTRL: 8 AD: 8 | CNTRL: healthy AD: sporadic/advanced | CNTRL: not provided AD: not provided | Updated on 18 September 2019 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva-Lucero, M.-d.-C.; Rivera-Osorio, J.; Gómez-Virgilio, L.; Lopez-Toledo, G.; Luna-Muñoz, J.; Montiel-Sosa, F.; Soto-Rojas, L.O.; Pacheco-Herrero, M.; Cardenas-Aguayo, M.-d.-C. Biomarker Candidates for Alzheimer’s Disease Unraveled through In Silico Differential Gene Expression Analysis. Diagnostics 2022, 12, 1165. https://doi.org/10.3390/diagnostics12051165
Silva-Lucero M-d-C, Rivera-Osorio J, Gómez-Virgilio L, Lopez-Toledo G, Luna-Muñoz J, Montiel-Sosa F, Soto-Rojas LO, Pacheco-Herrero M, Cardenas-Aguayo M-d-C. Biomarker Candidates for Alzheimer’s Disease Unraveled through In Silico Differential Gene Expression Analysis. Diagnostics. 2022; 12(5):1165. https://doi.org/10.3390/diagnostics12051165
Chicago/Turabian StyleSilva-Lucero, Maria-del-Carmen, Jared Rivera-Osorio, Laura Gómez-Virgilio, Gustavo Lopez-Toledo, José Luna-Muñoz, Francisco Montiel-Sosa, Luis O. Soto-Rojas, Mar Pacheco-Herrero, and Maria-del-Carmen Cardenas-Aguayo. 2022. "Biomarker Candidates for Alzheimer’s Disease Unraveled through In Silico Differential Gene Expression Analysis" Diagnostics 12, no. 5: 1165. https://doi.org/10.3390/diagnostics12051165