1. Introduction
Systemic sclerosis (SSc) is a systemic autoimmune disease characterized by vascular hyperreactivity, autoimmunity, and fibrosis affecting mainly the skin, the lungs, and the gastrointestinal tract [
1]. There are two main clinical phenotypes of SSc: limited SSc (lSSc) and diffuse SSc (dSSc), as defined by the extent of skin involvement [
2], which is difficult to assess [
3,
4,
5]. Such a dichotomy is useful in the clinics as limited SSc is associated with less extensive skin involvement, less frequent organ involvement, and a better survival than dSSc [
6], but SSc antibodies may also indicate specific involvement [
7].
In the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) set of classification criteria, the three following autoantibodies (Ab) were included: anti-topoisomerase I Ab (ATA), anti-centromere Ab (ACA), and anti-RNA polymerase III Ab. These main Ab are commonly associated with specific features of SSc. ACA are commonly found in lSSc patients and are associated with a better prognosis than dSSc. Still, ACA are also associated with late development of pulmonary hypertension (PH), as well as digital ulcers and gastrointestinal involvement [
8]. ATA are commonly associated with dSSc and a poorer prognosis [
9,
10,
11,
12]. Indeed, patients with ATA develop severe organ involvement such as severe interstitial lung disease (ILD), cardiac, or renal involvement [
8,
13]. Lastly, anti-RNA polymerase III Ab (POL3) are found in patients with dSSc and are associated with a high frequency of scleroderma renal crisis, rapidly progressive skin fibrosis, and little pulmonary and gastrointestinal involvement [
14]. Despite these common associations between Ab and clinical characteristics, inverted phenotypes of SSc (i.e., ACA dSSc and ATA lSSc) have been described in few studies. In these studies, authors reported that patients with inverted phenotypes had a mild course of SSc [
8,
15,
16,
17,
18,
19]. Still, little is known about the presentation and survival of patients with inverted phenotypes of SSc.
Therefore, the objective of the present study was to characterize for the first time ACA dSSc and ATA lSSc patients followed in a French national referral center for SSc.
2. Patients and Methods
2.1. Patients
We performed a monocentric retrospective case-control study, based on the database of the French national referral center for autoimmune and systemic disease, opened since 2000 [
20]. Inclusion criteria comprised a diagnosis of SSc according to ACR/EULAR classification criteria [
7], ≥18 years of age, with a single Ab (ATA or ACA).
2.2. Study Population
Patients were defined as having lSSc or dSSc based on Leroy and Medsger classification [
10]. Cases were defined as having ATA lSSc or ACA dSSc if they had lSSc with ATA Ab or dSSc with ACA Ab, respectively. For each case with ATA lSSc, a control with ATA dSSc and a control with ACA lSSc were randomly extracted from the same database of the national referral center for systemic autoimmune diseases of Ile de France and were also used as controls for patients with ACA dSSc.
2.3. Objectives of the Study
The primary objective of the study was to determine the prevalence of ACA dSSc and ATA lSSc. Secondary objectives were to describe the clinical characteristics and to study the survival rates of ACA dSSc and ATA lSSc patients.
2.4. Data Source
Data included demographic statement (age, gender), disease duration, and clinical profiles (modified Rodnan skin score (mRSS), presence of digital vasculopathy or other visceral impairment such as gastrointestinal tract, interstitial lung disease, pulmonary hypertension, cardiac, and renal). Results of biological samples, pulmonary function test, and echocardiography were also collected for patients and controls. Treatment usage including immunomodulating agents (mycophenolate mofetil, azathioprine, cyclophosphamide, and corticosteroids) were also collected.
Organ system involvement was defined as previously described [
21]. Respiratory failure was defined by one of the following three criteria: PaO
2 < 60 mmHg, or pCO
2 > 50 mmHg without supplemental oxygen, or resting O
2 saturation of <88% as determined by pulse oximetry. ILD was defined by pulmonary function tests showing a decrease of >15% in diffusing capacity of the lung of carbon monoxide (DLCO) or >10% in forced vital capacity (FVC) (actual change in % predicted units from baseline). The worsening of mRSS was defined as an increase of mRSS by ≥5 points for skin score or increase by >25% for baseline skin score > 20. Scleroderma renal crisis was defined by high blood pressure and one of the following 5 features: increases of ≥50% above baseline in serum creatinine; proteinuria: ≥2+ by dipstick confirmed by protein/creatinine ratio; hematuria: ≥2+ by dipstick or >10 red blood cells per high power field (without menstruation); thrombocytopenia: <100,000 platelets/mm
3; hemolysis: by blood smear or increased reticulocyte count. Cardiac involvement was defined by arrhythmia for >3 months, pericarditis, or cardiac heart failure. Myositis was defined by myositis (by elevated CPK and by electromyography (EMG) and/or biopsy). PH was defined as pulmonary artery peak systolic pressure (PAP) of ≥40 mmHg estimated by echocardiography. Joint involvement was defined as inflammatory arthralgias, arthritis, or tendon friction. Calcinosis was identified on hand by X-rays or when it was clinically obvious. Digital tip ulcers were based on physical observation. Gastrointestinal tract involvement was defined by one of the following features: distal esophageal dysmotility, hypomotility of the duodenum or small bowel intestine, malabsorption syndrome, or colon sacculations.
2.5. Statistical Analysis
Data for continuous variables are presented as the median and interquartile range (IQR). Data for qualitative variables are presented as the number and percentage. Fisher’s exact test was used to compare qualitative variables, the nonparametric Mann–Whitney U test was used to compare continuous variables, and the p-value was corrected by Bonferroni. Kaplan–Meier survival curves were used for the analysis of survival. P values less than 0.05 were considered significant. All analyses were performed using GraphPad Prism 8 or Stata.
3. Results
3.1. Prevalence
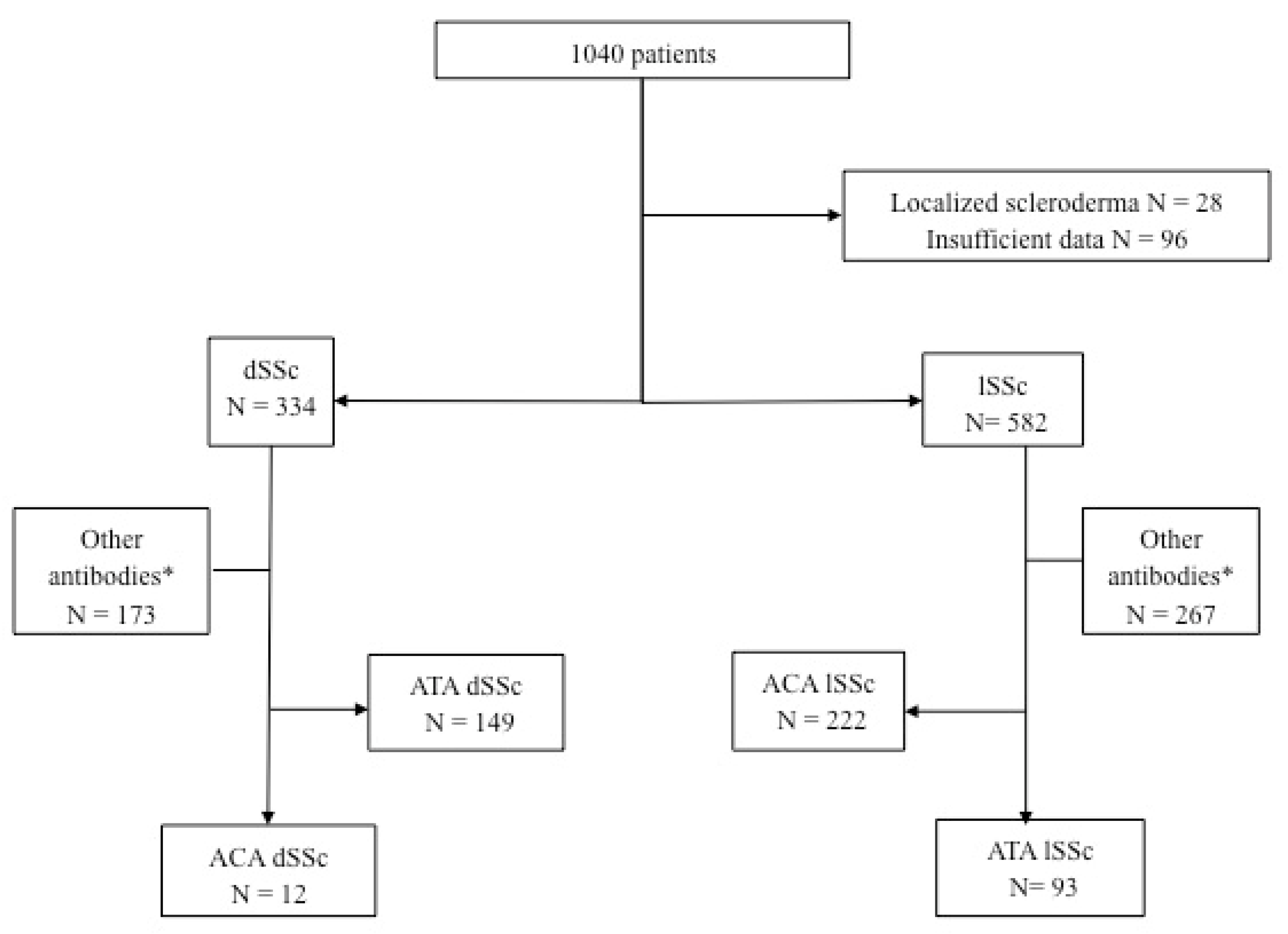
Between 2000 and 2019, 1040 patients with scleroderma were included (
Figure 1). Among them, 334 patients had dSSc and 582 patients had lSSc. Among patients with lSSc, 93 had ATA Ab, and among patients with dSSc, 12 had ACA.
In patients with scleroderma, the prevalence of ACA dSSc was 1.1% (12/1040) and 3.6% (12/334) in patients with dSSc. The prevalence of ATA lSSc was 8.9% (93/1040) in patients with SSc and 16% (93/582) in patients with lSSc (
Figure 1).
3.2. ACA dSSc Patients
Baseline characteristics of ACA dSSc patients are depicted in
Table 1 and
Table 2. Demographic characteristics of ACA dSSc subjects were similar to those of ACA lSSc patients and ATA dSSc patients. As expected, organ involvement differed between these three groups.
Compared to ACA lSSc patients, ACA dSSc patients were more severe as they more frequently had skin sclerosis, as evaluated by mRSS (26 [17–30] vs. 2 [7–25]; p < 0.01), calcinosis (58% vs. 20%; p < 0.05) gastrointestinal tract involvement (92% vs. 58%; p < 0.05), heart involvement (3 (25%) vs. 4 (4%); p < 0.05), muscle involvement (4 (33%) vs. 4 (8%); p < 0.05), inflammatory syndrome (CRP > 5 mg/l) (9 (75%) vs. 14 (5%); p < 0.001), and more frequently received immunosuppressants (5 (42%) vs. 5 (5%); p < 0.01).
Compared to ATA dSSc, ACA dSSc patients were quite similar patients but more frequently had calcinosis (58% vs. 16%; p < 0.01) and less frequently had ILD (42% vs. 80%; p < 0.01).
After a median follow up of 5 [5–9] years, three (43%) patients with ACA dSSc and eight (12%) patients with ACA lSSc died. The differences in organ involvement, identified at baseline, persisted during the follow-up with a higher mRSS in ACA dSSc than in ACA lSSc patients (23 [22–24] vs. 2 [0–5]; p < 0.0001). In ACA dSSc, ILD remained less frequently detected than in ATA dSSc patients (14% vs. 56%; p = 0.05).
3.3. ATA lSSc Patients
The baseline characteristics of ATA lSSc patients are depicted in
Table 3 and
Table 4. Compared to ATA dSSc patients, ATA lSSc patients more frequently had an older age at diagnosis of SSc (51 [41–61] vs. 43 [29–54];
p < 0.01) and digital ulcers (55 (59%) vs. 34 (37%);
p < 0.01) but less skin sclerosis as assessed by median (IQR) mRSS (4 [2–8] vs. 18 [10–27];
p < 0.0001) and less frequent gastrointestinal tract (59 (60%) vs. 75 (81%);
p < 0.01), joint (58 (62%) vs. 80 (86%);
p < 0.001), cardiac (6 (7%) vs. 19 (20%);
p < 0.01), and muscle involvement (15 (16%) vs. 54 (58%);
p < 0.0001).
Compared to ACA lSSc patients, ATA lSSc patients had less calcinosis (6 (7%) vs. 22 (23%); p < 0.01) and less telangiectasia (27 (29%) vs. 43 (46%); p < 0.05).
Interestingly, ILD was more prevalent in ATA lSSc than in ACA lSSc patients (67 (72%) vs. 31 (33%); p < 0.001), whereas it was equally prevalent in ATA dSSc patients (67 (72%) vs. 74 (80%); p = 0.30). Although, ATA dSSc more often had a decreased DLCO than ATA lSSc patients (43 (46%) vs. 59 (63%); p < 0.01), and the median (IQR) FCV was higher in ATA lSSc (86 [66–103] vs. 71 [60–90]; p < 0.01) than in ATA dSSc patients.
During a median (IQR) follow-up of 5 [3–9] years following inclusion, the median (IQR) mRSS remained lower in ATA lSSc patients than in ATA dSSc patients (4 [2–9] vs. 16 [2–22];
p < 0.05), without worsening (
p = 0.79). Oppositely, the median (IQR) mRSS was similar between ATA lSSc and ACA lSSc patients (4 [2–9] vs. 2 [0–5];
p = 0.19). ATA lSSc patients had more ILD than ACA lSSc patients (37 (64%) vs. 7 (10%);
p < 0.0001) but no less than ATA dSSc patients (37 (64%) vs. 39 (56%);
p = 0.37). Still, ATA dSSc patients had more severe ILD than ATA lSSc patients, as highlighted by the lower median (IQR) DLCO (42% [34–55] vs. 64% [44–73];
p < 0.001), FCV (68% [48–84] vs. 87 [67–99];
p < 0.01), and a lower total lung capacity (TLC) (77% [59–87] vs. 90% [71–101];
p < 0.001). Other differences during follow-up are depicted in
Table 5 and
Table 6.
3.4. Survival and Transition
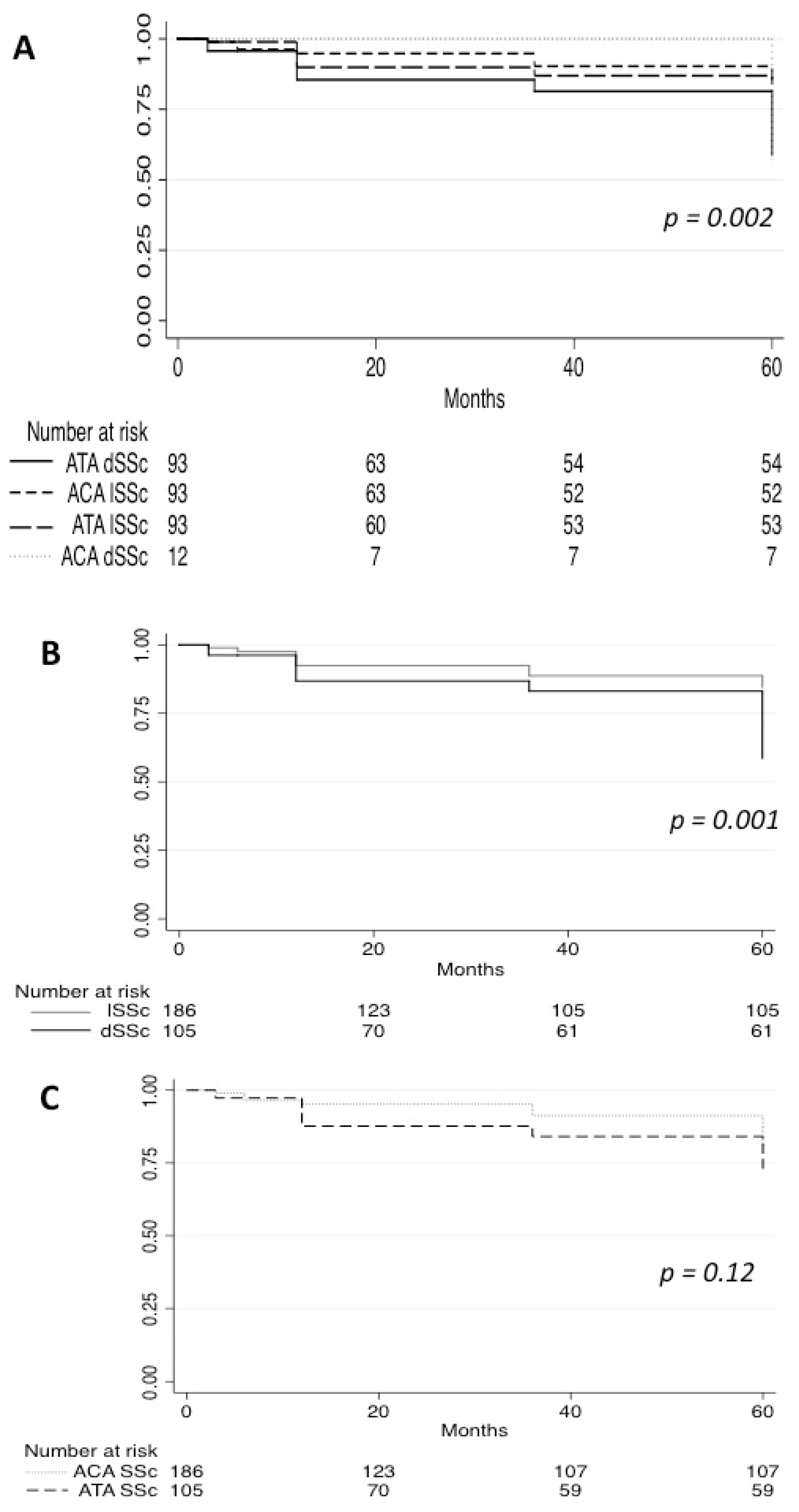
Survival analysis was undertaken among subjects with at least 1 year of follow-up data (n = 203 patients). The cumulative survival rate during 5 [3–9] years of follow-up was 71% in ACA dSSc and 95% in ATA lSSc compared with 84% (11 patients) in ACA lSSc and 66% (24 patients) in ATA dSSc (
Figure 2). Analysis of survival showed a statistically significant (
p < 0.001) difference between the four groups. Interestingly, survival did not differ significantly between patients with ATA and ACA Ab (
p = 0.12), whereas patients with dSSc had a worse survival than patients with lSSc (
p < 0.001).
Of note, none of the lSSc patients of our cohort experienced a transition from limited to diffuse skin involvement.
4. Discussion
The objectives of this study were to determine the prevalence and the main characteristics of SSc patients with inverted phenotypes in a French cohort of patients with SSc. According to our results, ACA dSSc and ATA lSSc exist and are rare SSc phenotypes and have their own specificities. Among inverted phenotypes, ACA dSSc is even rarer than ATA lSSc. Interestingly, our study highlights that ATA is specifically associated with ILD and confirms that dSSc has a worse prognosis both in term of organ involvement and mortality.
The prevalence of ACA dSSc and ATA lSSc varies in the literature. In comparison with the two only other published studies (
Table 7), we found a lower prevalence of patients with ATA lSSc and a higher prevalence of ACA dSSc [
15,
16]. At baseline, our patients with inverted phenotypes had a specific clinical phenotype: more ILD in ATA lSSc patients and more cardiac and muscle involvements in ACA dSSc. Regarding follow up, our patients seem to be similar to those reported in the two other studies. Our study also confirms that ACA dSSc and ATA lSSc have a mild course compared to usual phenotypes (ACA lSSc and ATA dSSc) over the 5 years of follow up. As previously reported, lung involvement of patients with ATA lSSc resembles those of ATA dSSc, but ATA lSSc patients have a better survival [
15,
16]. While the cardiac or muscle involvement or inflammatory syndrome of ACA dSSc patients is similar to those of ATA dSSc, ACA dSSc patients have a better survival than ATA dSSc patients in our study. This finding may reflect the protective role of ACA, suggested by Caetano et al. [
22] and also proposed by Srivastava et al., who did not find significative difference between the survival of ACA dSSc and ACA lSSc [
16].
Our findings confirmed a relationship between Ab and organ involvement. Our data suggest an association between ATA and ILD, whatever the skin subset confirming previous findings [
15,
16,
17,
23]. Kranenburg et al. have compared ATA patients with non-ATA patients and showed that ILD was more frequent in ATA dSSc and ATA lSSc than in non-ATA patients [
15]. Steen et al. and Walker et al. have suggested that Ab may be a predictor of organ involvement and disease outcome [
8,
9]. Steen et al. showed that PH typically occurs in patients with ACA and ILD in patients with ATA [
8]. This specific lung involvement might be the consequence of a mechanism involving B cells. Indeed, Dumoitier et al. showed a significant proportion of active lymphocyte B in patients with versus those without interstitial lung disease [
24], and Fava et al. showed an increased population of ATA-reactive T cells in patients with ILD compared to those without ILD [
25]. Other examples were described regarding Ab specificity and clinical presentation. Anti-PM/Scl antibodies are associated with myositis, calcinosis, acro-osteolysis, and interstitial lung disease [
26], and the anti-U1RNP antibodies characterize overlapping forms between SSc, systemic lupus erythematosus, and myositis [
8].
Furthermore, our study brings data to the relationships between prognosis, antibodies, and skin subsets. Old studies reported a gradient of clinical phenotypes in SSc based on skin involvement. For example, Cottrell et al. found an “intermediate” clinical phenotype (distal to elbow/knees without trunk involvement) with different autoantibody profiles and mild survival [
27]. Recent studies have tackled this issue showing that skin extension evaluation could not be sufficient to classify patients. Indeed, Sobanski et al. has determined six different clusters based on clinical features, autoantibody profiles, and survival [
28]. In other autoimmune diseases such as ANCA-associated vasculitis, it was showed that patients could be differentiated solely based on their antibody profiling [
29]. Although skin extension is a major issue in terms of survival in SSc patients, we believe that our work argues for an evaluation of SSc patients based on both mRSS and autoantibody profile.
Our study has some limitations. As a monocentric study, the number of patients with underrepresented SSc phenotypes such as ACA dSSc is low, and one has to be cautious when looking at characteristics of this subgroup. Since it is a retrospective study, missing data cannot be avoided. Lastly, as a study performed in a tertiary referral center, it included mostly patients with more than 5 years of disease evolution, which limited our ability to study transition patients in depth, as such a phenomenon is reported to occur in the first five years of evolution [
15,
17].
To conclude, ACA dSSc and ATA lSSc exist and represent 10% of our SSc patient cohort. They are rare phenotypes of SSc, have their own specificities, and are characterized by a mild course of evolution of SSc. Studying these phenotypes confirmed that antibodies assessment in SSc patients is mandatory and that antibodies may predict organ involvement, whereas the extent of skin sclerosis may predict survival. Overall, inverted phenotypes should be considered as a separate group and be assessed mainly combining Ab and skin subset.


 ,
, 


{kind=link}
{kind=link}