The Molecular Characterization of Genetic Abnormalities in Esophageal Squamous Cell Carcinoma May Foster the Development of Targeted Therapies


{kind=link}
Abstract
:1. Introduction
2. Precursor Lesions of ESCC
3. Genetic Alterations of ESCC
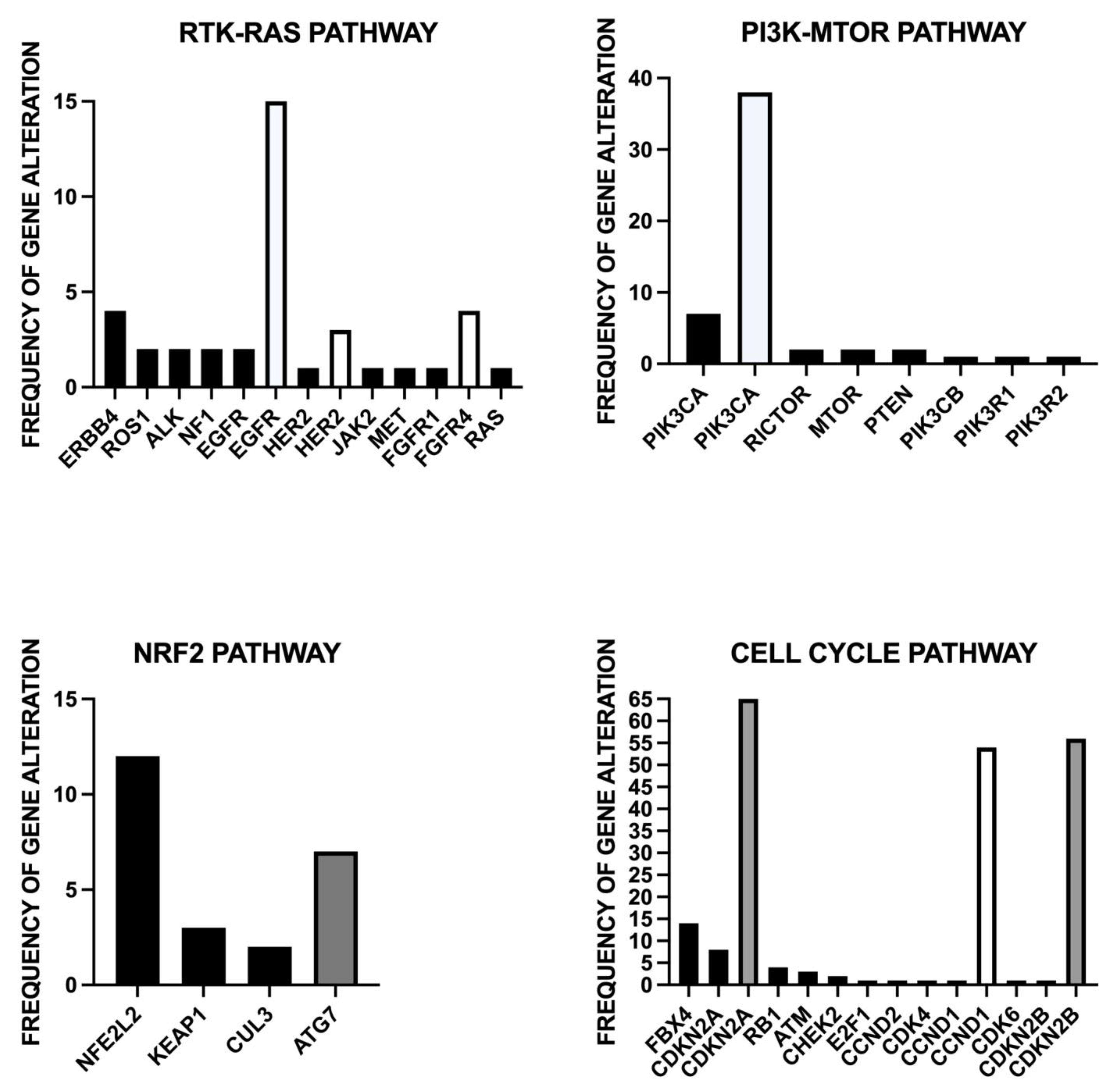
3.1. Genomic Alterations of ESCC
3.2. Mutational Signatures in ESCC
3.3. Molecular Classification of ESCC
3.4. ESCC Genetic Alterations and Response to Chemoradiotherapy
3.5. Intratumoral Heterogeneity of ESCC
3.6. Gene Expression Studies
4. Targeted Therapy in ESCC
4.1. Actionable Genetic Alterations in ESCC
4.2. Therapeutic Targeting of RTK-RAS Pathway
4.2.1. EGFR
4.2.2. FGFR
4.2.3. HER2
4.3. PI3K/AKT/mTOR
4.4. KEAP1-NRF2
4.5. CDK4/CDK6 Targeting
5. Immunotherapy of ESCC with Immune Checkpoint Inhibitors
5.1. Advanced/Metastatic Setting, Second-Line
5.2. Advanced/Metastatic Setting, First-Line
5.3. ICIs in Association with Neoadjuvant Chemoradiotherapy in Patients with Locally Advanced ESCC
5.3.1. Immunotherapy Studies in ESCC Patients with Resectable Tumors
5.3.2. Immunotherapy Studies in ESCC Patients with Unresectable Tumors
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Legergren, P.; Cunnigham, D. Oesophageal cancer. Nat. Rev. Dis. Prim. 2017, 3, 17048. [Google Scholar] [CrossRef] [PubMed]
- Lagergren, J.; Smyth, E.C.; Cunnigham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 25, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Castelli, G.; Pelosi, E. Esophageal cancer, genomic and molecular characterization, stem cell compartment and clonal evolution. Medicines 2017, 4, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, M. Individual and combined effects of environmental risk factors for esophageal cancer based from the Golestan cohort study. Gastroenterology 2019, 156, 1416–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, V.A. Informing etiologic research priorities for squamous cell esophageal cancer in Africa: A review of setting-specific exposures to known and putative risk factors. Int. J. Cancer 2017, 140, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Mello, F.W. The synergistic effect of tobacco and alcohol consumption on oral squamous cell carcinoma: A systematic review and meta-analysis. Clin. Oral Investig. 2019, 23, 2849–2859. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Abuet, C.C.; Dawsey, S.M. Squamous dysplasia-the precursor lesion for esophageal squamous cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2013, 22, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Lu, P.; Zhang, N.; Zhao, L.; Liu, J.; Yang, J.; Liu, J.; Zhao, D.; Wang, J. Progression of precancerous lesions of esophageal squamous cell carcinomas in a high-risk, rural Chinese population. Cancer Med. 2022; in press. [Google Scholar]
- Liu, X.; Zhang, M.; Ying, S.; Zhang, C.; Lin, R.; Zhang, J.; Zhang, G.; Tian, D.; Guo, Y.; Du, C.; et al. Genetic alterations in esophageal tissues from squamous dysplasia to carcinoma. Gastroenterology 2017, 153, 166–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.X.; Zhong, Q.; Liu, Y.; Yan, S.M.; Chen, Z.H.; Jin, S.Z.; Xia, T.L.; Lia, R.Y.; Zhou, A.J.; Su, Z.; et al. Genomic comparison of esophageal squamous cell carcinoma and its precursor lesions by multiple-region whole-exome sequence. Nat. Commun. 2017, 8, 524. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Wang, Y.; Cai, L.; Yan, J.; Chen, X.; Wu, X.; Chen, Q. Comparative genomic analysis reveals genetic variations in multiple primary esophageal squamous cell carcinoma of chinese population. Front. Oncol. 2022, 12, 869301. [Google Scholar] [CrossRef]
- Liu, M.; Liu, Y.; Zhou, R.; Liu, Z.; Guo, C.; Xu, R.; Li, F.; Liu, A.; Yang, H.; Zhang, S.; et al. Absence of NOTCH1 mutation and presence of CDKN2A deletion predict progression of esophageal lesions. J. Pathol. 2022, 258, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.; Abascal, F.; Hall, M.; Cagan, A.; Murai, K.; Mahbani, K.; Startton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef] [Green Version]
- Colom, B.; Alsolea, M.P.; Piedrafita, G.; Hall, M.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef] [PubMed]
- Colom, B.; Hewrems, A.; Hall, M.; Dentro, S.C.; King, C.; Sood, R.K.; Alcolea, M.P.; Piedrafita, G.; Fernandez-Antoran, D.; Ong, S.H.; et al. Mutant clones in normal epithelium outcompete and eliminate emerging tumora. Nature 2021, 598, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Yokoama, A.; Kakiuchi, N.; Yoshizato, T.; Nannya, Y.; Suzuki, A.; Takeuchi, Y.; Shiozawa, Y.; Sato, Y.; Aoki, K.; Kim, S.K.; et al. Age-related remodelling of oesophageal epithelia by mutated driver cancer genes. Nature 2019, 565, 312–317. [Google Scholar] [CrossRef] [Green Version]
- Abby, E.; Dentro, S.C.; Hall, M.; Fowler, J.C.; Ong, S.H.; Sood, R.; Siebel, C.W.; Gerstung, M.; Hall, B.A.; Jones, P.H. Notch1 mutation drives clonal expansion in normal esophageal epithelium but impairs tumor growth. bioRxiv 2021. [Google Scholar] [CrossRef]
- Murai, K.; Dentro, S.; Ong, S.H.; Sood, R.; Fernandez-Antoran, D.; Herms, A.; Kastan, V.; Hall, B.A.; Gerstung, M.; Jones, P.H. p53 mutation in normal esophagus promotes multiple strategies of carcinogenesis costrained by clonal competition. Nat. Commun. 2022, 13, 6206. [Google Scholar] [CrossRef]
- Fernandez-Antoran, D.; Piedrafita, G.; Murai, K.; Ong, S.H.; Herms, A.; Frezza, C.; Jones, P.H. Outcompeting p53-mutant cells in the normal esophagus by redox manipulation. Cell Stem Cell 2019, 25, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Jimenez, F.; Muinos, F.; Sentis, I.; Deu-Pons, T.; Reyes-Solazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; et al. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Lin, D.C.; Hao, J.J.; Nagata, Y.; Xu, L.; Shang, L.; Meng, X.; Sato, Y.; Ukuno, Y.; Varela, A.M.; Ding, L.W.; et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 467–473. [Google Scholar] [CrossRef]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y.; et al. Identification of genome alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Chen, Z.L.; Li, J.G.; Hu, X.D.; Shi, X.J.; Sun, Z.M.; Zhang, F.; Zho, Z.R.; Li, Z.T.; Liu, Z.Y.; et al. Genetic landscape of esophageal squamous cell carcinoma. Nat. Genet. 2014, 46, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, Y.; Cheng, C.; Cui, H.; Cheng, L.; Komg, P.; Wang, J.; Li, Y.; Chen, W.; Song, B.; et al. Genomic analyses reveal mutational signatures and frequenly altered genes in esophageal squamous cell carcinoma. Am. J. Hum. Genet. 2015, 96, 597–611. [Google Scholar] [CrossRef] [Green Version]
- Sawada, G.; Niida, A.; Uchi, R.; Hirata, H.; Shimamura, T.; Suzuki, Y.; Shiraishi, Y.; Chiba, K.; Imoto, S.; Takahashi, Y.; et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology 2016, 150, 1171–1182. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, P.; Huang, P.; Huang, X.; Li, X.; Feng, Z.; Li, F.; Liang, S.; Song, Y.; Stenvang, J.; Brunner, N.; et al. Comprehensive genomic analysis of oesophageal squamous cell carcinoma reveals clinical relevance. Sci. Rep. 2017, 7, 15324. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, M.; Yang, M.; Dai, H.; Zhang, B.; Wang, W.; Chu, X.; Wang, X.; Zheng, H.; Niu, R.; et al. A mutational signature associated with alcohol consumption and prognostically significantly mutated driver gene in esophageal squamous cell carcinoma. Ann. Oncol. 2018, 29, 938–944. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Meng, L. Comparative genomic analysis of esophageal squamous cell carcinoma and adenocarcinoma: New opportunities towards molecularly targeted therapy. Acta Pharm. Sin. B 2022, 12, 1054–1067. [Google Scholar] [CrossRef]
- Zou, B.; Guo, D.; Kong, P.; Wang, Y.; Cheng, X.; Cui, Y. Integrative genomic analyses of 1145 patient samples reveal new biomarkers in esophageal squamous cell carcinoma. Front. Mol. Biosci. 2022, 8, 792779. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Z.; Wang, Q.; Yi, Y.; Li, B. Integrated cohort of esophageal squamous cell cancer reveals genomic features underlying clinical characteristics. Nat. Commun. 2022, 13, 5268. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucab, J.E.; Zou, X.; Morganella, S.; Joel, M.; Nanda, A.S.; Nagy, E.; Gomez, C.; Degasperi, A.; Harris, R.; Jackson, S.P.; et al. A compendium of mutational signatures of environmental agents. Cell 2019, 177, 821–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Chen, H.; Xi, R.; Cui, H.; Zhao, Y.; Xu, E.; Yan, T.; Lu, X.; Huang, F.; Kong, P.; et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res. 2020, 30, 902–913. [Google Scholar] [CrossRef]
- Chang, J.; Tan, W.; Ling, Z.; Xi, R.; Shao, M.; Chen, M.; Luo, Y.; Zhao, Y.; Liu, Y.; Huang, X.; et al. Genomic analysis of oesophageal squamous cell carcinoma identifies alcohol drinking-related mutation signature and genomic alterations. Nat. Commun. 2017, 8, 15290. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Au, H.; Zhang, Y.; Sun, W.; Cheng, S.; Whang, R.; Wang, X.; Feng, L. Molecular analysis of Chinese oesophageal squamous cell carcinoma identifies novel subtypes associated with distinct clinical outcomes. EBioMedicine 2020, 57, 102831. [Google Scholar] [CrossRef]
- Liu, W.; Xie, L.; He, Y.-H.; Liu, L.-X.; Bai, X.-F.; Deng, D.-X.; Xu, X.-E.; Liao, L.-D.; Lin, W.; Heng, J.-H.; et al. Large-scale and high-resolution mass spectrometry-based proteomics profiling define molecular subtypes of esophageal cancer for therapeutic targeting. Nat. Commun. 2021, 12, 4961. [Google Scholar] [CrossRef]
- Mai, Z.; Yuan, J.; Yang, H.; Fang, S.; Xie, X.; Wang, X.; Xie, J.; Wen, J.; Fu, J. Inactivation of Hippo pathway characterizes a poor-prognosis subtype of esophageal cancer. JCI Insight 2022, 7, e155218. [Google Scholar] [CrossRef] [PubMed]
- Hirata, H.; Nida, A.; Kakiuchi, N.; Litch, R.; Sugimachi, K.; Masuda, T.; Saito, T.; Kageyama, S.L.; Motomura, Y.; Ito, S.; et al. The evolving genomic landscape of esophageal squamous cell carcinoma under under chemoradiotherapy. Cancer Res. 2021, 81, 4926–4938. [Google Scholar] [CrossRef]
- Huang, J.; Jiang, D.; Zhu, T.; Wang, Y.; Wang, H.; Wang, Q.; Tan, L.; Zhu, H.; Yao, J.; Hou, Y. Prognostic significance of c-MYC amplification in esophageal squamous cell carcinoma. Ann. Thorac. Surg. 2019, 107, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Weng, G.; Zhao, W.; Yun, Y.; Whang, S.; Du, L.; Lin, N.; Mu, D.; Yu, Q.; Yuan, S. Genomic alterations of whole exome sequencing in esophageal squamous cell carcinoma before and radiotherapy. J. Thorac. Dis. 2020, 12, 5945–5957. [Google Scholar] [CrossRef]
- Hao, J.J.; Lin, D.C.; Dinh, H.Q.; Mayakonda, A.; Jiang, Y.Y.; Chang, C.; Jiang, Y.; Lu, C.C.; Shi, Z.Z.; Xu, X.; et al. Spatial intratumoral heterogeneity and temporal clonal evolution in esophageal squamous cell carcinoma. Nat. Genet. 2016, 48, 1500–1507. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Cui, H.; Zhou, Y.; Yang, B.; Kang, P.; Zhang, Y.; Liu, Y.; Wang, B.; Cheng, Y.; Li, J.; et al. Multi-region sequencing unveils novel cationable targets and spatial heterogeneity in esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, Z.; Liu, Q.; Wang, X.; Xie, J.; Yuan, J.; Zhong, J.; Fang, S.; Xie, X.; Yang, H.; Wen, J.; et al. Integration of tumor heterogeneity for recurrence prediction in patients with esophageal squamous cell cancer. Cancers 2021, 13, 6084. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Hu, N.; Yang, H.H.; Wang, C.; Takikita, M.; Wang, Q.H.; Giffen, C.; Clifford, R.; Hewitt, S.M.; Shou, J.Z.; et al. Global gene expression profiling and validation in esophageal squamous cell carcinoma and its association with clinical phen otypes. Clin. Cancer Res. 2011, 17, 2955–2966. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Snell, J.M.; Jeck, W.R.; Hoadley, K.A.; Wilkerson, M.D.; Parker, J.S.; Mlombe, Y.B.; Mulima, G.; Lionuba, N.G.; Wolf, L.L.; et al. Subtyping sub-Saharan ESCC by comprehensive molecular analysis. JCJ Insight 2016, 1, e88755. [Google Scholar]
- Wang, F.; Yan, Z.; Lu, J.; Xin, J.; Dang, Y.; Sun, X.; An, Y.; Qi, Y.; Jing, Q.; Zhu, W.; et al. Gene expression profiling reveals distinct molecular subtypes of esophageal squamouis cell carcinoma in Asian population. Neoplasia 2019, 21, 571–581. [Google Scholar] [CrossRef]
- Zhang, H.; Zhong, J.; Tu, Y.; Liu, B.; Chen, Z.; Luo, Y.; Tang, Y.; Xiao, F.; Zhong, J. Integrated bioinformatics analysis identifies Hub genes associated with the pathogenesis and prognosis of esophageal squamous cell carcinoma. Biomed. Res. Int. 2019, 2019, 2615921. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Qu, J.; Liu, X.; Liang, J.; Li, Y.; Jiang, J.; Zhang, H.; Tian, H. Integrated bioinformatic analysis of differentially expressed genes and immune cell infiltration characteristics in ESCC. Sci. Rep. 2021, 21, 16616. [Google Scholar]
- Li, Y.; Xu, F.; Chen, F.; Chen, Y.; Ge, D.; Zhang, S.; Lu, C. Transcriptomics based multi-dimensional characterization and drug screen in esophageal squamous cell carcinoma. EBioMeidicne 2021, 70, 103510. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, C.; Fang, Y.; Li, S.; Wang, X.; Sun, L.; Zhou, G.; Ye, J. Development of a prognostic signature for esophageal cancer based on nine immune related genes. BMC Cancer 2021, 21, 113. [Google Scholar] [CrossRef]
- Liu, K.; Jiao, Y.L.; Shen, L.Q.; Chen, P.; Zhao, Y.; Li, M.X.; Gu, B.L.; Lan, Z.J.; Ruan, H.J.; Liu, Q.W.; et al. A prognostic model based on mRNA expression analysis of esophageal sqaumous cell carcinoma. Front. Bioing. Biotec. 2022, 10, 823619. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Yang, H.; Liu, M.Z.; Luo, K.J.; Liu, H.; Hu, Y.; Zhong, X.; Lai, R.C.; Lin, T.; Wang, H.Y.; et al. Gene expression analysis of pretreatment biopsies predicts the pathological response of es to noe-chemoradiotherapy. Ann. Oncol. 2014, 25, 1769–1774. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, S.; Kato, H.; Nagaoka, K.; Sun, C.; Imano, M.; Sato, T.; Johnson, T.A.; Fujita, M.; Majima, K.; Okawa, Y.; et al. Immuno-genomic profiling of biopsy specimens predicts neoadjuvant chemotherapy response in esophageal squamous cell carcinoma. Cell Rep. Med. 2022, 3, 100705. [Google Scholar] [CrossRef]
- Tungekar, A.; Mandarthi, S.; Mandarya, P.R.; Gadekar, V.P.; Tantry, A.; Kotian, S.; Reddy, J.; Prabha, D.; Bhat, S.; Sahay, S.; et al. ESCC ATLAS: A population wide compendium of biomarkers for esophageal squamous cell carcinoma. Sci. Rep. 2018, 8, 12715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Lee, H.; Wu, W.; Zaman, A.; McCorkle, S.; Yan, M.; Chen, J.; Xing, Q.; Sinnott-Armstrong, N.; Xu, H.; et al. Multi-faceted epigenetic dysregulation of gene expression promotes esophjageal squamous cell carcinoma. Nat. Commun. 2020, 11, 3675. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, X.; Liu, S.; Ma, T.; Cai, B.; Liang, L.; Qu, B.; Zhang, P.; Du, L.; Huang, X.; et al. Genomic profiling of esophageal squamous cell carcinoma to reveal actionable genetic alterations. J. Clin. Oncol. 2021, 39 (Suppl. S4), e160452. [Google Scholar] [CrossRef]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, M. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, H.; Liu, X.; Wang, Q.; Zhang, X.; He, J. Epidermal growth factor receptor is a prognosis predictor in patients with esophageal squamous cell carcinoma. Ann. Thorac. Surg. 2014, 98, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Hu, W.; Wang, J.; Deng, X.; Zhang, P.; Zhang, X. Phase II study of concurrent chemoradiation in combination with erlotinib for locally advanced esophageal carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 1407–1412. [Google Scholar] [CrossRef]
- Zhao, C.H.; Lin, L.; Liu, J.Z.; Liu, R.R.; Chen, Y.L.; Ge, F.J. A phase II study of concurrent chemoradiotherapy and erlotinib for inoperable esophageal squamous cell carcinoma. Oncotarget 2016, 7, 57310–57316. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Jing, Z.; Luo, H.; Jiang, W.; Ma, L.; Hu, W. Chemoradiotherapy with extended nodal irradiation and/or erlotinib in locally advanced esophageal squamous cell cancer: Long-term update of a randomized phase 3 trial. Br. J. Cancer 2020, 123, 1616–1624. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Fan, Q.; Lu, P.; Ying, J.; Ma, C.; Liu, W. Icotinib in patients with pretreated advanced esophageal squamous cell carcinoma with EGFR overexpression in EGFR gene amplification: A single-arm, multicenter phase II study. J. Thorac. Oncol. 2016, 11, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Jiang, W.; Ma, L.; Chen, R.; Fang, M.; Ding, L.; Hua, Y.; Du, D.; Jing, Z.; Xie, R.; et al. Icotinib with concurrent radiotherapy vs. radiotherapy alone in older adults with unresectable esophageal squamous cell carcinoma: A phase II randomized clinical trial. JAMA Netw. Open 2020, 3, e2019440. [Google Scholar] [CrossRef] [PubMed]
- Moehler, M.; Madener, A.; Thuss-Patience, P.; Brenner, B.; Meiler, J.; Ettrich, T.; Hofheinz, R.D.; Al-Batran, S.; Vogel, A.; Mueller, L.; et al. Cisplatin and 5-fluorouracil with or without epidermal growth factor receptor inhibition panitumumab for patients with non-resectable, advanced or metastatic esophageal squamous cell cancer: A prospective, open-label, randomised phase III AIO/EORTC trial (POWER). Ann. Oncol. 2020, 31, 228–235. [Google Scholar]
- Lu, Z.; Zhang, Y.; Fan, Q.; Pan, Y.; Jiang, D.; Lu, P.; Zhang, J.; Yuan, X.; Feng, Y.; Yang, S.; et al. Paclitaxel and cisplatin with or without cetuximab in metastatic esophageal squamous cell carcinoma: A randomized, multicenter phase II trial. Innovation 2022, 3, 10000239. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.; Song, X.; Jiang, B.; Gohgsun, X.; Song, L.; Qin, Q. Blocking FGFR4 exerts distinct anti-tumorigenic effects in esophageal squamous cell carcinoma. Thorac. Cancer 2018, 9, 1687–1698. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Quan, J.; Xiao, H.; Luo, J.; Zhang, Q.; Pi, G. FGFR inhibitor AZD4547 can enhance sensitivity of esophageal squamous cell carcinoma cells with epithelial-mesenchymal transition to gefitinib. Oncol. Rep. 2018, 39, 2270–2278. [Google Scholar] [CrossRef]
- Maehara, O.; Suda, G.; Natsuizaka, M.; Ohnishi, S.; Komatsu, Y.; Sato, F.; Nakai, M.; Sho, T.; Morikawa, K.; Ogawa, K.; et al. Fibroblast growth factor-2-mediated FGFR/ERK signaling supports maintenance of cancer stem-like cells in esophageal squamous cell carcinoma. Carcinogenesis 2017, 38, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Banclay, C.; Li, A.W.; Geldenhuys, L.; Naguna-Nibasheka, M.; Porter, G.A.; Veugelers, P.; Murphy, P.R.; Casson, A.G. Basic fibroblast growth factor (FGF-2) overexpression is a risk factor for esophageal cancer recurrence and reduced survival, which is ameliorated by coexpression of the FGF-2 antisense gene. Clin. Cancer Res. 2005, 11, 7683–7691. [Google Scholar] [CrossRef] [Green Version]
- Malehara, O.; Suda, G.; Natsuizaka, M. FGFR2 maintains cance cell differentiation via AKT signaling in esophageal squamous cell carcinoma. Cancer Biol. Ther. 2021, 22, 372–380. [Google Scholar] [CrossRef]
- Li, X.; Nie, C.; Tian, B. miR-671-5p blocks the progression of human ESCC by suppressing EGFR. Int. J. Biol. Sci. 2019, 15, 1892–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, L.; Wang, B.; Guo, L.; Liu, X.; Wang, B.; Yiang, J.; Xue, L.; Lu, N. HER2 expression and relevant clinicopathologic features in esophageal squamous cell carcinoma in a Chinese population. Diagn. Pathol. 2020, 24, 27. [Google Scholar] [CrossRef] [Green Version]
- Egebjerg, K.; Garbyal, R.S.; Hasselby, J.P.; Baeksgaard, L.; Mau-Sorensen, M. Prevalence of HER2 overexpression and amplification in squamous cell carcinoma of the esophagus: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2021, 16, 103339. [Google Scholar] [CrossRef]
- Shigaki, H.; Baba, Y.; Watanabe, M.; Murata, A.; Ishimoto, T.; Iwatsuki, M. PIK3CA mutation is associated with a favorable prognosis among patients with curatively resected esophageal squamous cell carcinoma. Clin. Cancer Res. 2013, 19, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Shan, L.; Zhang, S.; Ying, J.; Xue, L.; Yuan, Y.; Xie, Y.; Lu, N. PIK3CA gene mutations and overexpression: Implications for prognostic biomarker and therapeutic target in Chinese esophageal squamouas cell carcinoma. PLoS ONE 2014, 9, e103021. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, Y.; Wang, B.; Zhao, Y.; Zhang, F.; Ding, B. Mutational landscape of pan-cancer patients with PIK3CA alterations in Chinese population. BMC Med. Genom. 2022, 15, 146. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, S.E.; Bae, Y.S.; Kim, D.J.; Lee, C.G.; Hur, J.; Chung, H. PIK3CA amplification is associated with poor prognosis among patients with curatively resected esophageal squamous cell carcinoma. Oncotarget 2016, 7, 3691–3700. [Google Scholar] [CrossRef] [Green Version]
- Kojima, T.; Kato, K.; Hara, H.; Takahashi, S.; Muro, K.; Nishina, T.; Wakabayashi, M.; Nomura, S.; Sato, A.; Ohtsu, A.; et al. Phase II study of BKM 120 in patients with advanced esophageal squamous cell carcinoma (EPOC 1303). Esophagus 2022, 19, 702–710. [Google Scholar] [CrossRef]
- Hirose, W.; Oshikiri, H.; Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 system and esophageal cancer. Cancers 2022, 14, 4702. [Google Scholar] [CrossRef]
- Kim, Y.R.; Oh, J.E.; Kim, M.S.; Kang, M.R.; Park, S.W.; Han, J.Y.; Eom, H.S.; Yoo, N.J.; Lee, S.H. Oncogenic NRF2 mutations in squamous cell carcinomas of esophagus and skin. J. Pathol. 2010, 220, 446–451. [Google Scholar] [CrossRef]
- Kawasaki, Y.; Okumura, H.; Uchikado, Y.; Kita, Y.; Sasaki, K.; Owaki, T.; Ishigami, S.; Natsugoe, S. Nrf2 is useful for predicting the effect of chemoradiation therapy on esophageal squamous cell carcinoma. Am. Surg. Oncol. 2014, 21, 2347–2352. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Li, M.; Kong, L.; Yu, J. The expression of p-p62 and nuclear Nrf2 in esophageal squamous cell carcinoma and association with radioresistance. Thorac. Cancer 2020, 11, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.H.; Kuan, W.H.; Chuang, W.L.; Chuo, I.Y.; Liu, H.; Shieh, D.B.; Liu, H.; Tan, B.; Wang, Y.C. Dysregulation of SOX17/NRF2 axis confers chemoradiotherapy and emerges as a novel therapeutic target in esophageal squamous cell carcinoma. J. Biomed. Sci. 2022, 29, 90. [Google Scholar] [CrossRef]
- Jiang, X.; Zhou, X.; Yu, X.; Chen, X.; Hu, X.; Lu, J.; Zhao, H.; Cao, Q.; Gu, Y.; Yang, Y.; et al. High expression of nuclear NRF2 combined with NFE2L” alterations predicts poor prognosis in esophageal squamous cell carcinoma patients. Mod. Pathol. 2022, 35, 929–937. [Google Scholar] [CrossRef]
- Horiuchi, M.; Taguchi, K.; Hirose, W.; Tsuchida, K.; Suzuki, M.; Taniyama, Y.; Kamei, T.; Yamamoto, M. Cellular Nrf2 levels determine cell fate during chemical carcinogenesis in esophageal epithelium. Mol. Cell Biol. 2021, 41, e00536-20. [Google Scholar] [CrossRef] [PubMed]
- Hirose, W.; Horiuchi, M.; Li, D.; Motoike, I.N.; Zhang, L.; Nishi, H.; Taniyama, Y.; Kamei, T.; Suzuki, M.; Kinoshita, K.; et al. Selective elimination of NRF2-activated cells by competition with neighboring cells in the esophageal epithelium. Cell. Mol. Gastroent. Hepatol. 2022; in press. [Google Scholar]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gaighate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, J.; Xiong, Z.; Huang, C.; Li, J.; Yang, W.; Han, Y.; Paiboonnruungruan, C.; Major, M.B.; Chen, K.N.; Kang, X.; et al. Hyperactivity of the transcriptional factor Nrf2 causes metabolic reprogramming in mouse esophagus. J. Biol. Chem. 2019, 294, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Galan-Cobo, A.; Sitthideatphailboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019, 79, 3251–3267. [Google Scholar] [CrossRef]
- Binkley, M.S.; Jeon, Y.J.; Nesselbush, M.; Moding, E.; Nabet, B.Y.; Alamanza, D.; Kunder, C.; Stehr, H.; Yoo, C.H.; Rhee, S.; et al. KEAP1/NFE2L2 mutations predict lung cancer radiation resistance that can be targeted by glutaminase inhibition. Cancer Discov. 2020, 10, 1826–1841. [Google Scholar] [CrossRef]
- Qie, S.; Yoshida, A.; Parnham, S.; Oleinik, N.; Boeson, G.C.; Boeson, C.C.; Ogretmen, B.; Bass, A.J.; Wong, K.K.; Rustgi, A.K.; et al. Targeting glutamine-sddiction and overcoming CDK4/6 inhibitor resistance in human esophageal squamous cell carcinoma. Nat. Commun. 2019, 10, 1296. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wu, Z.; Wong, G.; Pectasides, E.; Nagaraja, A.; Stachler, M.; Zhang, H.; Chen, T.; Zhang, H.; Liu, J.B.; et al. CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in esophageal squamous cell carcinoma. Nat. Commun. 2017, 8, 13897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.J.; Su, Y.G.; Ding, X.L.; Zhao, Z.J.; Wang, Y.Y. CDK4/6 inhibitor enhances the radiosensitization of esophageal squamous cell carcinoma (ESCC) by activating autophagy signaling via the suppression of mTOR. Am. J. Transl. Med. 2022, 14, 1616–1627. [Google Scholar]
- Chen, L.; Pan, J. Dual cyclin-dependent kinase 4/6 inhibition by PD-0332991 induces apoptosis and senescence in esophageal squamous cell carcinoma cells. Brit. J. Pharmacol. 2017, 174, 2427–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, T.; Piha-Paul, S.; Jalal, S.I.; Lunceford, J.; Kosiji, M.; Bennouna, J. Safety and antitumor activity of the anti-Programmed Death-1 antibody pembrolizumab in patients with advanced esophageal carcinoma. J. Clin. Oncol. 2018, 36, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.H.; Doi, T.; Moriwaki, T.; Kim, S.B.; Lee, S.H.; et al. Randomized phase III KEYNOTE-181 study of pembrolizumab versus chemotherapy in advanced esophageal cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Qin, S.; Ren, Z.; Meng, Z.; Chen, Z.; Chai, X.; Xiong, J.; Bai, Y.; Yang, L.; Zhu, H.; Fang, W.; et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: A multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 571–580. [Google Scholar] [CrossRef]
- Cao, Y.; Qin, S.; Luo, S.; Li, Z.; Cheng, Y.; Fan, Y.; Sun, Y.; Yin, X.; Yuan, X.; Li, W.; et al. Pembrolizumab versus chemotherapy for patients with esophageal squamous cell carcinoma enrolled in the randomised KEYNOTE-181 trial in Asia. ESMO Open 2022, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Doki, Y.; Ura, T.; Hamamoto, Y.; Kojima, T.; Tsushima, T.; Hironaka, S.; Hara, H.; Satoh, T.; Iwasa, S.; et al. Nivolumab in advanced squamous cell carcinoma (ATTRACTION-1/ONO-4538-07): Minimum of five years of follow-up. J. Clin. Oncol. 2021, 39, 207. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.Y.; Chin, K.; Kadowaki, S.; Ahn, M.J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus chemotherapy in patients with advanced esophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Zhou, Y.X.; Chen, P.; Sun, Y.T.; Zhang, B.; Qiu, M.Z. Comparison of PD-1 inhibitors in patients with advanced esophageal squamous cell carcinoma in the second-line setting. Front. Oncol. 2021, 11, 698732. [Google Scholar] [CrossRef]
- Shen, L.; Kato, K.; Kim, S.B.; Ajani, J.A.; Zhao, K.; He, Z.; Yu, X.; Shu, Y.; Luo, Q.; Wang, J.; et al. Tisellizumab versus chemotherapy as second-line treatment for advanced or metastatic esophageal squamous cell carcinoma (RATIONALE-302): A randomized phase III study. J. Clin. Oncol. 2022, 40, 3065–3076. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, Y.; Fan, Q.; Shu, Y.; Yang, L.; Cui, T.; Gu, K.; Tao, M.; Wang, X.; Cui, C.; et al. Clinical and biomarker analyses of sintilimab versus chemotherapy as second-line therapy for advanced or metastatic esophageal squamous cell carcinoma: A randomized, open-label phase 2 study (ORIENT-2). Nat. Commun. 2022, 13, 857. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.P.; Meidt-Beinker, N.M.; Gutting, T.; Maenz, M.; Betge, J.; Schulte, N.; Zhan, T.; Weidner, P.; Burgermeister, E.; Hofheinz, H.; et al. Second-line therapy with nivolumab and ipilimumab for older patients with esophageal squamous cell cancer (RAMONA): A multicenter, open-label phase 2 trial. Lancet Health Longev. 2022, 3, e417–e427. [Google Scholar] [CrossRef]
- He, Y.; Li, C.; Zhang, F.; Hu, B.; Sun, Y.; Xia, X. Clinical study on the second-line treatment of advanced esophageal squamous cell carcinoma with camrelizumab combined with apatinib and irinotecan: A single-arm, multicenter, phase II study. J. Clin. Oncol. 2022, 40 (Suppl. S4), 319. [Google Scholar] [CrossRef]
- Huang, J.; Xu, B.; Mo, H.; Zhang, W.; Chen, X.; Wu, D.; Qu, O.; Wang, X.; Lan, B.; Yang, B.; et al. Safety, activity, and biomarkers of SHR-1210, an anti-PD-1 antibody, for patients with advanced esophageal carcinoma. Clin. Cancer Res. 2018, 24, 1296–1304. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.P.; Li, Z.; Kim, S.B.; et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced esophageal cancer (KEYNOTE-590): A randomised, placebo-controlled, phase 3 study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Metges, J.P.; Kato, K.; Sun, J.M.; Shah, M.A.; Enzinger, P.C.; Adenis, A.; Doi, T.; Kojima, T.; Li, Z.; Kim, S.B.; et al. First-line pembrolizumab plus chemotherapy versus chemotherapy in advanced esophageal cancer: Longer-term efficacy, safety, and quality-of-life results from the phase 3 KEYNOTE-590 study. J. Clin. Oncol. 2022, 40 (Suppl. S4), 241. [Google Scholar] [CrossRef]
- Luo, H.; Lu, J.; Bai, Y.; Mao, T.; Wang, J.; Fan, Q.; Zhang, Y.; Zhao, K.; Chen, Z.; Gao, S.; et al. ESCORT-1st Investigators. Effect of camrelizumab vs. placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: The ESCORT-1st randomized clinical trial. JAMA 2021, 326, 916–925. [Google Scholar] [CrossRef]
- Doki, Y.; Ajani, J.A.; Kato, K.; Xu, J.; Wyrwicz, L.; Motoyama, S.; Ogata, T.; Kawakami, H.; Hsu, C.H.; Adenis, A.; et al. CheckMate 648 trial investigators. Nivolumab combination therapy in advanced esophageal squamous cell carcinoma. N. Engl. J. Med. 2022, 386, 449–462. [Google Scholar] [CrossRef]
- Lu, Z.; Wang, J.; Shu, Y.; Liu, L.; Kong, L.; Yang, L.; Wang, B.; Sun, G.; Ji, Y.; Cao, G.; et al. Sintilimab versus placebo in combination with chemotherapy as first line treatment for locally advanced or metastatic esophageal squamous cell carcinoma (ORIENT-15): Multicentre, randomised, double blind, phase 3 trial. BMJ 2022, 377, e068714. [Google Scholar] [CrossRef]
- Wang, Z.X.; Cui, C.; Yao, J.; Zhang, Y.; Li, M.; Feng, J.; Yang, S.; Fan, Y.; Shi, J.; Zhang, X.; et al. Toripilimab plus chemotherapy in treatment-naive, advanced esophageal squamous cell carcinoma (JUPITER-06): A multi-center phase 3 trial. Cancer Cell 2022, 40, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Song, Y.; Zhao, K.; Liu, Y.; Fan, Q.; Wang, X.; Li, Q.; Wang, X.; Huang, J. SHR-1316, an anti-PD-L1 antibody, plus chemotherapy as the first line treatment for advanced esophageal squamous cell carcinoma. A multicentre, phase 2 study. Thorac. Cancer 2021, 12, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, H.; Chen, Y.; Zhu, C.; Fang, W.; Yu, Z.; Mao, W.; Xiang, J.; Han, Y.; Chen, Z.; et al. Neoadjuvant chemoradiotherapy followed by surgery versus surgery alone for locally advanced squamous cell carcinoma of the esophagus (NEOCRTEC5010): A phase III multicenter, randomized, open-label clinical trial. J. Clin. Oncol. 2018, 36, 2796–2803. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.H.; Kim, H.; Park, S.Y.; Kim, D.J.; Lee, C.G.; Cho, J.; Kim, J.H.; Kim, H.R.; Kim, Y.H.; Park, S.R.; et al. Phase II trial of preoperative chemoradiotherapy and pembrolizumab for locally advanced esophageal squamous cell carcinoma (ESCC). J. Clin. Oncol. 2019, 37 (Suppl. S15), 4027. [Google Scholar] [CrossRef]
- Li, C.; Zhao, S.; Zheng, Y.; Han, Y.; Chen, X.; Cheng, Z.; Wu, Y.; Feng, X.; Qi, W.; Chen, K.; et al. Preoperative pembrolizumab combined with chemoradiotherapy for esophageal squamous cell carcinoma (PALACE-1). Eur. J. Cancer 2021, 144, 232–241. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, C.; Yu, B.; Zhao, S.; Li, J.; Chen, X.; Li, H. Preoperative pembroluzumab combined with chemoradiotherapy for esophageal squamous cell carcinoma: Trial design. JTCVS Open 2022, 9, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Sun, Z.; Zhang, Y.; Shen, L.; Liu, Q.; Zhang, C.; Wu, H.; Lin, H.; Bai, Y.; Ma, X.; et al. Neoadjuvant chemoradiotherapy combined with perioperative toripilimab in locally advanced esophageal cancer. J. Clin. Oncol. 2022, 40 (Suppl. S16), e16065. [Google Scholar] [CrossRef]
- Jiang, N.; Jiang, M.; Zhu, X.; Ren, B.; Zhang, J.; Guo, Z.; Wu, Y.; Song, X.; Zhao, L.; Kong, C.; et al. SCALE-1: Safety and efficacy of short course neoadjuvant chemoradiotherapy plus toripilimab for locally advanced resectable squamous cell carcinoma of esophagus. J. Clin. Oncol. 2022, 40 (Suppl. S16), 4063. [Google Scholar]
- Yang, P.; Zhou, X.; Yang, X.; Wang, Y.; Sun, T.; Feng, S.; Ma, X. Neoadjuvant camrelizumab plus chemotherapy in treating locally advanced esophageal squamous cell carcinoma patients: A pilot study. World J. Surg. Oncol. 2021, 19, 333. [Google Scholar] [CrossRef]
- Yang, W.; Xing, X.; Yeung, S.C.; Wang, S.; Chen, W.; Bao, Y.; Wang, F.; Feng, S.; Peng, F.; Wang, X.; et al. Neoadjuvant programmed cell death 1 blockade combined with chemotherapy for resectable esophageal squamous cell carcinoma. J. Immunother. Cancer 2022, 10, e003497. [Google Scholar] [CrossRef]
- Li, Z.; Liu, J.; Zhang, M.; Shao, J.; Yang, Y.; Li, H.; Liu, Z.; Zhang, R.; Yun, B.; Chen, H. A phase II study of neoadjuvant immunotherapy combined with chemotherapy (camrelizumab plus albumin paclitaxel and carboplatin) in resectable thoracic esophageal squamous cell carcinoma patients: Interim results. J. Clin. Oncol. 2021, 39 (Suppl. S15), 4060. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, L.; Cheng, Y.; Liu, Z.; Cai, X.; Shao, J.; Zhang, M.; Liu, J.; Sun, Y.; Li, Y.; et al. Three-arm phase II trial comparing camrelizumab plus chemotherapy versus camrelizumab plus chemoradiation versus chemoradiation as preoperative treatment for locally advanced esophageal squamous cell carcinoma (NICE-2 study). BMC Cancer 2022, 22, 506. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Kato, K.; Daiko, H.; Kojima, T.; Hara, H.; Abe, T.; Tsubosa, Y.; Nagashima, K.; Aoki, K.; Mizoguchi, Y.; et al. Feasibility study of nivolumab as neoadjuvant chemotherapy for locally esophageal carcinoma: FRONTiER (JCOG1804E). Future Oncol. 2020, 16, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Kato, K.; Daiko, H.; Kojima, T.; Hara, H.; Abe, T.; Tsubosa, Y.; Kawakubo, H.; Fujita, T.; Fukuda, T.; et al. FRONTiER: A feasibility trial of nivolumab with neoadjuvant CF or DCF, FLOT therapy for locally advanced esophageal carcinoma (JCOG1804E)—The short term results of cohort A and B. J. Clin. Oncol. 2021, 39 (Suppl. S3), 202. [Google Scholar] [CrossRef]
- Matsuda, S.; Yamamoto, S.; Kato, K.; Daiko, H.; Kojima, T.; Hara, H.; Abe, T.; Tsubosa, Y.; Kawakubo, H.; Nagashima, K.; et al. FRONTiER: A feasibility trial of nivolumab with neoadjuvant CF or DCF, FLOT therapy for locally advanced esophageal carcinoma (JCOG1804E)—Short term results for cohort C and D. J. Clin. Oncol. 2022, 40 (Suppl. S4), 286. [Google Scholar] [CrossRef]
- Shang, X.; Zhang, C.; Zhao, G.; Zhang, W.; Liu, L.; Duan, X.; Yue, J.; Ma, Z.; Chen, C.; Meng, B.; et al. LBA3—Safety and efficacy of pembrolizumab combined with paclitaxel and cisplatin as neoadjuvant treatment for locally advanced resectable (stage III) squamous cell carcinoma (Keystone-001): Interim analysis of a prospective, single-arm, single-center phase II trial. Ann. Oncol. 2021, 32 (Suppl. S7), S1428–S1457. [Google Scholar]
- Shang, X.; Zhang, W.; Zhao, G.; Liang, F.; Zhang, C.; Yue, J.; Duan, X.; Ma, Z.; Chen, C.; Pang, Q.; et al. Pembrolizumab combined with neoadjuvant chemotherapy versus neoadjuvant chemoradiotherapy followed by surgery for locally advanced esophageal squamous cell carcinoma: Protocol for a multicentre, prospective, randomized-controlled, phase III clinical study (Keystone-002). Front. Oncol. 2022, 12, 831345. [Google Scholar]
- Xing, W.; Zhao, L.; Zheng, Y.; Liu, B.; Liu, X.; Li, T.; Zhang, Y.; Ma, B.; Yang, Y.; Shang, Y.; et al. The sequence of chemotherapy and toripalimab might influence the efficacy of neoadjuvant chemoimmunotherapy in locally advanced esophageal squamous cell cancer—A phase II study. Front. Immunol. 2021, 12, 772450. [Google Scholar] [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdal, J.; Zander, T.; Van Gustern, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N. Engl. J. Med. 2021, 384, 1191–1203. [Google Scholar] [CrossRef]
- Park, S.; Sun, J.M.; Choi, Y.L.; Oh, D.; Kim, H.K.; Lee, T.; Chi, S.A.; Lee, S.H.; Choi, Y.S.; Jung, S.H.; et al. Adjuvant durvalumab for esophageal squamous cell carcinoma after neoadjuvant chemoradiotherapy: A placebo-controlled, randomized, double blind, phase II study. ESMO Open 2022, 7, 100385. [Google Scholar] [CrossRef]
- Jing, Z.; Du, D.; Zhang, N.; Dai, H.; Wang, X.; Hua, Y.; Jiang, M.; Wu, C. Combination of radiation therapy and anti-PD1 antibody SHR-1210 in treating patients with esophageal squamous cell cancer. Int. J. Radiat. Oncol. 2018, 102, e31. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yan, C.; Gao, X.; Li, X.; Cao, F.; Zhao, G.; Zhao, J.; Er, P.; Zhang, T.; Che, X.; et al. Safety and feasibility of radiotherapy plus camrelizumab for locally advanced esophageal squamous cell carcinoma. Oncologist 2021, 26, e1110–e1124. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Oh, D.; Choi, Y.L.; Chi, S.A.; Kim, K.; Ahn, M.J. Durvalumab and tremelimumab with definitive chemoradiotherapy for locally advanced esophageal squamous cell carcinoma. Cancer 2022, 128, 2148–2158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, C.; Zhang, T.; Chen, X.; Dong, J.; Zhao, J.; Han, H.; Wang, J.; Zhao, G.; Cao, F.; et al. Addition of camrelizumab to docetaxel, cisplatin, and radiation therapy in patients with locally advanced esophageal squamous cell carcinoma: A phase 1b study. Oncoimmunology 2021, 10, e1971418. [Google Scholar] [CrossRef]
- Wei, T.; Ti, W.; Song, Q.; Cheng, Y. Study of PD-1 inhibitors in combination with chemoradiotherapy/chemotherapy in patients with esophageal squamous carcinoma. Curr. Oncol. 2022, 29, 2920–2927. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Testa, U.; Castelli, G.; Pelosi, E. The Molecular Characterization of Genetic Abnormalities in Esophageal Squamous Cell Carcinoma May Foster the Development of Targeted Therapies. Curr. Oncol. 2023, 30, 610-640. https://doi.org/10.3390/curroncol30010048
Testa U, Castelli G, Pelosi E. The Molecular Characterization of Genetic Abnormalities in Esophageal Squamous Cell Carcinoma May Foster the Development of Targeted Therapies. Current Oncology. 2023; 30(1):610-640. https://doi.org/10.3390/curroncol30010048
Chicago/Turabian StyleTesta, Ugo, Germana Castelli, and Elvira Pelosi. 2023. "The Molecular Characterization of Genetic Abnormalities in Esophageal Squamous Cell Carcinoma May Foster the Development of Targeted Therapies" Current Oncology 30, no. 1: 610-640. https://doi.org/10.3390/curroncol30010048