
Effect of Geometrical Confinement on Ordering of Thermoplastic Polyurethanes with Crystallizable Hard and Soft Blocks


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
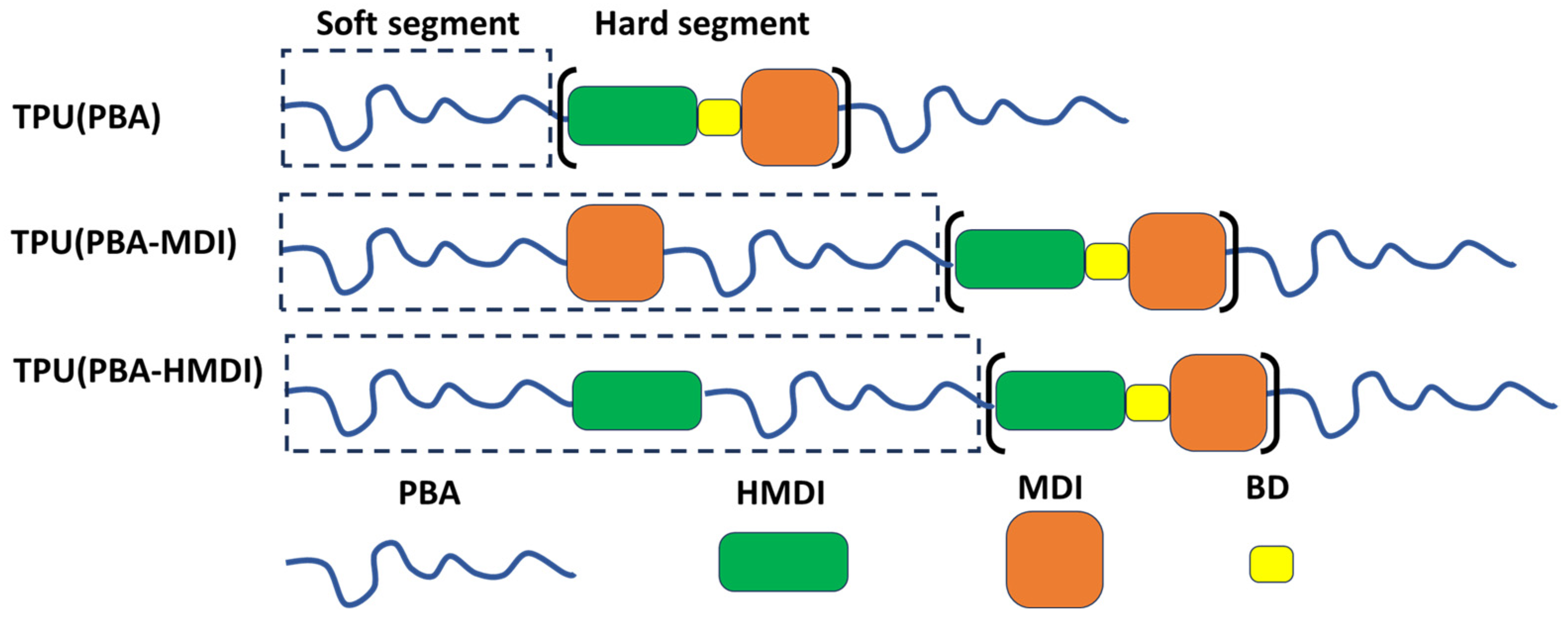
2.2. Synthesis of Multi-Block Thermoplastic Polyurethane (TPU)
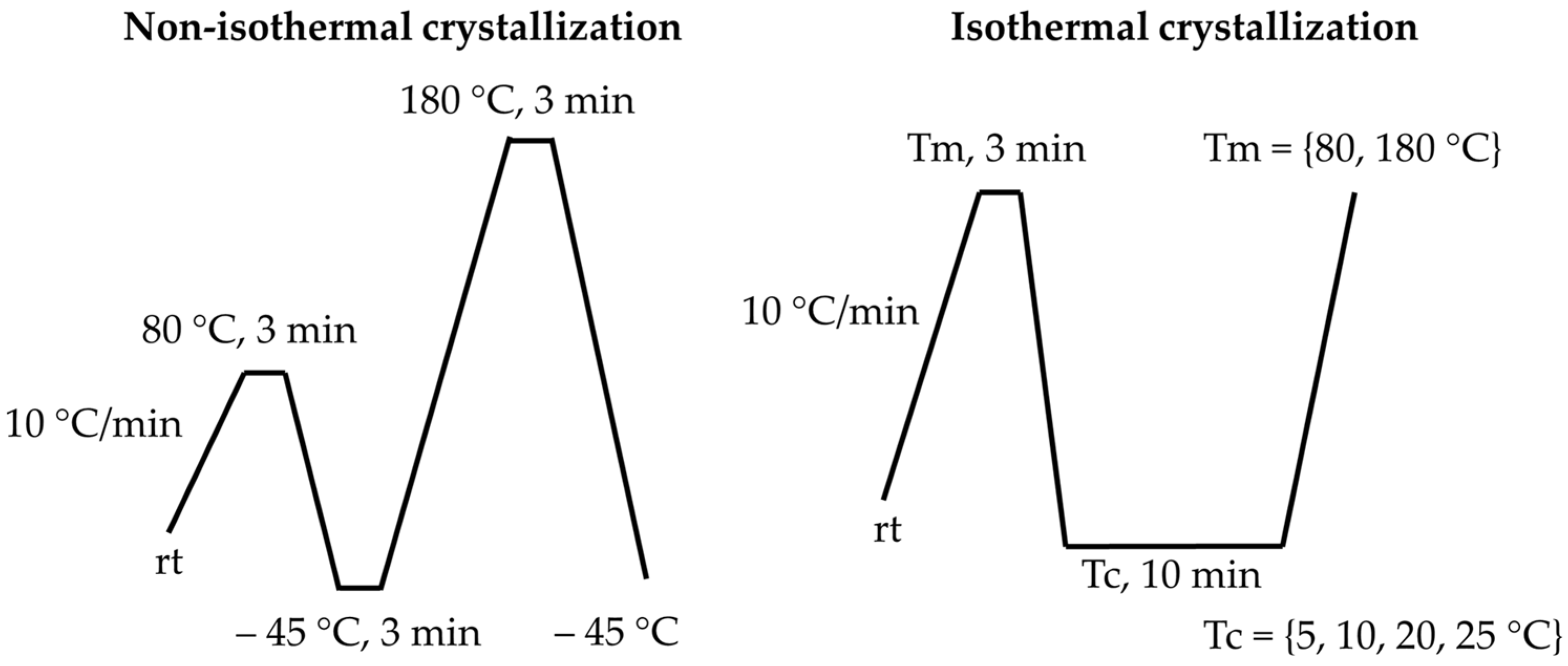
2.3. Characterization
3. Results
3.1. Thermal Analysis
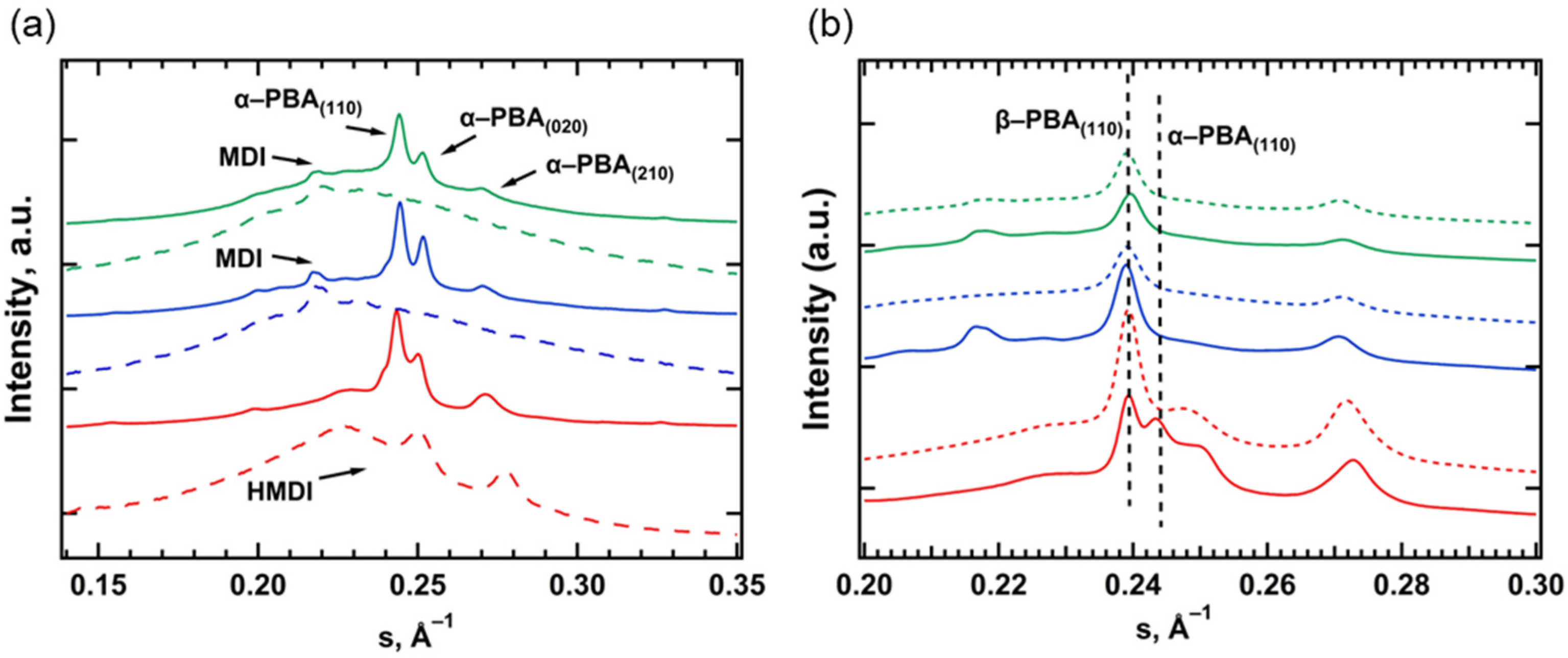
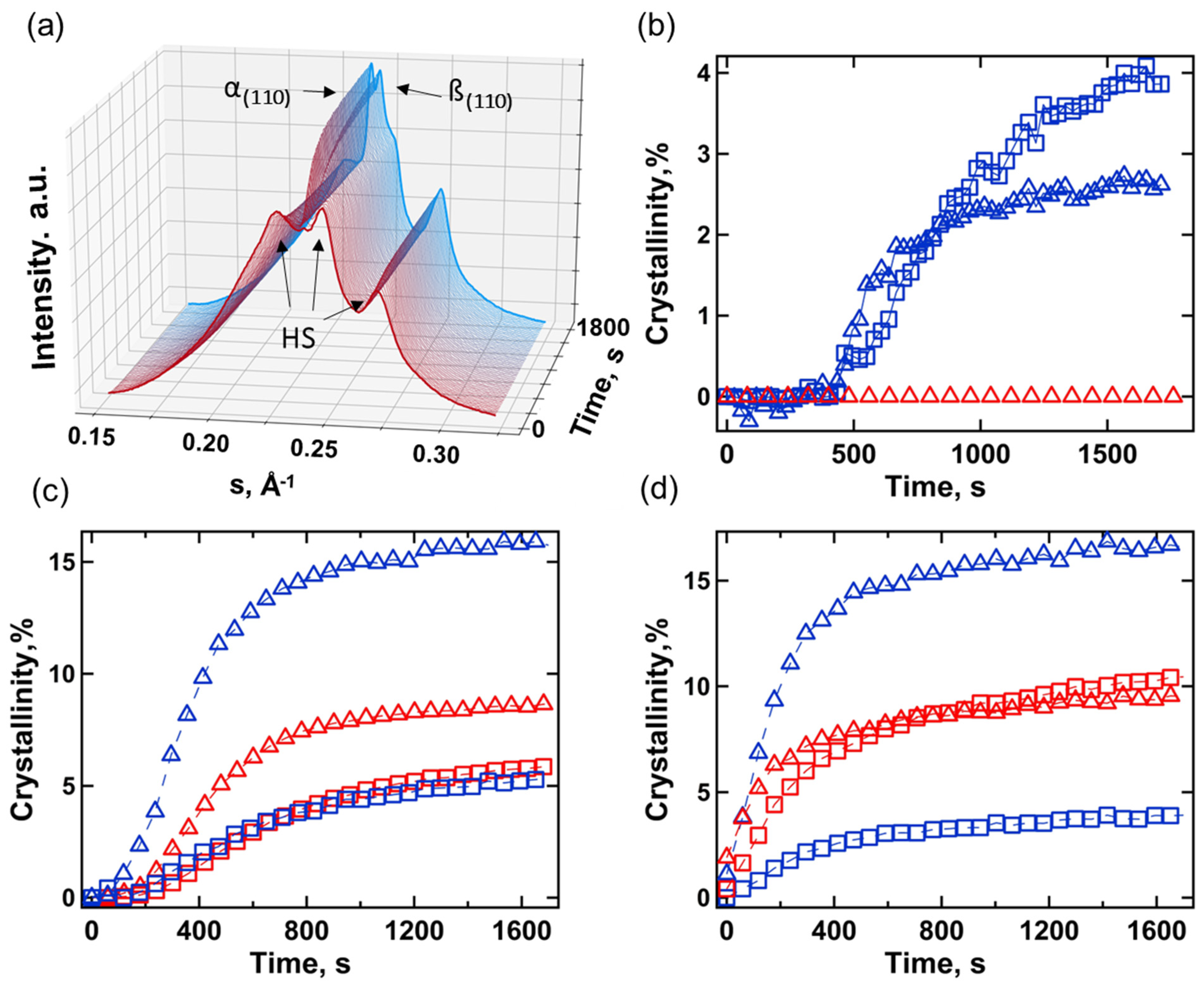
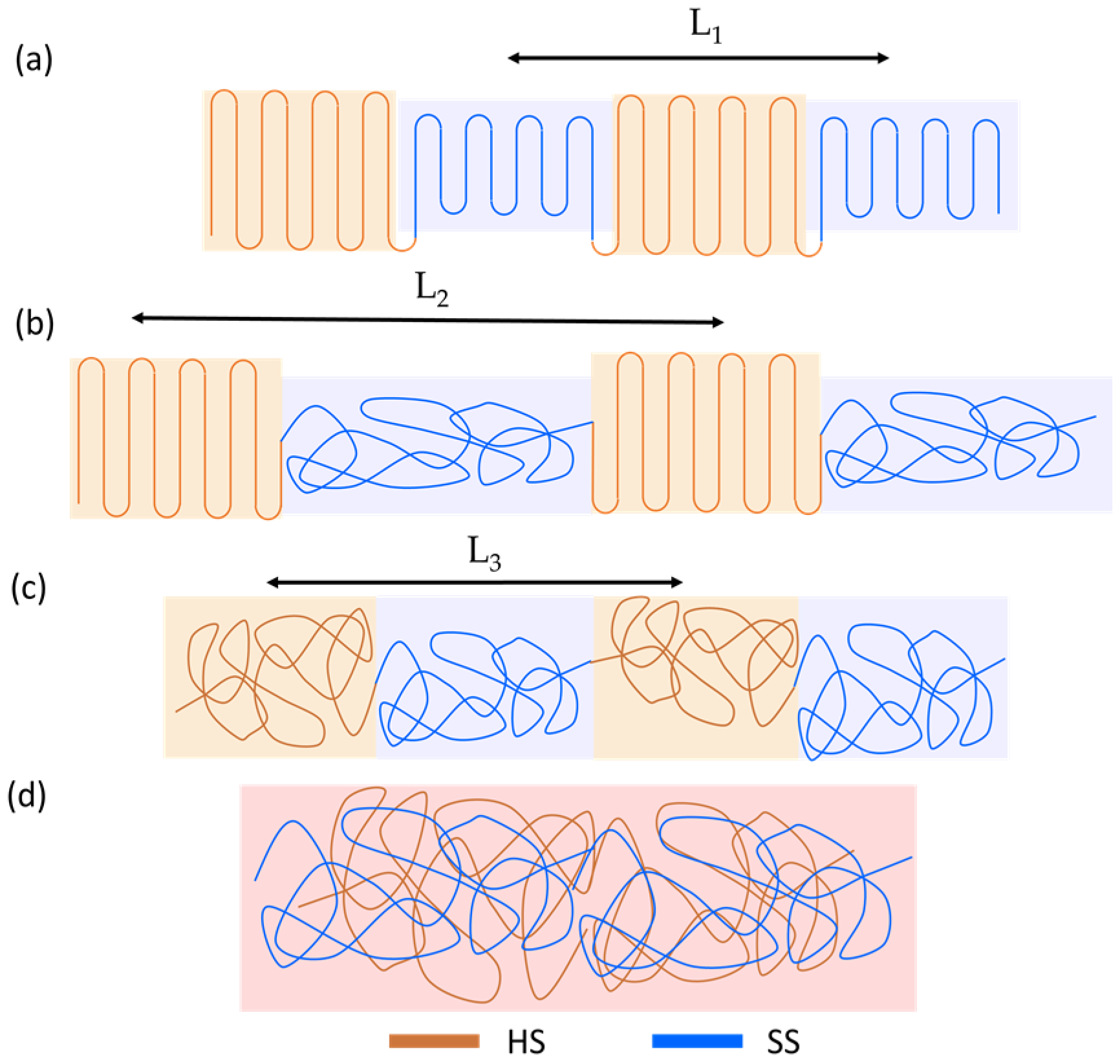
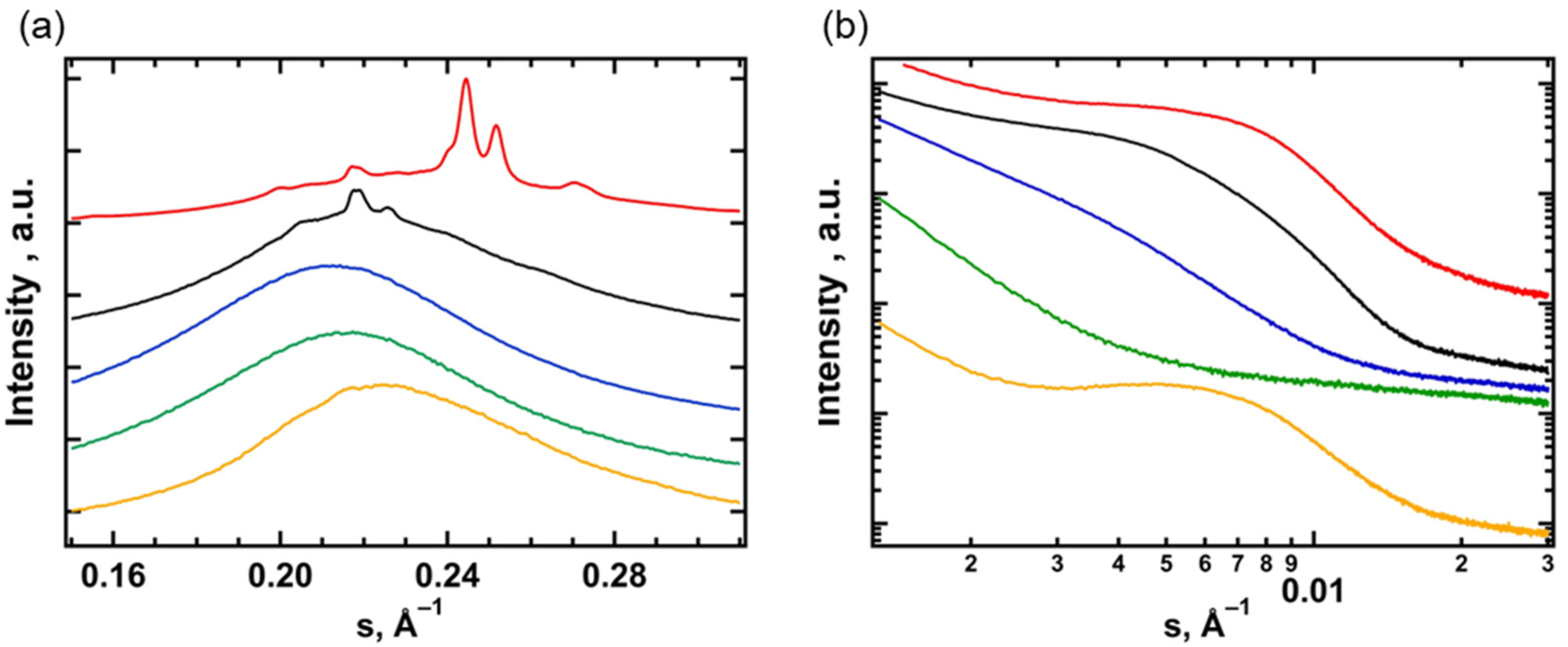
3.2. Small- and Wide-Angle X-ray Scattering
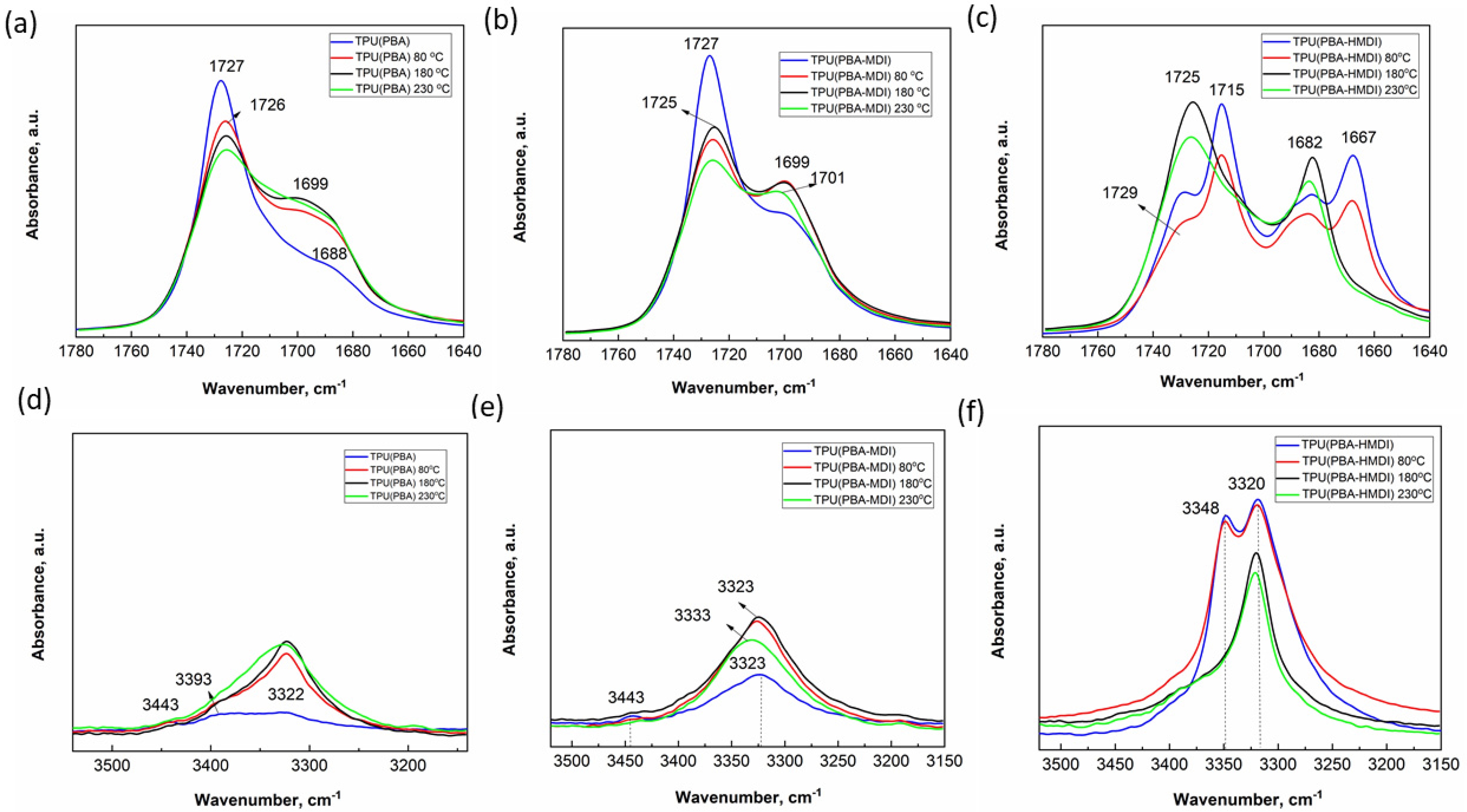
3.3. Study of Morphology of TPUs with FTIR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lendlein, A.; Langer, R. Biodegradable, elastic shape-memory polymers for potential biomedical applications. Science 2002, 296, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Hayashi, N.; Hayashi, S. Structure and properties of shape-memory polyurethane block copolymers. J. Appl. Polym. Sci. 1996, 60, 1061–1069. [Google Scholar] [CrossRef]
- Wang, Y.; Cheng, Z.; Liu, Z.; Kang, H.; Liu, Y. Cellulose nanofibers/polyurethane shape memory composites with fast water-responsivity. J. Mater. Chem. B 2018, 6, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Fan, F.; Sun, C.; Yu, Y.; Xu, H. Visible light-induced plasticity of shape memory polymers. ACS Appl. Mater. Interfaces 2017, 9, 33169–33175. [Google Scholar] [CrossRef] [PubMed]
- Lendlein, A.; Jiang, H.; Junger, O.; Langer, R. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chen, H.; Zhou, S.; Zhao, K.; Wang, B.; Hu, P. Thermo- and pH-sensitive shape memory polyurethane containing carboxyl groups. Polym. Chem. 2016, 7, 1739–1746. [Google Scholar] [CrossRef]
- Li, J.; Viveros, J.A.; Wrue, M.H.; Anthamatten, M. Shape-Memory Effects in Polymer Networks Containing Reversibly Associating Side-Groups. Adv. Mater. 2007, 19, 2851–2855. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, R.; Huang, M.; Dong, X.; Xu, W.; Wang, Y.; Hu, G.H.; Zhu, J. Synthesis and shape memory property of segmented poly(ester urethane) with poly(butylene 1,4-cyclohexanedicarboxylate) as the soft segment. RSC Adv. 2016, 6, 95527–95534. [Google Scholar] [CrossRef]
- Ahmad, M.; Xu, B.; Purnawali, H.; Fu, Y.; Huang, W.; Miraftab, M.; Luo, J. High Performance Shape Memory Polyurethane Synthesized with High Molecular Weight Polyol as the Soft Segment. Appl. Sci. 2012, 2, 535–548. [Google Scholar] [CrossRef]
- Yuan, X.; Sang, Z.; Zhao, J.; Zhang, Z.; Zhang, J.; Cheng, J. Synthesis and properties of non-isocyanate aliphatic thermoplastic polyurethane elastomers with polycaprolactone soft segments. J. Polym. Res. 2017, 24, 88. [Google Scholar] [CrossRef]
- Sabahi, N.; Roohani, I.; Wang, C.H.; Farajzadeh, E.; Li, X. Thermoplastic polyurethane-based shape memory polymers with potential biomedical application: The effect of TPU soft-segment on shape memory effect and cytocompatibility. Polymer 2023, 283, 121689. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Peh, K.W.E.; Yu, Q.; Lu, Y.; Hua, W.; Men, Y. Effect of Phase Separation and Crystallization on Enthalpy Relaxation in Thermoplastic Polyurethane. Macromolecules 2022, 55, 8566–8576. [Google Scholar] [CrossRef]
- Frick, A.; Rochman, A. Characterization of TPU-elastomers by thermal analysis (DSC). Polym. Test. 2004, 23, 413–417. [Google Scholar] [CrossRef]
- Minke, R.; Blackwell, J. Polymorphic Structures of Poly(tetramet hylene Adipate). J. Macromol. Sci.-Phys. B 1979, 16, 407–417. [Google Scholar] [CrossRef]
- Zheng, Y.; Pan, P. Crystallization of biodegradable and biobased polyesters: Polymorphism, cocrystallization, and structure-property relationship. Prog. Polym. Sci. 2020, 109, 101291. [Google Scholar] [CrossRef]
- Lotz, B.; Miyoshi, T.; Cheng, S. 50th anniversary perspective: Polymer crystals and crystallization: Personal journeys in a challenging research field. Macromolecules 2017, 50, 5995–6025. [Google Scholar] [CrossRef]
- Prasannan, A.; Bichtram, T.; Hsu, D.-Y.; Hong, P.-D.; Pan, G.-R. Nucleation effects of α-cyclodextrin inclusion complexes on the crystallization behavior of biodegradable poly(1,4-butylene adipate). Cryst. Eng. Comm. 2013, 15, 5119–5126. [Google Scholar] [CrossRef]
- Wang, H.; Gao, Z.; Yang, X.; Liu, K.; Zhang, M.; Qiang, X.; Wang, X. Epitaxial Crystallization Behavior of Poly(butylene adipate) on Orientated Poly(butylene succinate) Substrate. Polymer 2018, 10, 110. [Google Scholar] [CrossRef]
- Song, Y.; Ye, H.; Xu, J.; Hou, K.; Zhou, Q.; Lu, G. Stretch-induced bidirectional polymorphic transformation of crystals in poly(butylene adipate). Polymer 2014, 55, 3054–3061. [Google Scholar] [CrossRef]
- Bothe, M.; Emmerling, F.; Pretsch, T. Poly(ester urethane) with Vary-ing Polyester Chain Length: Polymorphism and Shape-Memory Behavior. Macromol. Chem. Phys. 2013, 214, 2683–2693. [Google Scholar] [CrossRef]
- Sun, X.; Fang, Q.; Li, H.; Ren, Z.; Yan, S. Effect of Anodic Alumina Oxide Pore Diameter on the Crystallization of Poly(butylene adipate). Langmuir 2016, 32, 3269–3275. [Google Scholar] [CrossRef] [PubMed]
- Chan-Chan, L.; Chan-Chan, L.; Solis-Correa, R.; Vargas-Coronado, R.; Cervantes-U, J.; Cauich-Rodríguez, J.; Quintana, P.; Bartolo-Pérez, P. Degradation studies on segmented polyurethanes prepared with HMDI, PCL and different chain extenders. Acta Biomater. 2010, 6, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Pielichowski, K.; Słotwińska, D.; Dziwiński, E. Segmented MDI/HMDI-based polyurethanes with lowered flammability. J. Appl. Polym. Sci. 2004, 91, 3214–3224. [Google Scholar] [CrossRef]
- Herrera, M.; Matuschek, G.; Kettrup, A. Thermal degradation of thermoplastic polyurethane elastomers (TPU) based on MDI. Polym. Degrad. Stab. 2002, 78, 323–331. [Google Scholar] [CrossRef]
- Ji, F.; Hu, J.; Li, T.; Wong, Y. Morphology and shape memory effect of segmented polyurethanes. Part I: With crystalline reversible phase. Polymer 2007, 48, 5133–5145. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, L.; Liu, H.; He, S.; Lui, X.; Li, W.; Huang, M.; Chen, Z. Polyurethane as smart biocoatings: Effects of hard segments on phase structures and properties. Prog. Org. Coat. 2021, 150, 106000. [Google Scholar] [CrossRef]
- Quiram, D.J.; Register, R.A.; Marchand, G.R.; Adamson, D.H. Chain Orientation in Block Copolymers Exhibiting Cylindrically Confined Crystallization. Macromolecules 1998, 31, 4891–4898. [Google Scholar] [CrossRef]
- Li, Y.J.; Ren, Z.Y.; Zhao, M.; Yang, H.C.; Chu, B. Multiphase structure of segmented polyurethanes—Effects of hard-segment flexibility. Macromolecules 1993, 26, 612–622. [Google Scholar] [CrossRef]
- Gorbunova, M.; Anokhin, D.V.; Abukaev, A.; Ivanov, D. The Influence of Long-Time Storage on the Structure and Properties of Multi-Block Thermoplastic Polyurethanes Based on Poly(butylene adipate) Diol and Polycaprolactone Diol. Materials 2023, 16, 818. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Anokhin, D.V.; Abukaev, A.F.; Ivanov, D.A. Impact of Soft Segment Composition on Phase Separation and Crystallization of Multi-Block Thermoplastic Polyurethanes Based on Poly(butylene adipate) Diol and Polycaprolactone Diol. Crystals 2023, 13, 1447. [Google Scholar] [CrossRef]
- Gorbunova, M.A.; Komov, E.V.; Grunin, L.Y.; Ivanova, M.S.; Abukaev, A.F.; Imamutdinova, A.M.; Ivanov, D.A.; Anokhin, D.V. Effect of separation of blocks on crystallization kinetics and phase composition of poly(butylene adipate) in multi-block thermoplastic polyurethanes. Phys. Chem. Chem. Phys. 2022, 24, 902–913. [Google Scholar] [CrossRef]
- Tarasov, A.E.; Lodygina, V.P.; Komratova, V.V.; Gorbunova, M.A.; Badamshina, E.R. New IR-Spectroscopic Methods for Determining the Hydroxyl Content in Oligomers. J. Appl. Spectrosc. 2017, 84, 211–216. [Google Scholar] [CrossRef]
- He, Y.; Xie, D.; Zhang, X. The Structure, Microphase-Separated Morphology, and Property of Polyurethanes and Polyureas. J. Mater. Sci. 2014, 49, 7339–7352. [Google Scholar] [CrossRef]
- Gan, Z.; Kuwabara, K.; Abe, H.; Iwata, H.; Doi, Y. Metastability and Transformation of Polymorphic Crystals in Biodegradable Poly(butylene adipate). Biomacromolecules 2004, 5, 371–378. [Google Scholar] [CrossRef]
- Gan, Z.; Abe, H.; Doi, Y. Temperature-Induced Polymorphic Crystals of Poly(butylene adipate). Macromol. Chem. Phys. 2002, 203, 2369–2374. [Google Scholar] [CrossRef]
- Mukhametzyanov, T.; Schmelzer, J.; Yarko, E.; Abdullin, A.; Ziganshin, M.; Sedov, I.; Schick, C. Crystal Nucleation and Growth in Cross-Linked Poly(ε-caprolactone) (PCL). Polymers 2021, 13, 3617. [Google Scholar] [CrossRef]
- D’hollander, S.; Gommes, C.J.; Mens, R.; Adriaensens, P.; Goderis, B.; Prez, F.D. Modeling the morphology and mechanical behavior of shape memory polyurethanes based on solid-state NMR and synchrotron SAXS/WAXD. J. Mater. Chem. 2010, 20, 3475–3486. [Google Scholar] [CrossRef]
- Koberstein, J.T.; Galambos, A.F. Multiple Melting in Segmented Polyurethane Block Copolymers. Macromolecules 1992, 25, 5618–5624. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N° | Sample | Polydiol | Diisocyanate | Chain Extender |
|---|---|---|---|---|
| 1 | TPU(PBA) | PBA | HMDI, MDI | BD |
| 2 | TPU(PBA-MDI) | PBA-MDI-PBA | ||
| 3 | TPU(PBA-HMDI) | PBA-HMDI-PBA |
| Sample | T(onset), °C | T(50%), °C | T(endset), °C | T2, °C | T2, °C | T3, °C |
|---|---|---|---|---|---|---|
| TPU(PBA) | 297 | 385 | 487 | 361 | 395 | 478 |
| TPU(PBA-MDI) | 283 | 385 | 494 | 358 | 495 | 470 |
| TPU(PBA-HMDI) | 296 | 380 | 488 | 361 | 409 | 450 |
| Sample | First Heating | First Cooling | Second Heating | Second Cooling | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tonset, °C | Tmaxc, °C | χDSC, % | Tonset, °C | Tmaxc, °C | χDSC, % | Tonset, °C | Tmaxc, °C | χDSC, % | Tonset, °C | Tmaxc, °C | χDSC, % | |
| TPU-PBA | 40.7 | 46.6 | 14 | 11.2 | 4.6 | 25 | 30.7 | 47.1 | 37 | 2.6 | −4.9 | 18 |
| TPU-PBA-MDI | 41.0 | 45.5 | 21 | 10.6 | 2.6 | 15 | 24.8 | 46.1 | 28 | −4.5 | −13.4 | 11 |
| TPU-PBA-HMDI | 40.4 | 44.5 | 27 | 12.7 | 6.6 | 33 | 29.7 | 24.2 | 36 | 15.6 | 8.7 | 33 |
| Sample | Melt Temperature, °C | χWAXS, % | ||
|---|---|---|---|---|
| Total | α-PBA | β-PBA | ||
| TPU(PBA) | native | 31 | 28 | 3 |
| 80 | 7 | 4 | 3 | |
| 180 | Amorphous | |||
| 230 | Amorphous | |||
| TPU(PBA-MDI) | native | 21 | 20 | 1 |
| 80 | 21 | 5 | 16 | |
| 180 | 15 | 6 | 9 | |
| 230 | Amorphous | |||
| TPU(PBA-HMDI) | native | 11 | 11 | 0 |
| 80 | 21 | 4 | 17 | |
| 180 | 20 | 11 | 9 | |
| 230 | 10 | 5 | 5 | |
| Temperature | SAXS Distance, nm | ||
|---|---|---|---|
| TPU(PBA) | TPU(PBA-MDI) | TPU(PBA-HMDI) | |
| 25 °C | 11.5 (L1) | 12.3 (L1) | 12.1 (L1) |
| 80 °C | 14.6 (L2) | 15.6 (L2) | 15.2 (L2) |
| 25 °C 30 min after cooling from 80 °C | 12.6 (L1) | 13.1 (L1) | 12.4 (L1) |
| 180 °C | 26.3 (L3) | 25.2 (L3) | 22.6 (L3) |
| 25 °C after cooling from 180 °C | 21.3 (L2/L3) | 15.2 (L2) | |
| 25 °C 30 min after cooling from 180 °C | 20.1 (L2/L3) | 16.1 (L1/L2) | 14.2 (L1) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abukaev, A.F.; Gorbunova, M.A.; Anokhin, D.V.; Ivanov, D.A. Effect of Geometrical Confinement on Ordering of Thermoplastic Polyurethanes with Crystallizable Hard and Soft Blocks. Crystals 2023, 13, 1662. https://doi.org/10.3390/cryst13121662
Abukaev AF, Gorbunova MA, Anokhin DV, Ivanov DA. Effect of Geometrical Confinement on Ordering of Thermoplastic Polyurethanes with Crystallizable Hard and Soft Blocks. Crystals. 2023; 13(12):1662. https://doi.org/10.3390/cryst13121662
Chicago/Turabian StyleAbukaev, Ainur F., Marina A. Gorbunova, Denis V. Anokhin, and Dimitri A. Ivanov. 2023. "Effect of Geometrical Confinement on Ordering of Thermoplastic Polyurethanes with Crystallizable Hard and Soft Blocks" Crystals 13, no. 12: 1662. https://doi.org/10.3390/cryst13121662