

Kinetic Optimization of the Batch Crystallization of an Active Pharmaceutical Ingredient in the Presence of a Low-Solubility, Precipitating Impurity

Abstract
:1. Introduction
2. Materials and Methodology
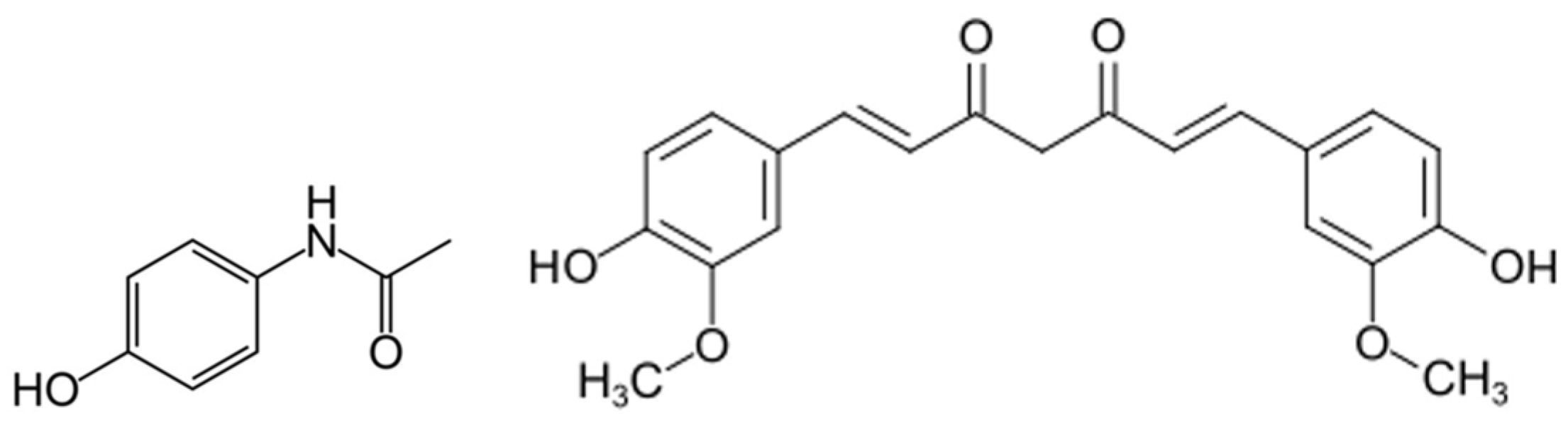
2.1. Materials
2.2. Solubility Measurements
2.3. Seeded Batch Crystallization Experiments
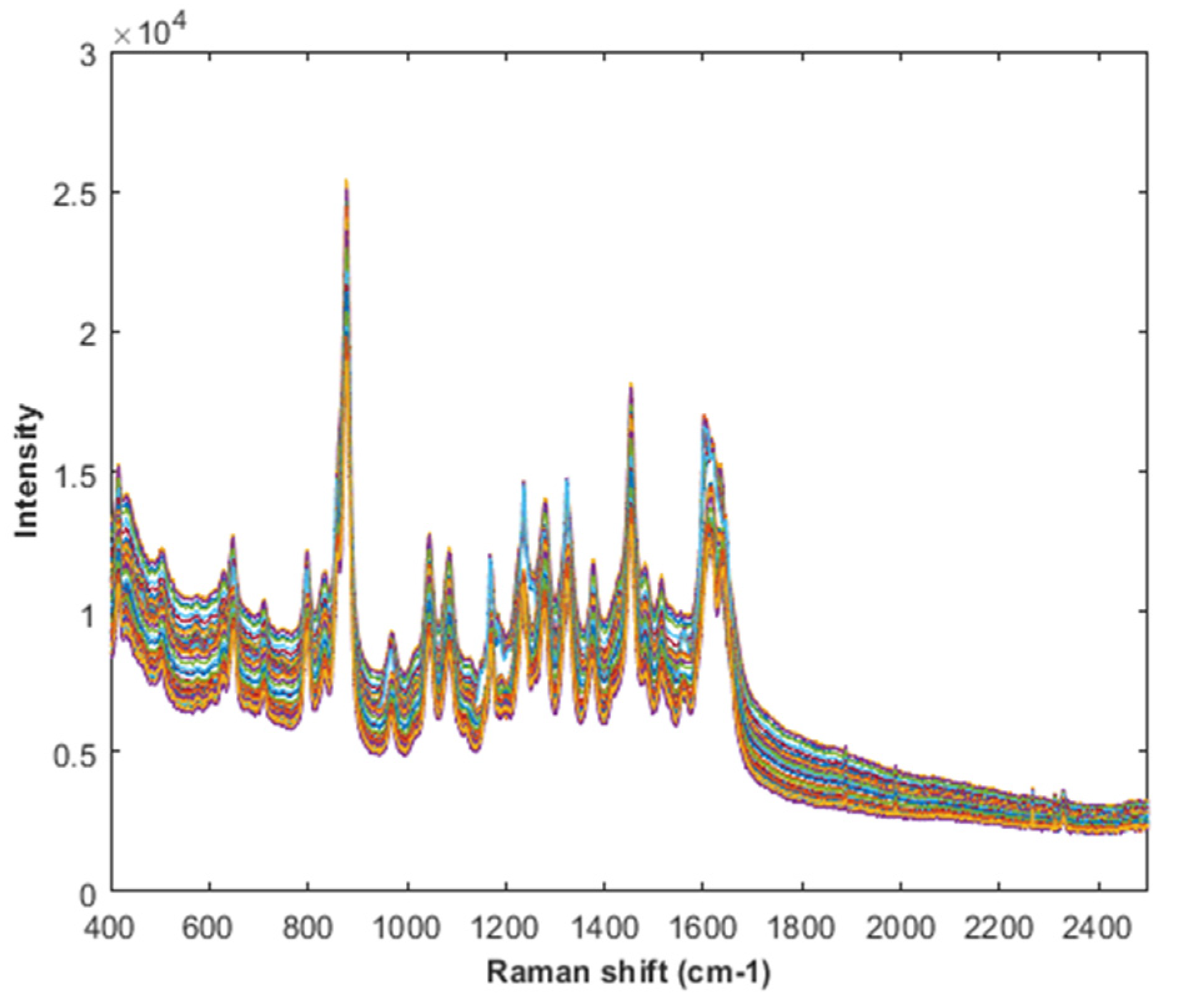
2.4. Raman Spectroscopy
2.5. HPLC Method
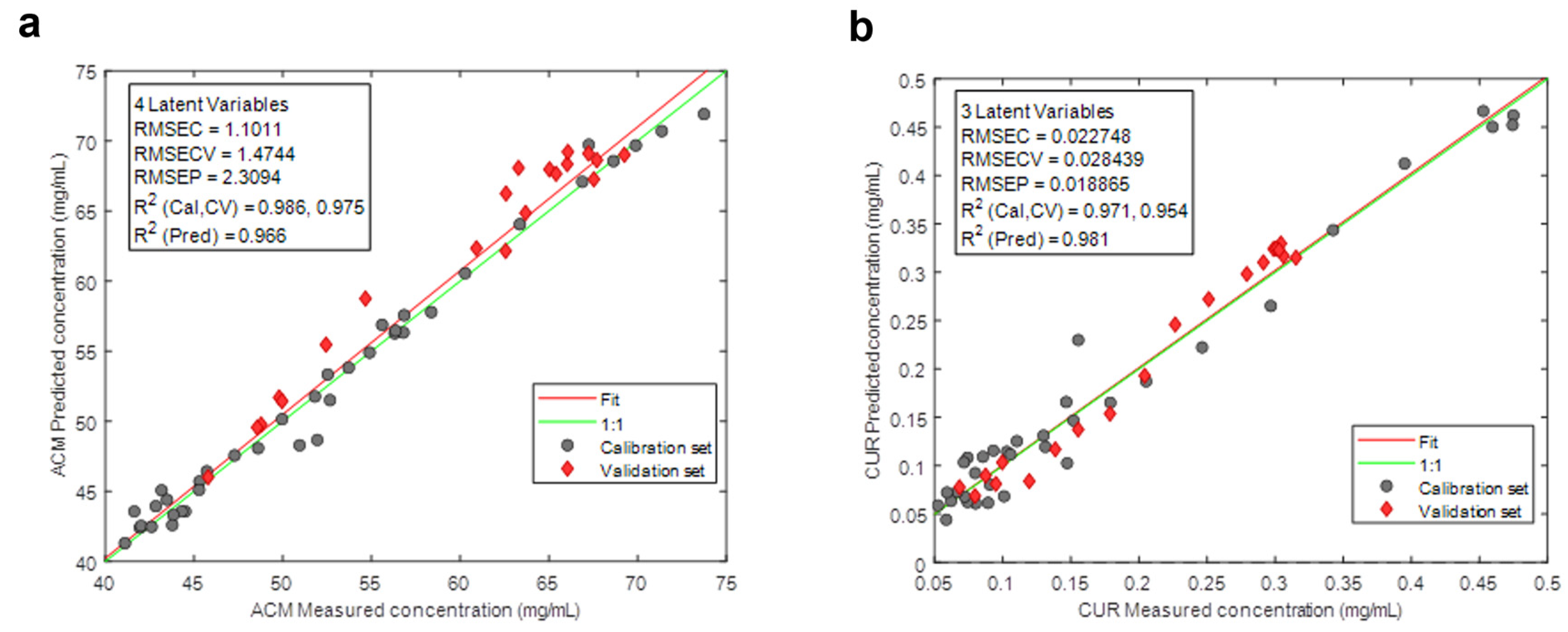
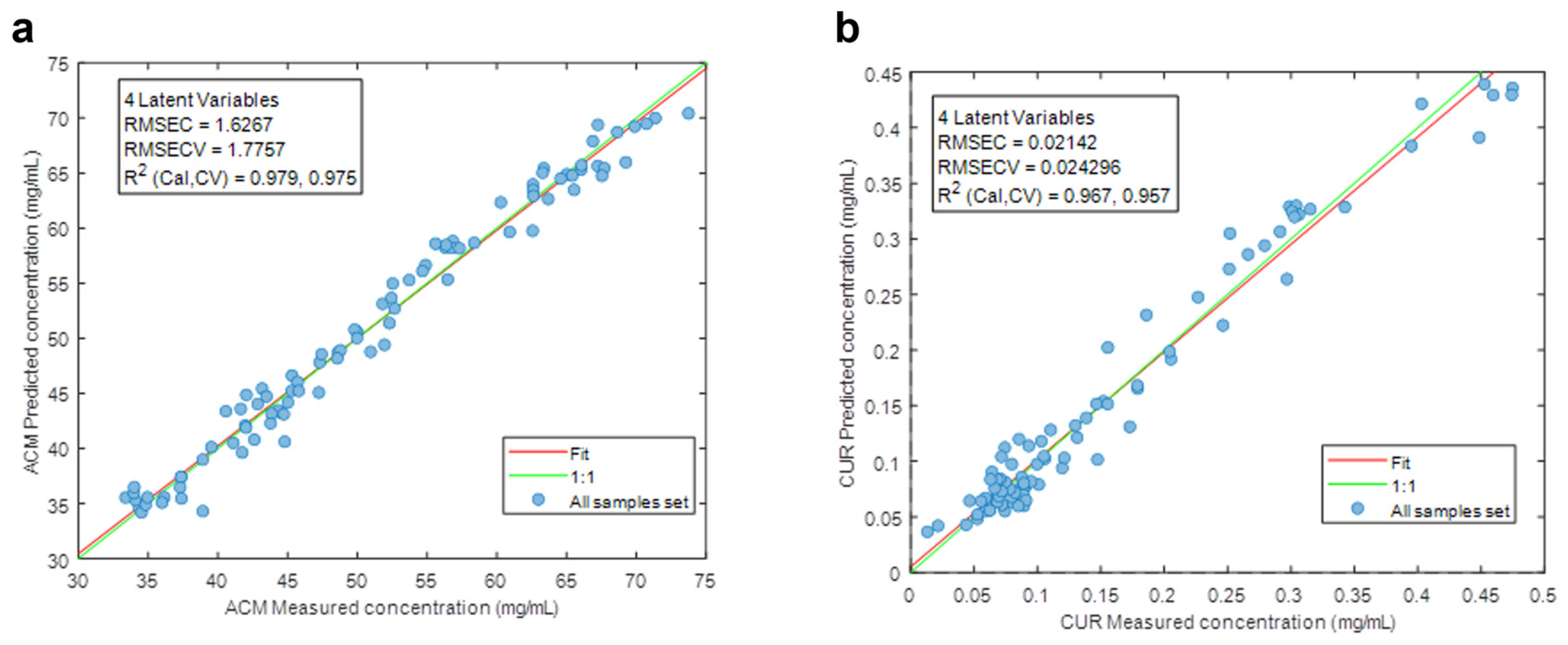
2.6. PLS Calibration
2.7. Parameter Estimation in gPROMS
3. Crystallization Model
3.1. Phase Balances
3.2. Yield and Purity
3.3. Supersaturation
3.4. Batch Crystallization Kinetics
3.5. Population Balance
3.6. PLS Equations
4. Results and Discussion
4.1. PLS Results
4.2. Particle Imaging
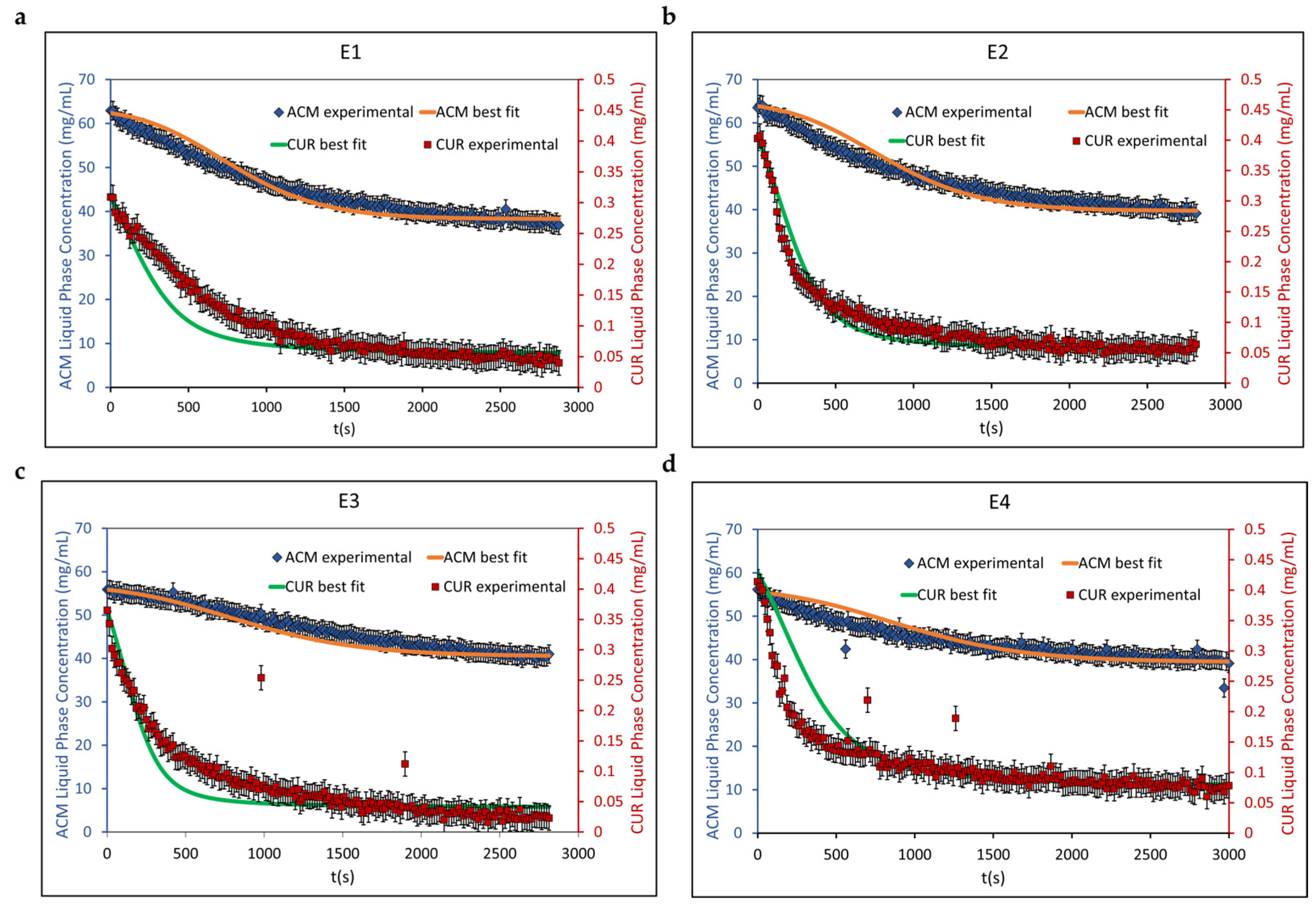
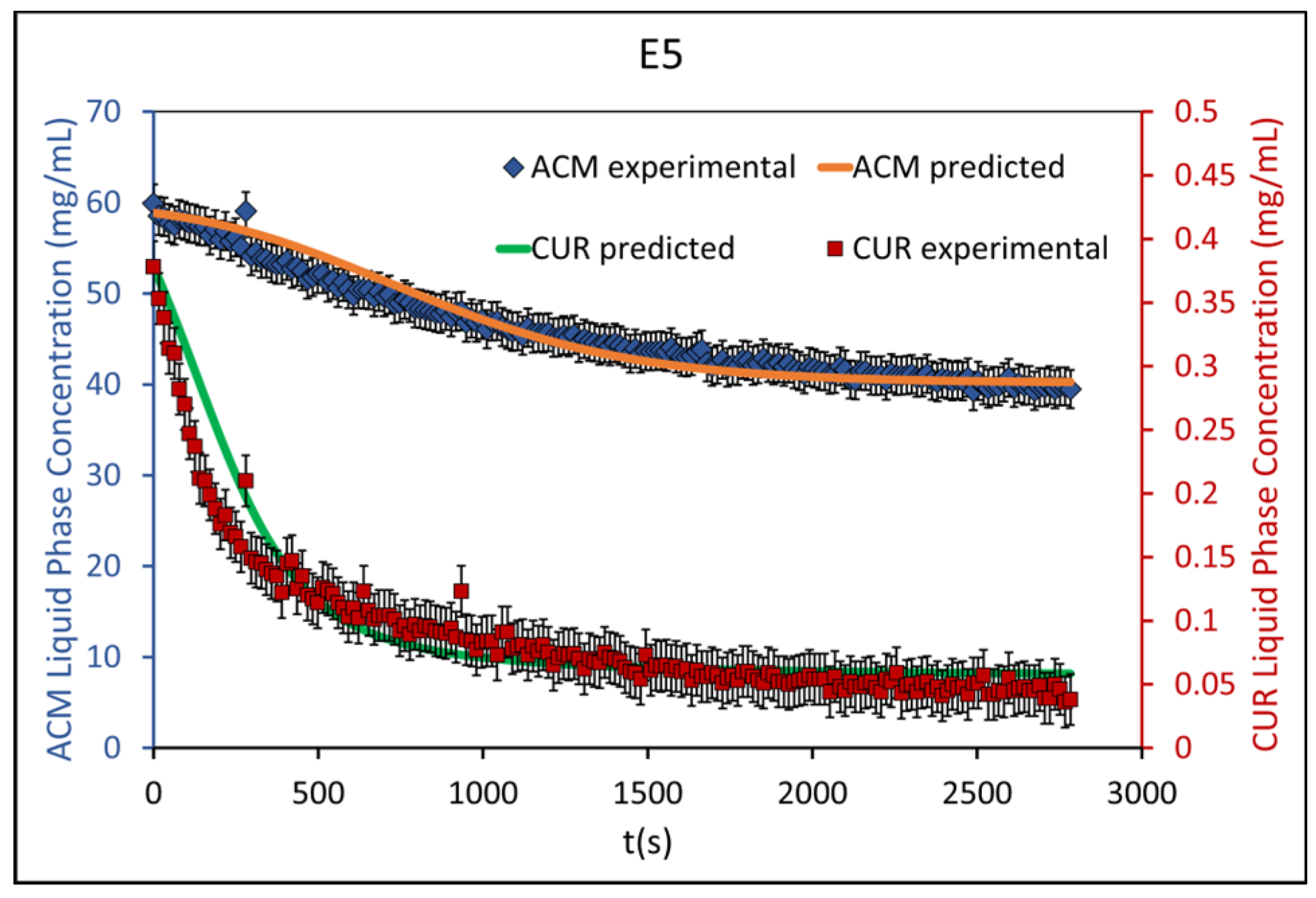
4.3. Population Balance Model Calibration and Validation Results
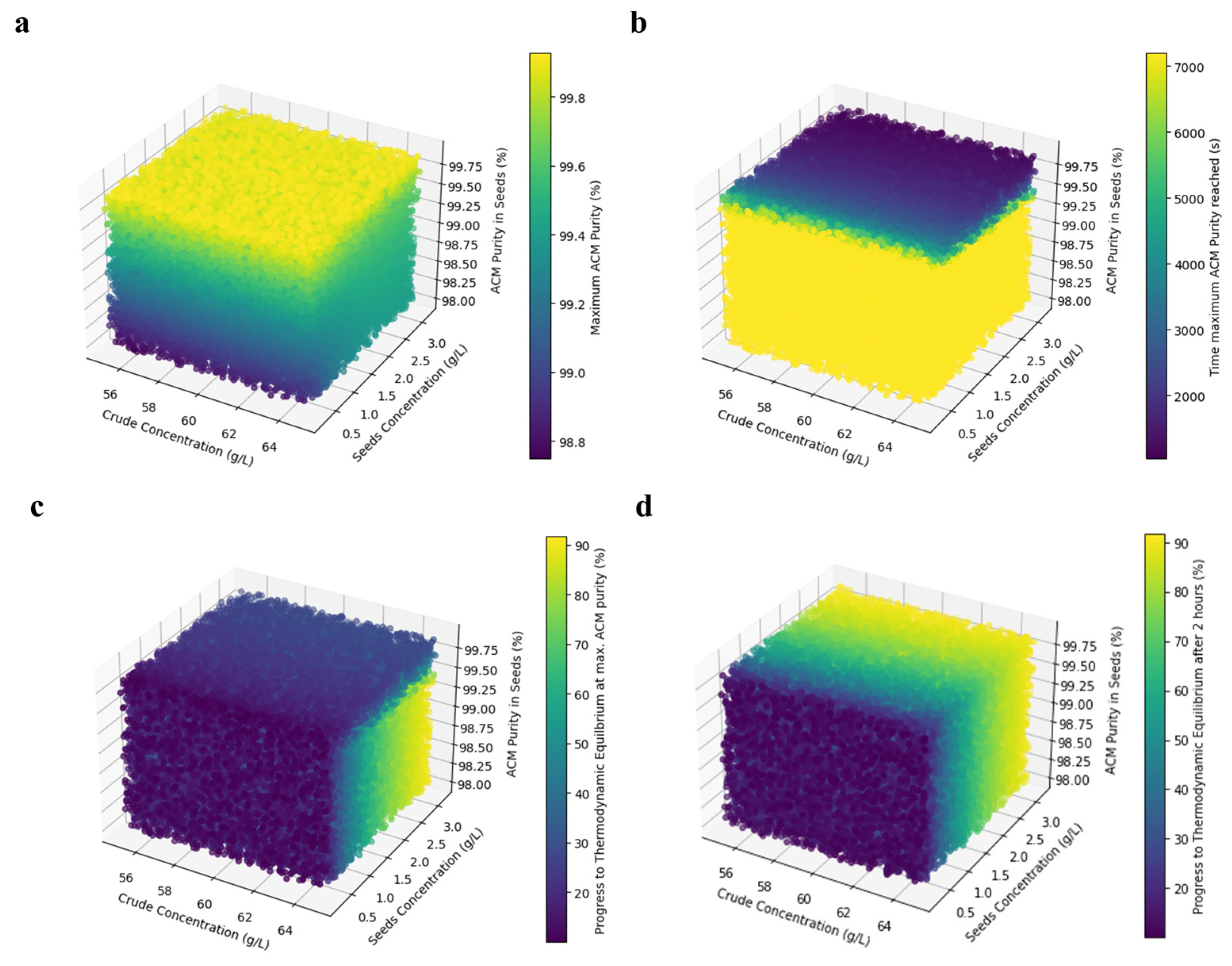
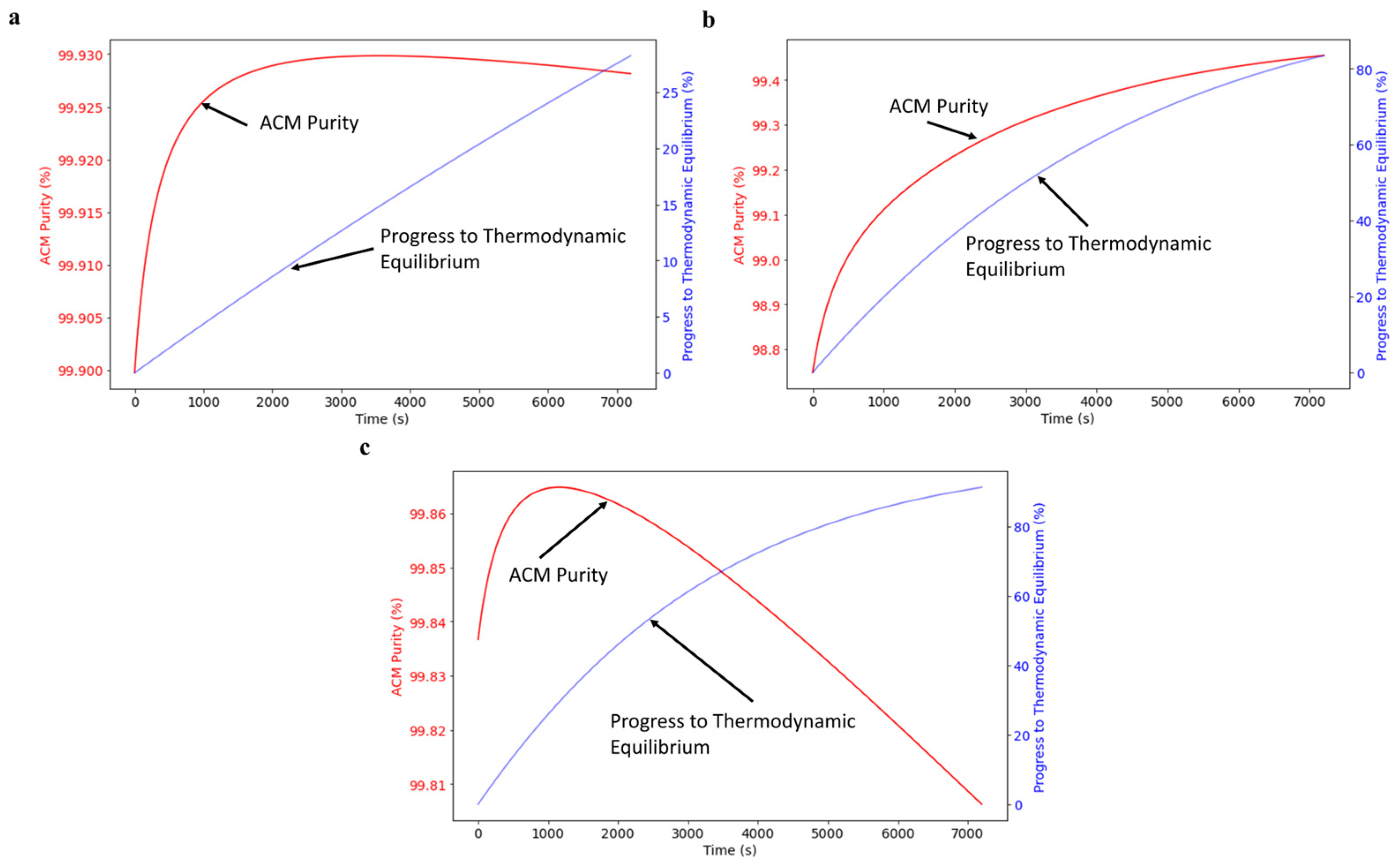
4.4. Process Optimization and Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Myerson, A.S.; Erdemir, D.; Lee, A.Y. Handbook of Industrial Crystallization, 3rd ed.; Cambridge University Press: Cambridge, UK, 2019. [Google Scholar]
- Erdemir, D.; Lee, A.Y.; Myerson, A.S. Nucleation of Crystals from Solution: Classical and Two-Step Models. Acc. Chem. Res. 2009, 42, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Moynihan, H.; Horgan, D. Impurity Occurrence and Removal in Crystalline Products from Process Reactions. Org. Process Res. Dev. 2017, 21, 689–704. [Google Scholar] [CrossRef]
- Li, J.; Lai, T.C.; Trout, B.L.; Myerson, A.S. Continuous Crystallization of Cyclosporine: Effect of Operating Conditions on Yield and Purity. Cryst. Growth Des. 2017, 17, 1000–1007. [Google Scholar] [CrossRef]
- Rogers, L.; Briggs, N.; Achermann, R.; Adamo, A.; Azad, M.; Brnacazio, D.; Capellades, G.; Hammersmith, G.; Hart, T.; Imbrogno, J.; et al. Continuous Production of Five Active Pharmaceutical Ingredients in Flexible Plug-and-Play Modules: A Demonstration Campaign. Org. Process Res. Dev. 2020, 24, 2183–2196. [Google Scholar] [CrossRef]
- Kubota, N. Effect of Impurities on the Growth Kinetics of Crystals. Cryst. Res. Technol. 2001, 36, 749–769. [Google Scholar] [CrossRef]
- Capellades, G.; Bonsu, J.O.; Myerson, A.S. Impurity incorporation in solution crystallization: Diagnosis, prevention, and control. CrystEngComm 2022, 24, 1989–2001. [Google Scholar] [CrossRef]
- Capellades, G.; Wiemeyer, H.; Myerson, A.S. Mixed-Suspension, Mixed-Product Removal Studies of Ciprofloxacin from Pure and Crude Active Pharmaceutical Ingredients: The Role of Impurities on Solubility and Kinetics. Cryst. Growth Des. 2019, 19, 4008–4018. [Google Scholar] [CrossRef]
- Berkovitch-Yellin, Z.; Addadi, L.; Idelson, M.; Lahav, M.; Leiserowitz, L. Controlled Modification of Crystal Habit by “Tailor-Made” Impurities: Application to Benzamide. Angew. Chem. Int. Ed. Engl. 1982, 21, 631–632. [Google Scholar] [CrossRef]
- Addadi, L.; Berkovitch-Yellin, Z.; Weissbuch, I.; Van Mil, J.; Shimon, L.J.W.; Lahav, M.; Leiserowitz, L. Growth and Dissolution of Organic Crystals with “Tailor-Made” Inhibitors- Implications in Stereochemistry and Materials Science. Angew. Chem. Int. Ed. Engl. 1985, 24, 466–485. [Google Scholar] [CrossRef]
- Görög, S. The importance and the challenges of impurity profiling in modern pharmaceutical analysis. TrAC Trends Anal. Chem. 2006, 25, 755–757. [Google Scholar]
- Statement Alerting Patients and Health Care Professionals of NDMA Found in Samples of Ranitidine. Available online: https://www.fda.gov/news-events/press-announcements/statement-alerting-patients-and-health-care-professionals-ndma-found-samples-ranitidine (accessed on 13 September 2019).
- FDA Requests Removal of All Ranitidine Products (Zantac) from the Market. Available online: https://www.fda.gov/news-events/press-announcements/fda-requests-removal-all-ranitidine-products-zantac-market (accessed on 1 April 2020).
- Nordstrom, F.L.; Sirota, E.; Hartmanshenn, C.; Kwok, T.T.; Paolello, M.; Li, H.; Abeyta, V.; Bramante, T.; Madrigal, E.; Behre, T.; et al. Prevalence of Impurity Retention Mechanisms in Pharmaceutical Crystallizations. Org. Process Res. Dev. 2023, 27, 723–741. [Google Scholar] [CrossRef]
- Nordstrom, F.L.; Linehan, B.; Teerakapibal, R.; Li, H. Solubility-Limited Impurity Purge in Crystallization. Cryst. Growth Des. 2019, 19, 1336–1346. [Google Scholar] [CrossRef]
- Tung, H.-H.; Paul, E.L.; Midler, M.; McCauley, J.A. Crystallization of Organic Compounds: An Industrial Perspective, 1st ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Heffernan, C.; Soto, R.; Hodnett, B.K.; Rasmuson, A.C. Growth Kinetics of curcumin form I. CrystEngComm 2020, 22, 3505–3518. [Google Scholar] [CrossRef]
- Gornicka, J.; Mika, M.; Wroblewska, O.; Siudem, P.; Paradowska, K. Methods to Improve the Solubility of Curcumin from Turmeric. Life 2023, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Svärd, M.; Hippen, P.; Rasmuson, A.C. Solubility and Crystal Nucleation in Organic Solvents of Two Polymorphs of Curcumin. J. Pharm. Sci. 2015, 104, 2183–2189. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, T.; Ma, Y.; Xue, F.; Gao, Z.; Hou, B.; Gong, J. Application of PAT-Based Feedback Control Approaches in Pharmaceutical Crystallization. Crystals 2021, 11, 221. [Google Scholar] [CrossRef]
- Simone, E.; Zhang, W.; Nagy, Z.K. Application of Process Analytical Technology-Based Feedback Control Strategies To Improve Purity and Size Distribution in Biopharmaceutical Crystallization. Cryst. Growth Des. 2015, 15, 2908–2919. [Google Scholar] [CrossRef]
- Howard, K.S.; Nagy, Z.K.; Saha, B.; Robertson, A.L.; Steele, G.; Martin, D. A Process Analytical Technology Based Investigation of the Polymorphic Transformations during the Antisolvent Crystallization of Sodium Benzoate from IPA/Water Mixture. Cryst. Growth Des. 2009, 9, 3964–3975. [Google Scholar] [CrossRef]
- Sheng, F.; Chow, P.S.; Yu, Z.Q.; Tan, R.B.H. Online Classification of Mixed Co-Crystal and Solute Suspensions using Raman Spectroscopy. Org. Process Res. Dev. 2016, 20, 1068–1074. [Google Scholar] [CrossRef]
- Nicoud, L.; Licordari, F.; Myerson, A.S. Polymorph control in batch seeded crystallizers. A case study with paracetamol. CrystEngComm 2019, 21, 2105–2118. [Google Scholar] [CrossRef]
- Kachrimanis, K.; Braun, D.E.; Griesser, U.J. Quantitative analysis of paracetamol polymorphs in powder mixtures by FT-Raman spectroscopy and PLS regression. J. Pharm. Biomed. Anal. 2007, 43, 407–412. [Google Scholar] [CrossRef]
- Simone, E.; Saleemi, A.N.; Nagy, Z.K. Application of quantitative Raman spectroscopy for the monitoring of polymorphic transformation in crystallization processes using a good calibration practice procedure. Chem. Eng. Res. Des. 2014, 92, 594–611. [Google Scholar] [CrossRef]
- Hu, Y.; Liang, J.K.; Myerson, A.S.; Taylor, L.S. Crystallization Monitoring by Raman Spectroscopy: Simultaneous Measurement of Desupersaturation Profile and Polymorphic Form in Flufenamic Acid Systems. Ind. Eng. Chem. Res. 2005, 44, 1233–1240. [Google Scholar] [CrossRef]
- Simone, E.; Saleemi, A.N.; Tonnon, N.; Nagy, Z.K. Active Polymorphic Feedback Control of Crystallization Processes Using a Combined Raman and ATR-UV/Vis Spectroscopy Approach. Cryst. Growth Des. 2014, 14, 1839–1850. [Google Scholar] [CrossRef]
- Ündey, C.; Ertunc, S.; Mistretta, T.; Looze, B. Applied advanced process analytics in biopharmaceutical manufacturing: Challenges and prospects in real-time monitoring and control. J. Process Control 2010, 20, 1009–1018. [Google Scholar] [CrossRef]
- Schall, J.M.; Capellades, G.; Myerson, A.S. Methods for estimating supersaturation in antisolvent crystallization systems. CrystEngComm 2019, 21, 5811–5817. [Google Scholar] [CrossRef]
- Na, H.-S.; Arnold, S.; Myerson, A.S. Water activity in supersaturated aqueous solutions of organic solutes. J. Cryst. Growth 1995, 149, 229–235. [Google Scholar] [CrossRef]
- Schall, J.M.; Mandur, J.S.; Braatz, R.D.; Myerson, A.S. Nucleation and Growth Kinetics for Combined Cooling and Antisolvent Crystallization in a Mixed-Suspension, Mixed-Product Removal System: Estimating Solvent Dependency. Cryst. Growth Des. 2018, 18, 1560–1570. [Google Scholar] [CrossRef]
- Omar, H.M.; Rohani, S. Crystal Population Balance Formulation and Solution Methods: A Review. Cryst. Growth Des. 2017, 17, 4028–4041. [Google Scholar] [CrossRef]
- Borsos, Á.; Lakatos, B.G. Investigation and simulation of crystallization of high aspect ratio crystals with fragmentation. Chem. Eng. Res. Des. 2014, 92, 1133–1141. [Google Scholar] [CrossRef]
- Szilágy, B.; Lakatos, B.G. Model-based analysis of stirred cooling crystallizer of high aspect ratio crystals with linwar and nonlinear breakage. Comput. Chem. Eng. 2017, 98, 180–196. [Google Scholar] [CrossRef]
- Fytopoulos, A.A.; Kavousanakis, M.E.; Van Gerven, T.; Boudouvis, A.G.; Stefanidis, G.D.; Xiouras, C. Crystal Growth, Dissolution, and Agglomeration Kinetics of Sodium Chlorate. Ind. Eng. Chem. Res. 2021, 60, 7367–7384. [Google Scholar] [CrossRef]
- Schöll, J.; Lindenberg, C.; Vicum, L.; Borzio, J.; Mazzotti, M. Precipitation of α-glutamic acid: Determination of growth kinetics. Faraday Discuss. 2007, 136, 247–264. [Google Scholar] [CrossRef] [PubMed]
- Capellades, G.; Joshi, P.U.; Dam-Johansen, K.; Mealy, M.J.; Christensen, T.V.; Kiil, S. Characterization of a Multistage Continuous MSMPR Crystallization Process Assisted by Image Analysis of Elongated Crystals. Cryst. Growth Des. 2018, 18, 6455–6469. [Google Scholar] [CrossRef]
- Geladi, P.; Kowalski, B.R. Partial least-squares regression: A tutorial. Anal. Chim. Acta 1986, 185, 1–17. [Google Scholar] [CrossRef]
- Rosipal, R.; Krämer, N. Overview and recent advances in partial least squares. In Subspace, Latent Structure and Feature Selection; Springer: Berlin/Heidelberg, Germany, 2006; pp. 34–51. [Google Scholar]
- Quillo, G.L.; Bhonsdale, S.; Gielen, B.; Van Impe, J.F.; Collas, A.; Xiouras, C. Crystal Growth Kinetics of an Industrial Active Pharmaceutical Ingredient: Implications of Different Representations of Supersaturation and Simultaneous Growth Mechanisms. Cryst. Growth Des. 2021, 21, 5403–5420. [Google Scholar] [CrossRef]
- Mitchell, N.A.; Ó’Ciardhá, C.T.; Frawley, P.J. Estimation of the growth kinetics for the cooling crystallization of paracetamol and ethanol solutions. J. Cryst. Growth 2011, 328, 39–49. [Google Scholar] [CrossRef]
- Soto, R.; Rasmuson, A.C. Crystal Growth Kinetics of Piracetam Polymorphs in Ethanol and Isopropanol. Cryst. Growth Des. 2019, 19, 4273–4286. [Google Scholar] [CrossRef]
- Parambil, J.V.; Heng, J.Y.Y. Seeding in Crystallisation. In Engineering Crystallography: From Molecule to Crystal to Functional Form. NATO Science for Peace and Security Series A: Chemistry and Biology; Roberts, K., Docherty, R., Tamura, R., Eds.; Springer: Dordrecht, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Teerakapibal, R.; Li, H.; Linehan, B.; Nordstrom, F.L. Material Impurity Distribution of Lattice-Incorporated Impurities in Salicylic Acid. Cryst. Growth Des. 2020, 20, 1716–1728. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp Setting | Solute | E1 | E2 | E3 | E4 | E5 |
|---|---|---|---|---|---|---|
| Feed Concentration, pre seed (g/L) | ACM | 60.9 | 60.9 | 54.0 | 54.0 | 57.5 |
| CUR | 0.29 | 0.43 | 0.29 | 0.36 | 0.36 | |
| Initial supersaturation, pre seed (t = 0) | ACM | 0.57 | 0.57 | 0.45 | 0.45 | 0.52 |
| CUR | 2.17 | 2.57 | 2.17 | 2.39 | 2.39 |
| Provided Variable | Solute | E1 | E2 | E3 | E4 | E5 |
|---|---|---|---|---|---|---|
| Cc (g/L) | - | 63.37 | 64.23 | 56.21 | 55.97 | 60.24 |
| Cs (g/L) | - | 1.59 | 1.59 | 1.60 | 1.60 | 1.59 |
| Pc (wt. %) | ACM | 99.51 | 99.37 | 99.39 | 99.23 | 99.36 |
| CUR | 0.49 | 0.63 | 0.61 | 0.77 | 0.64 |
| Input (Units) | Variable |
|---|---|
| kg,ACM (μm/min) | 3.46 |
| kg,CUR (μm/min) | 0.45 |
| gACM | 1 |
| gCUR | 2 |
| kv,ACM | 1 |
| kv,CUR | 1 |
| ρACM (g/mL) | 1.293 |
| ρCUR (g/mL) | 1.293 |
| Csat,ACM (g/L) | 34.3 |
| Csat,CUR (g/L) | 0.033 |
| Process Conditions | Results for Max. Purity | ||||||
|---|---|---|---|---|---|---|---|
| Variable | Ps,acm | Cs | Cc | Max. Pp,acm | t, Max. Pp,acm Attained | E,acm at Max. Pp,acm | Pp,acm at Eq. |
| Units | (%) | (g/L) | (g/L) | (%) | (s) | (%) | (%) |
| S1 | 99.90 | 0.51 | 64.91 | 99.93 | 3557 | 12.5 | 99.58 |
| S2 | 98.75 | 2.45 | 61.46 | 99.49 | Eq. | Eq. | 99.49 |
| S3 | 99.84 | 3.12 | 56.43 | 99.87 | 1157 | 29.4 | 99.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paolello, M.; Bichari, I.; Brouckaert, D.; Francis, M.; Yang, D.; Capellades, G. Kinetic Optimization of the Batch Crystallization of an Active Pharmaceutical Ingredient in the Presence of a Low-Solubility, Precipitating Impurity. Crystals 2023, 13, 1569. https://doi.org/10.3390/cryst13111569
Paolello M, Bichari I, Brouckaert D, Francis M, Yang D, Capellades G. Kinetic Optimization of the Batch Crystallization of an Active Pharmaceutical Ingredient in the Presence of a Low-Solubility, Precipitating Impurity. Crystals. 2023; 13(11):1569. https://doi.org/10.3390/cryst13111569
Chicago/Turabian StylePaolello, Mitchell, Ilyes Bichari, Davinia Brouckaert, Mirvatte Francis, Dawn Yang, and Gerard Capellades. 2023. "Kinetic Optimization of the Batch Crystallization of an Active Pharmaceutical Ingredient in the Presence of a Low-Solubility, Precipitating Impurity" Crystals 13, no. 11: 1569. https://doi.org/10.3390/cryst13111569