
TET2 Mutation and High miR-22 Expression as Biomarkers to Predict Clinical Outcome in Myelodysplastic Syndrome Patients Treated with Hypomethylating Therapy

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
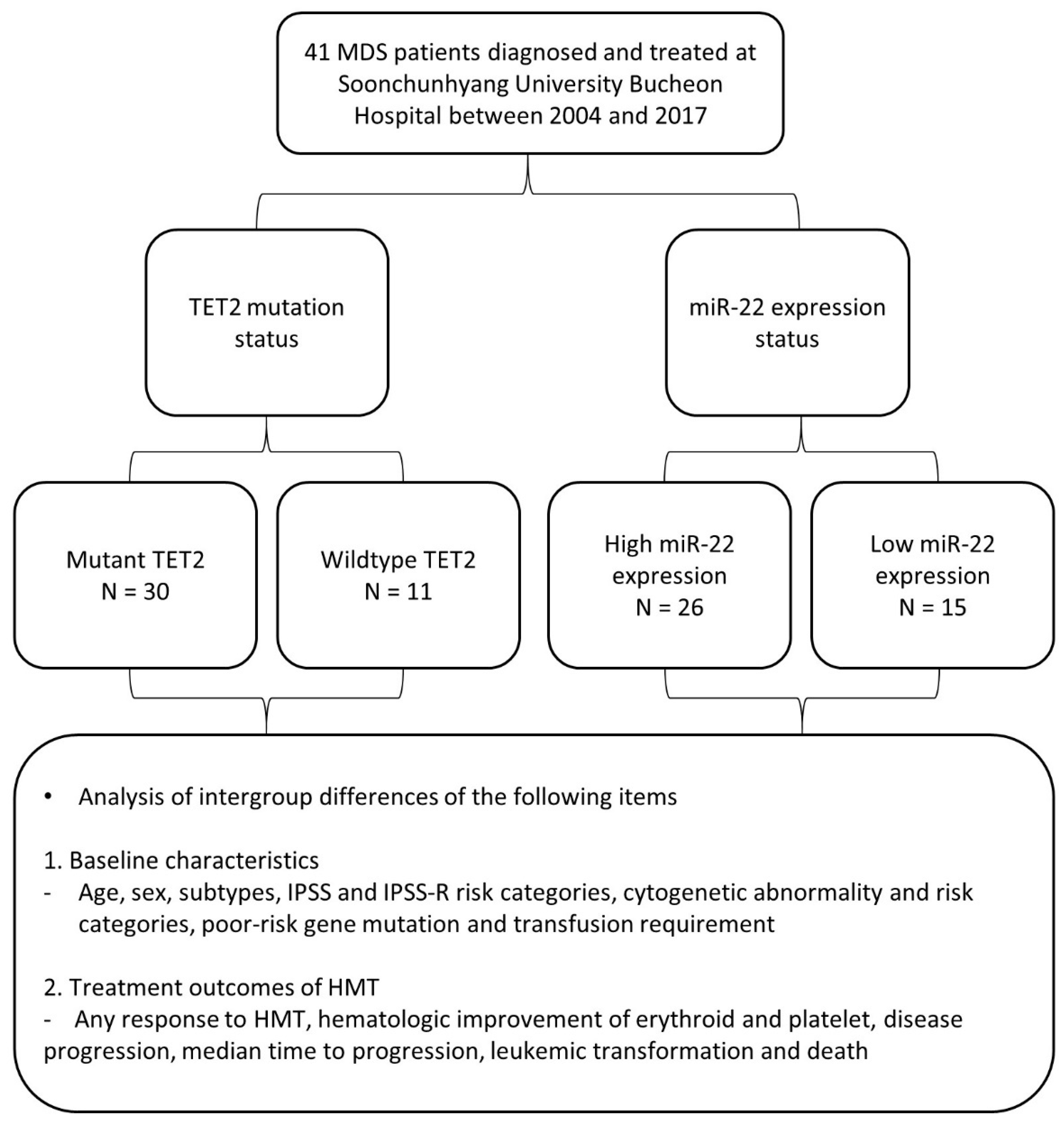
2.1. Patient Characteristics and Sample Preparations
2.2. Next-Generation Sequencing (NGS) for the Detection of Mutations in TET2 and Other Genes
2.3. Detection of TET2 Deletion Using Fluorescence In Situ Hybridization (FISH)
2.4. Real-Time Quantitative PCR of miR-22
2.5. Statistical Analysis
3. Results
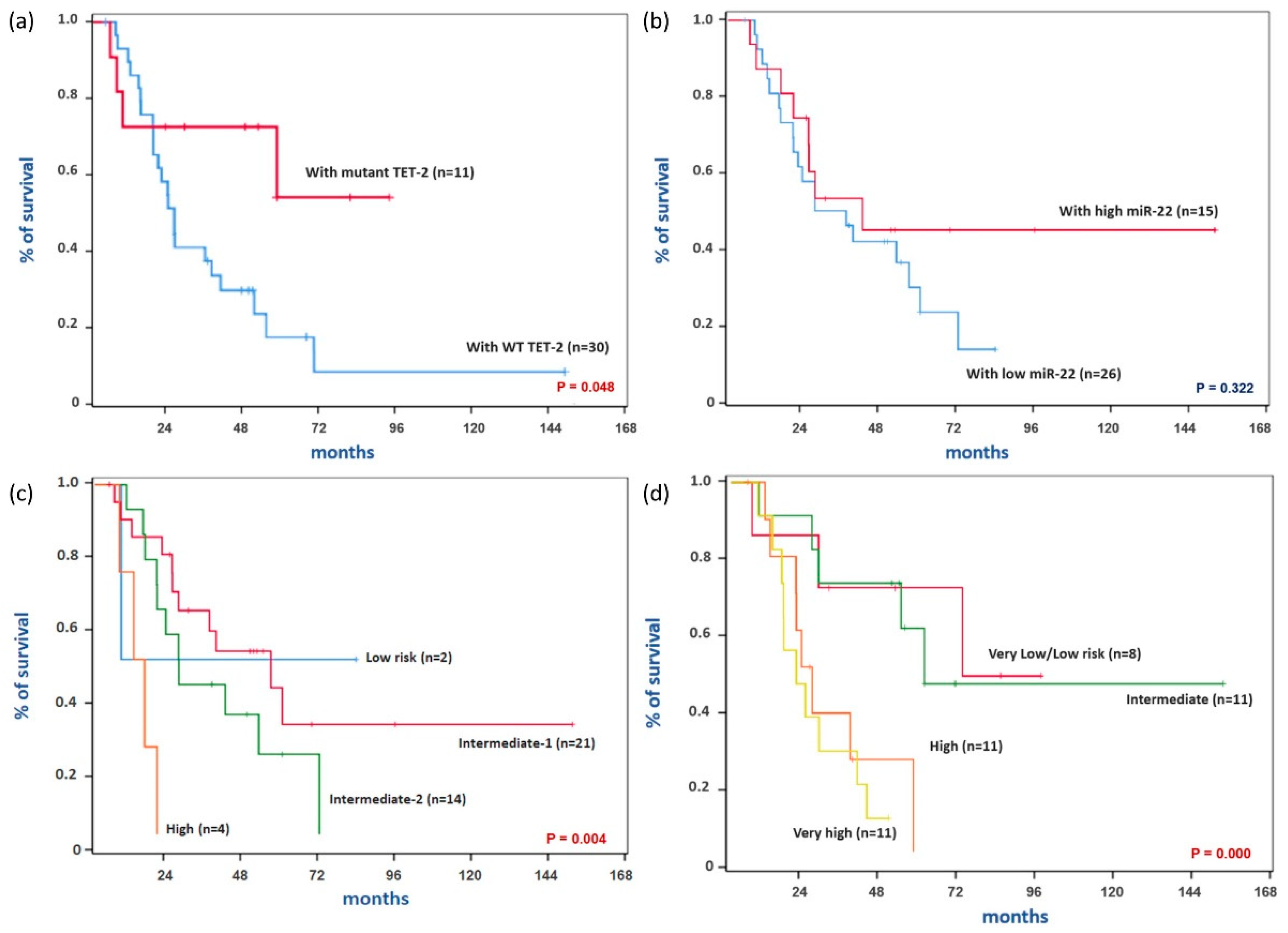
3.1. Comparison of Baseline Characteristics and Treatment Outcomes According to TET2 Mutation Status
3.2. Comparison of Baseline Characteristics and Treatment Outcomes of MDS Patients According to miR-22 Expression Status
3.3. Association of Clinical Factors with Responsiveness to Hypomethylating Therapy, Leukemic Transformation, and Overall Survival
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Visconte, V.; Tiu, R.V.; Rogers, H.J. Pathogenesis of myelodysplastic syndromes: An overview of molecular and non-molecular aspects of the disease. Blood Res. 2014, 49, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Park, E.H.; Lee, H.; Won, Y.J.; Ju, H.Y.; Oh, C.M.; Ingabire, C.; Kong, H.J.; Park, B.K.; Yoon, J.Y.; Eom, H.S.; et al. Nationwide statistical analysis of myeloid malignancies in Korea: Incidence and survival rate from 1999 to 2012. Blood Res. 2015, 50, 204–217. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S. Genetics of MDS. Blood 2019, 133, 1049–1059. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6587668/ (accessed on 3 August 2021). [CrossRef] [Green Version]
- Itzykson, R.; Fenaux, P. Epigenetics of myelodysplastic syndromes. Leukemia 2014, 28, 497–506. [Google Scholar] [CrossRef]
- Woods, B.A.; Levine, R.L. The role of mutations in epigenetic regulators in myeloid malignancies. Immunol. Rev. 2015, 263, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Jang, J.H.; Kwak, J.Y.; Lee, J.H.; Kim, H.J. Use of azacitidine for myelodysplastic syndromes: Controversial issues and practical recommendations. Blood Res. 2013, 48, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.K.; Han, D.H.; Ji, Y.S.; Lee, M.S.; Min, C.W.; Park, S.K.; Kim, S.H.; Yun, J.; Kim, H.J.; Kim, K.H.; et al. Biomarkers of angiogenesis as prognostic factors in myelodysplastic syndrome patients treated with hypomethylating agents. Leuk. Res. 2016, 50, 21–28. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Jia, G.; Johansen, J.V.; Pedersen, M.T.; Rapin, N.; Bagger, F.O.; Porse, B.T.; Bernard, O.A.; Christensen, J.; Helin, K. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev. 2015, 29, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, J.; Jelinek, J.; Lu, Y.; Cesaroni, M.; Madzo, J.; Neumann, F.; He, R.; Taby, R.; Vasanthakumar, A.; Macrae, T.; et al. TET2 mutations affect Non-CpG island DNA methylation at enhancers and transcription factor-binding sites in chronic myelomonocytic Leukemia. Cancer Res. 2015, 75, 2833–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Guo, S.; Chen, S.; Mastriano, S.J.; Liu, C.; D’Alessio, A.C.; Hysolli, E.; Guo, Y.; Yao, H.; Megyola, C.M.; et al. An Extensive Network of TET2-Targeting MicroRNAs Regulates Malignant Hematopoiesis. Cell Rep. 2013, 5, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The Oncogenic MicroRNA miR-22 Targets the TET2 Tumor Suppressor to Promote Hematopoietic Stem Cell Self-Renewal and Transformation. Cell Stem Cell 2013, 13, 87–101. Available online: https://linkinghub.elsevier.com/retrieve/pii/S1934590913002610 (accessed on 3 August 2021). [CrossRef] [PubMed] [Green Version]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejar, R.; Lord, A.; Stevenson, K.; Bar-Natan, M.; Pérez-Ladaga, A.; Zaneveld, J.; Wang, H.; Caughey, B.; Stojanov, P.; Getz, G.; et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014, 124, 2705–2712. [Google Scholar] [CrossRef]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Cheson, B.D.; Greenberg, P.L.; Bennett, J.M.; Lowenberg, B.; Wijermans, P.W.; Nimer, S.D.; Pinto, A.; Beran, M.; De Witte, T.M.; Stone, R.M.; et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood 2006, 108, 419–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myelodysplastic Syndromes. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology-V1.2021, National Comprehensive Cancer Network. Available online: https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf (accessed on 11 September 2020).
- Hou, H.A.; Tsai, C.H.; Lin, C.C.; Chou, W.C.; Kuo, Y.Y.; Liu, C.Y.; Tseng, M.H.; Peng, Y.L.; Liu, M.C.; Liu, C.W.; et al. Incorporation of mutations in five genes in the revised International Prognostic Scoring System can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018, 8, 39. [Google Scholar] [CrossRef]
- Braun, T.; Itzykson, R.; Renneville, A.; de Renzis, B.; Dreyfus, F.; Laribi, K.; Bouabdallah, K.; Vey, N.; Toma, A.; Recher, C.; et al. Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: A phase 2 trial. Blood 2011, 118, 3824–3831. [Google Scholar] [CrossRef] [Green Version]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef]
- Jung, S.H.; Kim, Y.J.; Yim, S.H.; Kim, H.J.; Kwon, Y.R.; Hur, E.H.; Goo, B.K.; Choi, Y.S.; Lee, S.H.; Chung, Y.J.; et al. Somatic mutations predict outcomes of hypomethylating therapy in patients with myelodysplastic syndrome. Oncotarget 2016, 7, 55264–55275. [Google Scholar] [CrossRef] [Green Version]
- Reilly, B.; Tanaka, T.N.; Diep, D.; Yeerna, H.; Tamayo, P.; Zhang, K.; Bejar, R. DNA methylation identifies genetically and prognostically distinct subtypes of myelodysplastic syndromes. Blood Adv. 2019, 3, 2845–2858. [Google Scholar] [CrossRef]
- Smith, A.E.; Mohamedali, A.M.; Kulasekararaj, A.; Lim, Z.; Gäken, J.; Lea, N.C.; Przychodzen, B.; Mian, S.A.; Nasser, E.E.; Shooter, C.; et al. Next-generation sequencing of the TET2 gene in 355 MDS and CMMLpatients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood 2010, 116, 3923–3932. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Pellagatti, A.; Karimi, M.; Sato-Otsubo, A.; Sato, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood 2017, 130, 2642–2653. [Google Scholar] [CrossRef]
- Ko, M.; Huang, Y.; Jankowska, A.M.; Pape, U.J.; Tahiliani, M.; Bandukwala, H.S.; An, J.; Lamperti, E.D.; Koh, K.P.; Ganetzky, R.; et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010, 468, 839–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Cai, X.; Cai, C.L.; Wang, J.; Zhang, W.; Petersen, B.E.; Yang, F.C.; Xu, M. Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 2011, 118, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.S.; Kim, H.J.; Kim, Y.K.; Jung, S.H.; Yang, D.H.; Lee, J.J.; Lee, I.K.; Kim, N.Y.; Minden, M.D.; Jung, C.W.; et al. Adverse prognostic effect of homozygous TET2 mutation on the relapse risk of acute myeloid leukaemia in patients of normal karyotype. Haematologica 2015, 100, e351. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| WT TET2 (n = 30) | Mutant TET2 (n = 11) | p-Value | |
|---|---|---|---|
| Median age | 63 (23–86) | 65 (38–82) | 0.835 |
| Sex (M/F) | 18/12 | 8/3 | 0.716 |
| Disease subtypes | 0.434 | ||
| MDS-SLD | 2 (6.7) | 0 (0.0) | |
| MDS-MLD | 6 (20.0) | 4 (36.4) | |
| MDS-EB1 | 9 (30.0) | 2 (18.2) | |
| MDS-EB2 | 8 (26.7) | 3 (27.3) | |
| MDS-U | 1 (3.3) | 0 (0.0) | |
| CMML | 4 (13.3) | 1 (9.1) | |
| 5q-syndrome | 0 (0.0) | 1 (9.1) | |
| IPSS risk categories Low Intermediate-1 Intermediate-2 High Lower-risk disease * Higher-risk disease * | 1 (3.3) 13 (43.3) 12 (40.0) 4 (13.3) 14 (46.7) 16 (53.3) | 1 (9.1) 8 (72.7) 2 (18.2) 0 (0.0) 9 (81.8) 2 (18.2) | 0.039 0.075 |
| IPSS-R risk categories Very low Low Intermediate High Very high Lower-risk group * Higher-risk group * | 1 (3.3) 3 (10.0) 6 (20.0) 9 (30.0) 11 (36.7) 10 (33.3) 20 (66.7) | 0 (0.0) 4 (36.4) 5 (45.5) 2 (18.2) 0 (0.0) 9 (81.8) 2 (18.2) | 0.009 0.006 |
| Cytogenetic abnormality | 13 (43.3) | 2 (18.2) | 0.168 |
| Cytogenetic risk categories | |||
| Very good | 2 (6.7) | 0 (0.0) | 0.102 |
| Good | 19 (63.3) | 11 (100.0) | |
| Intermediate | 2 (6.7) | 0 (0.0) | |
| Poor | 2 (6.7) | 0 (0.0) | |
| Very poor | 5 (16.7) | 0 (0.0) | |
| Lower-risk karyotype ◊ | 21 (70.0) | 11 (100.0) | 0.083 |
| Higher-risk karyotype ◊ | 9 (30.0) | 0 (0.0) | |
| Poor-risk gene mutation | 13 (43.3) | 4 (36.4) | 0.736 |
| Transfusion requirement | 26 (86.7) | 8 (72.7) | 0.361 |
| Variables | WT TET2 (n = 30) | Mutant TET2 (n = 11) | p-Value |
|---|---|---|---|
| Any response to HMT | 25 (83.3) | 9 (81.8) | 1.000 |
| Hematologic improvement | |||
| 20 (66.7) | 6 (54.5) | 0.491 |
| 21 (70.0) | 5 (45.5) | 0.272 |
| Disease progression | 19 (63.3) | 3 (27.3) | 0.040 |
| Median time to progression (m) * | 19.3 (5.5–47.8) | 49.8 (6.7–95.9) | 0.007 |
| Leukemic transformation | 17 (56.7) | 2 (18.2) | 0.029 |
| Death | 23 (76.7) | 4 (36.4) | 0.026 |
| Low miR-22 Expression (n = 26) | High miR-22 Expression (n = 15) | p-Value | |
|---|---|---|---|
| Median age | 63 (42–86) | 60 (23–79) | 0.422 |
| Sex (M/F) | 16/10 | 10/5 | 0.743 |
| Disease subtypes | 0.061 | ||
| MDS-SLD | 1 (3.8) | 1 (6.7) | |
| MDS-MLD | 4 (15.4) | 6 (40.0) | |
| MDS-EB1 | 5 (19.2) | 6 (40.0) | |
| MDS-EB2 | 9 (34.6) | 2 (13.3) | |
| MDS-U | 1 (3.8) | 0 (0.0) | |
| CMML | 5 (19.2) | 0 (0.0) | |
| 5q-syndrome | 1 (3.8) | 0 (0.0) | |
| IPSS risk categories Low Intermediate-1 Intermediate-2 High Lower-risk disease * Higher-risk disease * | 2 (7.7) 10 (38.5) 10 (38.5) 4 (15.4) 12 (46.2) 14 (53.8) | 0 (0.0) 11 (73.3) 4 (26.7) 0 (0.0) 11 (73.3) 4 (26.7) | 0.149 0.091 |
| IPSS-R risk categories | 0.429 | ||
| Very low | 1 (3.8) | 0 (0.0) | |
| Low | 3 (11.5) | 4 (26.7) | |
| Intermediate | 6 (23.1) | 5 (33.3) | |
| High | 9 (34.6) | 2 (13.3) | |
| Very high | 7 (26.9) | 4 (26.7) | 0.183 |
| Lower-risk disease * | 10 (38.5) | 9 (60.0) | |
| Higher-risk disease * | 16 (61.5) | 6 (40.0) | |
| Cytogenetic abnormality | 9 (34.6) | 6 (40.0) | 0.730 |
| Cytogenetic risk categories | |||
| Very good | 1 (3.8) | 1 (6.7) | 0.757 |
| Good | 20 (76.9) | 10 (66.7) | |
| Intermediate | 1 (3.8) | 1 (6.7) | |
| Poor | 1 (3.8) | 1 (6.7) | |
| Very poor | 3 (11.5) | 2 (13.3) | |
| Lower-risk karyotype ◊ | 21 (80.8) | 11 (73.3) | 0.701 |
| Higher-risk karyotype ◊ | 5 (19.2) | 4 (26.7) | |
| Poor-risk gene mutation | 11 (42.3) | 6 (40.0) | 0.885 |
| Transfusion requirement | 23 (88.5) | 11 (73.3) | 0.390 |
| Variables | Low miR-22 Expression (n = 26) | High miR-22 Expression (n = 15) | p-Value |
|---|---|---|---|
| Any response to HMT | 21 (80.8) | 13 (86.7) | 1.000 |
| Hematologic improvement | |||
| 15 (57.7) | 11 (73.3) | 0.317 |
| 17 (65.4) | 9 (60.0) | 0.730 |
| Disease progression | 16 (61.5) | 6 (40.0) | 0.183 |
| Median time to progression (m) * | 23.1 (5.5–67.1) | 25.9 (6.7–95.9) | 0.155 |
| Leukemic transformation | 15 (57.7) | 4 (26.7) | 0.055 |
| Death | 19 (73.1) | 8 (53.3) | 0.199 |
| Variables | Any Response to HMT | |
|---|---|---|
| Odds Ratio (95% CI) | p-Value a | |
| TET2 mutation | 0.900 (0.148–5.489) | 0.909 |
| High miR-22 expression | 1.548 (0.261–9.175) | 0.631 |
| IPSS (higher-risk to lower-risk disease) b | 2.222 (0.377–13.082) | 0.377 |
| IPSS-R (higher-risk to lower-risk disease) b | 1.689 (0.327–8.732) | 0.532 |
| Cytogenetic abnormality | 4.200 (0.454–38.843) | 0.206 |
| Cytogenetic risk categories | 1.846 (0.193–17.700) | 0.595 |
| (higher-risk to lower-risk karyotype) c | ||
| Poor-risk gene mutation | 0.933 (0.180–4.838) | 0.935 |
| Elderly (≥70) | 0.933 (0.180–4.838) | 0.935 |
| Variables | Leukemic Transformation | |
|---|---|---|
| Odds Ratio (95% CI) | p-Value a | |
| TET2 mutation | 0.170 (0.031–0.924) | 0.040 |
| High miR-22 expression | 0.267 (0.067–1.064) | 0.061 |
| IPSS (higher-risk to lower-risk disease) b | 7.367 (1.836–29.554) | 0.005 |
| IPSS-R (higher-risk to lower-risk disease) b | 4.900 (1.282–18.725) | 0.020 |
| Cytogenetic abnormality | 2.400 (0.654–8.811) | 0.187 |
| Cytogenetic risk categories | 5.833 (1.038–32.797) | 0.045 |
| (higher-risk vs. lower-risk karyotype) c | ||
| Poor-risk gene mutation | 17.733 (3.619–86.885) | 0.000 |
| Response to HMT | 0.592 (0.115–3.061) | 0.532 |
| Elderly (≥70) | 1.575 (0.451–5.504) | 0.477 |
| Variables | Overall Survival | |
|---|---|---|
| Hazard Ratio (95% CI) | p-Value a | |
| TET2 mutation | 0.354 (0.121–1.039) | 0.059 |
| High miR-22 expression | 0.661 (0.289–1.513) | 0.327 |
| IPSS (higher-risk vs. lower-risk disease) b | 2.093 (0.972–4.507) | 0.059 |
| IPSS-R (higher-risk vs. lower-risk disease) b | 4.310 (1.724–10.776) | 0.002 |
| Cytogenetic abnormality | 1.454 (0.666–3.173) | 0.348 |
| Cytogenetic risk categories | 2.136 (0.905–5.041) | 0.083 |
| (higher-risk vs. lower-risk karyotype) c | ||
| Poor-risk gene mutation | 2.366 (1.093–5.120) | 0.029 |
| Response to HMT | 0.490 (0.196–1.228) | 0.128 |
| Elderly (≥70) | 1.639 (0.762–3.525) | 0.206 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, J.; Ji, Y.S.; Jang, G.H.; Lim, S.H.; Kim, S.H.; Kim, C.K.; Bae, S.B.; Won, J.H.; Park, S.K. TET2 Mutation and High miR-22 Expression as Biomarkers to Predict Clinical Outcome in Myelodysplastic Syndrome Patients Treated with Hypomethylating Therapy. Curr. Issues Mol. Biol. 2021, 43, 917-931. https://doi.org/10.3390/cimb43020065
Yun J, Ji YS, Jang GH, Lim SH, Kim SH, Kim CK, Bae SB, Won JH, Park SK. TET2 Mutation and High miR-22 Expression as Biomarkers to Predict Clinical Outcome in Myelodysplastic Syndrome Patients Treated with Hypomethylating Therapy. Current Issues in Molecular Biology. 2021; 43(2):917-931. https://doi.org/10.3390/cimb43020065
Chicago/Turabian StyleYun, Jina, Young Sok Ji, Geum Ha Jang, Sung Hee Lim, Se Hyung Kim, Chan Kyu Kim, Sang Byung Bae, Jong Ho Won, and Seong Kyu Park. 2021. "TET2 Mutation and High miR-22 Expression as Biomarkers to Predict Clinical Outcome in Myelodysplastic Syndrome Patients Treated with Hypomethylating Therapy" Current Issues in Molecular Biology 43, no. 2: 917-931. https://doi.org/10.3390/cimb43020065