
Innovative Family-Based Genetically Informed Series of Analyses of Whole-Exome Data Supports Likely Inheritance for Grammar in Children with Specific Language Impairment
 , , , and
, , , and
Abstract
:1. Introduction
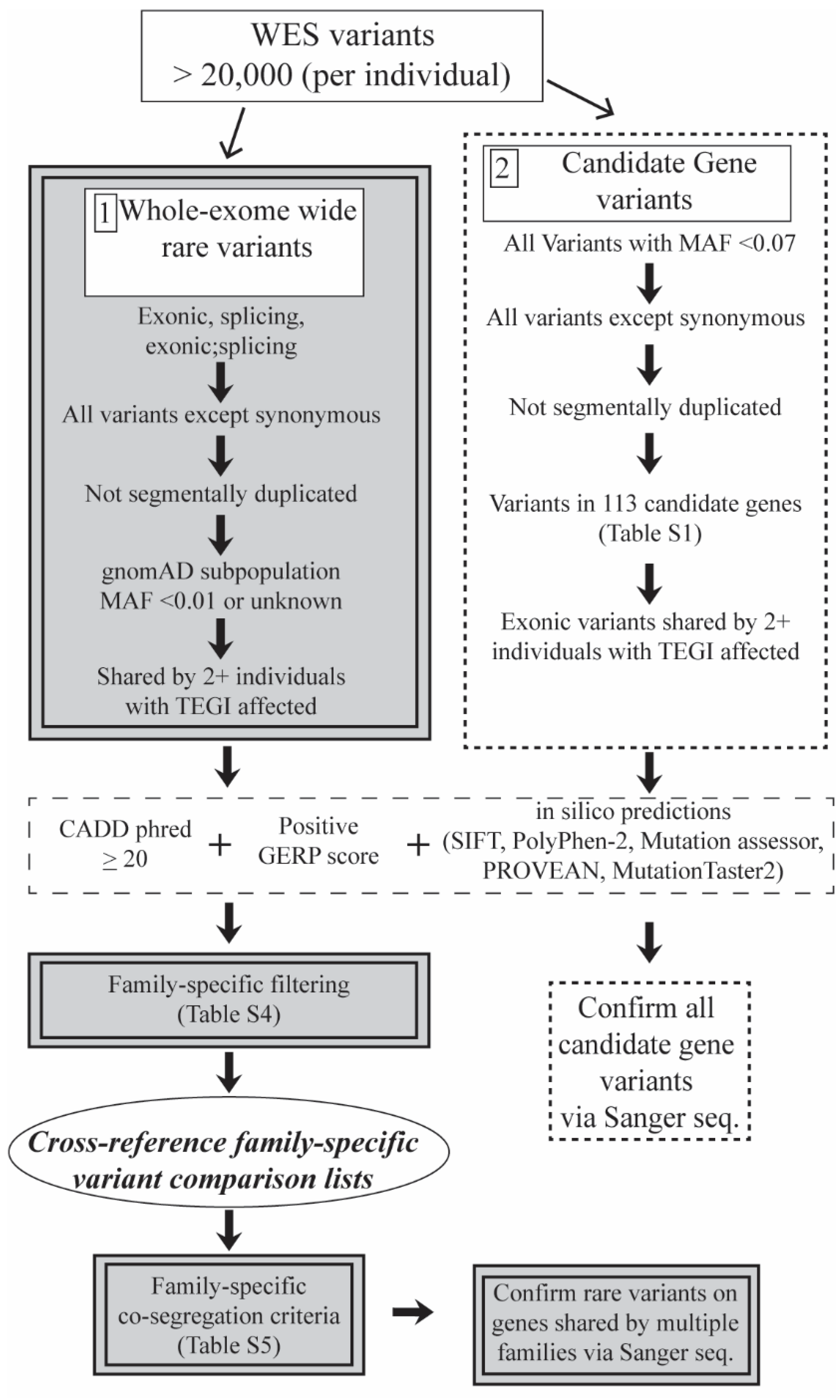
2. Materials and Methods
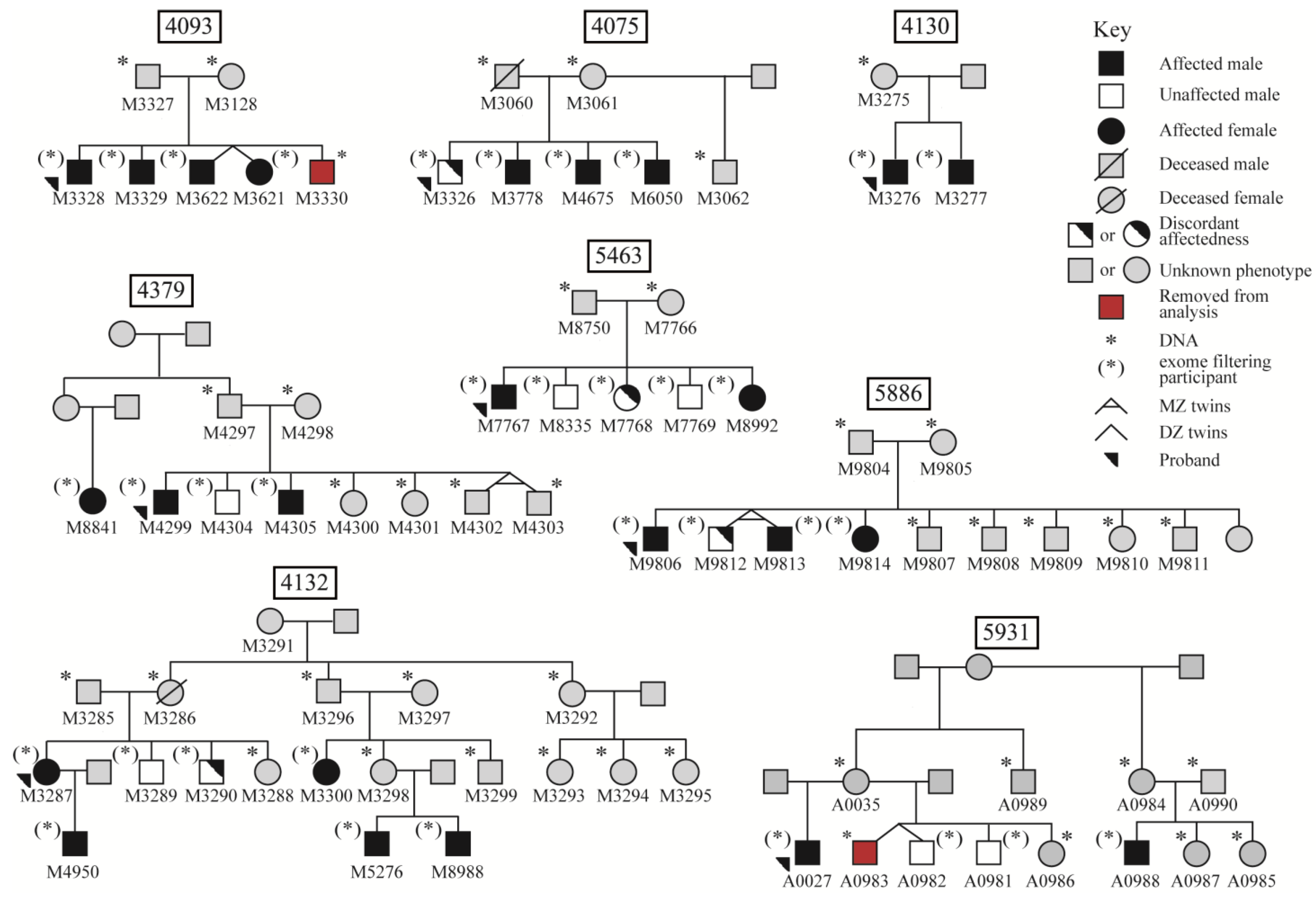
3. Results
3.1. Variant Prioritization Workflow 1: Whole-Exome Wide Rare Variants
3.2. Variant Prioritization Workflow 2: Candidate Gene Variants
3.3. Significance of Identified Variants in Additional Unrelated Probands
4. Discussion
4.1. Limitations
4.2. Future Directions
4.3. Implications of Family-Based Genetic Study for Understanding Factors Involved in Language Development
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tallal, P.; Hirsch, L.S.; Realpe-Bonilla, T.; Miller, S.; Brzustowicz, L.M.; Bartlett, C.W.; Flax, J.F. Familial Aggregation in Specific Language Impairment. J. Speech Lang. Hear. Res. 2001, 44, 1172–1182. [Google Scholar] [CrossRef]
- Stromswold, K. Genetics of Spoken Language Disorders. Hum. Biol. 1998, 70, 297–324. [Google Scholar]
- Bishop, D.V.M.; Adams, C.V.; Norbury, C.F. Distinct Genetic Influences on Grammar and Phonological Short-term Memory Deficits: Evidence from 6-year-old Twins. Genes Brain Behav. 2006, 5, 158–169. [Google Scholar] [CrossRef]
- Dale, P.S.; Rice, M.L.; Rimfeld, K.; Hayiou-Thomas, M.E. Grammar Clinical Marker Yields Substantial Heritability for Language Impairments in 16-Year-Old Twins. J. Speech Lang. Hear. Res. 2018, 61, 66–78. [Google Scholar] [CrossRef]
- Rice, M.L.; Taylor, C.L.; Zubrick, S.R.; Hoffman, L.; Earnest, K.K. Heritability of Specific Language Impairment and Nonspecific Language Impairment at Ages 4 and 6 Years across Phenotypes of Speech, Language, and Nonverbal Cognition. J. Speech Lang. Hear. Res. 2020, 63, 793–813. [Google Scholar] [CrossRef]
- Rice, M.L.; Haney, K.R.; Wexler, K. Family Histories of Children with SLI Who Show Extended Optional Infinitives. J. Speech Lang. Hear. Res. 1998, 41, 419–432. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Deafness and Other Communication Disorders Developmental Language Disorder. Available online: https://www.nidcd.nih.gov/health/developmental-language-disorder (accessed on 8 May 2023).
- Tomblin, J.B.; Records, N.L.; Buckwalter, P.; Zhang, X.; Smith, E.; O’Brien, M. Prevalence of Specific Language Impairment in Kindergarten Children. J. Speech Lang. Hear. Res. 1997, 40, 1245–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norbury, C.F.; Gooch, D.; Wray, C.; Baird, G.; Charman, T.; Simonoff, E.; Vamvakas, G.; Pickles, A. The Impact of Nonverbal Ability on Prevalence and Clinical Presentation of Language Disorder: Evidence from a Population Study. J. Child. Psychol. Psychiatry 2016, 57, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.V.M.; Snowling, M.J.; Thompson, P.A.; Greenhalgh, T.; Consortium, C. CATALISE: A Multinational and Multidisciplinary Delphi Consensus Study. Identifying Language Impairments in Children. PLoS ONE 2016, 11, e0158753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, D.V.M.; Snowling, M.J.; Thompson, P.A.; Greenhalgh, T.; CATALISE-2 consortium. Phase 2 of CATALISE: A Multinational and Multidisciplinary Delphi Consensus Study of Problems with Language Development: Terminology. J. Child Psychol. Psychiatry 2017, 58, 1068–1080. [Google Scholar] [CrossRef] [Green Version]
- Leonard, L.B. Children with Specific Language Impairment, 2nd ed.; MIT Press: Cambridge, MA, USA, 2014; ISBN 9780262324021. [Google Scholar]
- Developmental Language Disorders: From Phenotypes to Etiologies; Lawrence Erlbaum Associates Publishers: Mahwah, NJ, USA, 2004; pp. xi–411. ISBN 978-0-8058-4662-1.
- Mountford, H.S.; Braden, R.; Newbury, D.F.; Morgan, A.T. The Genetic and Molecular Basis of Developmental Language Disorder: A Review. Children 2022, 9, 586. [Google Scholar] [CrossRef]
- Rice, M.L.; Wexler, K. Rice/Wexler Test of Early Grammatical Impairment Examiner’s Manual; The Psychological Corporation: San Antonio, TX, USA, 2001. [Google Scholar]
- Rice, M.L.; Wexler, K.; Hershberger, S. Tense Over Time: The Longitudinal Course of Tense Acquisition in Children with Specific Language Impairment. J. Speech Lang. Hear. Res. 1998, 41, 1412–1431. [Google Scholar] [CrossRef]
- Rice, M.L.; Wexler, K. Toward Tense as a Clinical Marker of Specific Language Impairment in English-Speaking Children. J. Speech Lang. Hear. Res. 1996, 39, 1239–1257. [Google Scholar] [CrossRef]
- Andres, E.M.; Earnest, K.K.; Smith, S.D.; Rice, M.L.; Raza, M.H. Pedigree-Based Gene Mapping Supports Previous Loci and Reveals Novel Suggestive Loci in Specific Language Impairment (SLI). J. Speech Lang. Hear. Res. 2020, 63, 4046–4061. [Google Scholar] [CrossRef]
- Andres, E.M.; Earnest, K.K.; Zhong, C.; Rice, M.L.; Raza, M.H. Family-Based Whole-Exome Analysis of Specific Language Impairment (SLI) Identifies Rare Variants in BUD13, a Component of the Retention and Splicing (RES) Complex. Brain Sci. 2022, 12, 47. [Google Scholar] [CrossRef]
- Peter, B.; Bruce, L.; Finestack, L.; Dinu, V.; Wilson, M.; Klein-Seetharaman, J.; Lewis, C.R.; Braden, B.B.; Tang, Y.-Y.; Scherer, N.; et al. Precision Medicine as a New Frontier in Speech-Language Pathology: How Applying Insights from Behavior Genomics Can Improve Outcomes in Communication Disorders. Am. J. Speech-Lang. Pathol. 2023, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Eising, E.; Mirza-Schreiber, N.; de Zeeuw, E.L.; Wang, C.A.; Truong, D.T.; Allegrini, A.G.; Shapland, C.Y.; Zhu, G.; Wigg, K.G.; Gerritse, M.L.; et al. Genome-Wide Analyses of Individual Differences in Quantitatively Assessed Reading- and Language-Related Skills in up to 34,000 People. Proc. Natl. Acad. Sci. USA 2022, 119, e2202764119. [Google Scholar] [CrossRef] [PubMed]
- Nudel, R.; Christensen, R.V.; Kalnak, N.; Schwinn, M.; Banasik, K.; Dinh, K.M.; Erikstrup, C.; Pedersen, O.B.; Burgdorf, K.S.; Ullum, H.; et al. Developmental Language Disorder—a Comprehensive Study of More than 46,000 Individuals. Psychiatry Res. 2023, 323, 115171. [Google Scholar] [CrossRef] [PubMed]
- Toseeb, U.; Vincent, J.; Oginni, O.A.; Asbury, K.; Newbury, D.F. The Development of Mental Health Difficulties in Young People with and without Developmental Language Disorder: A Gene–Environment Interplay Study Using Polygenic Scores. J. Speech Lang. Hear. Res. 2023, 66, 1639–1657. [Google Scholar] [CrossRef]
- Ash, A.C.; Redmond, S.M. Using Finiteness as a Clinical Marker to Identify Language Impairment. Perspect. Lang. Learn. Educ. 2014, 21, 148–158. [Google Scholar] [CrossRef]
- Weiler, B.; Schuele, C.M. Tense Marking in the Kindergarten Population: Testing the Bimodal Distribution Hypothesis. J. Speech Lang. Hear. Res. 2021, 64, 593–612. [Google Scholar] [CrossRef] [PubMed]
- Weiler, B.; Schuele, C.M.; Feldman, J.I.; Krimm, H. A Multiyear Population-Based Study of Kindergarten Language Screening Failure Rates Using the Rice Wexler Test of Early Grammatical Impairment. Lang. Speech Hear. Serv. Sch. 2018, 49, 248–259. [Google Scholar] [CrossRef]
- Wexler, K. Optional Infinitives, Head Movement and the Economy of Derivations. In Verb Movement; Lightfoot, D., Hornstein, N., Eds.; Cambridge University Press: Cambridge, UK, 1994. [Google Scholar]
- Rice, M.L.; Wexler, K.; Cleave, P.L. Specific Language Impairment as a Period of Extended Optional Infinitive. J. Speech Lang. Hear. Res. 1995, 38, 850–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, M.L.; Earnest, K.K.; Hoffman, L. Longitudinal Grammaticality Judgments of Tense Marking in Complex Questions in Children with and without Specific Language Impairment, Ages 5–18 Years. J. Speech Lang. Hear. Res. in press.
- Rice, M.L.; Zubrick, S.R.; Taylor, C.L.; Hoffman, L.; Gayán, J. Longitudinal Study of Language and Speech of Twins at 4 and 6 Years: Twinning Effects Decrease, Zygosity Effects Disappear, and Heritability Increases. J. Speech Lang. Hear. Res. 2018, 61, 79–93. [Google Scholar] [CrossRef]
- Rice, M.L.; Smith, S.D.; Gayán, J. Convergent Genetic Linkage and Associations to Language, Speech and Reading Measures in Families of Probands with Specific Language Impairment. J. Neurodev. Disord. 2009, 1, 264–282. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.S.; Reader, R.H.; Hoischen, A.; Veltman, J.A.; Simpson, N.H.; Francks, C.; Newbury, D.F.; Fisher, S.E. Next-Generation DNA Sequencing Identifies Novel Gene Variants and Pathways Involved in Specific Language Impairment. Sci. Rep. 2017, 7, 1–17. [Google Scholar] [CrossRef]
- Villanueva, P.; Nudel, R.; Hoischen, A.; Fernández, M.A.; Simpson, N.H.; Gilissen, C.; Reader, R.H.; Jara, L.; Echeverry, M.M.; Francks, C.; et al. Exome Sequencing in an Admixed Isolated Population Indicates NFXL1 Variants Confer a Risk for Specific Language Impairment. PLoS Genet. 2015, 11, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Nudel, R. An Investigation of NFXL1, a Gene Implicated in a Study of Specific Language Impairment. J. Neurodev. Disord. 2016, 8, 13. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for Investigating Causality of Sequence Variants in Human Disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, A.; Rice, M.L.; Talcott, J.B.; Diaz, R.; Smith, S.D.; Raza, M.H.; Snowling, M.J.; Hulme, C.; Stein, J.; Hayiou-Thomas, M.E.; et al. A Rare Missense Variant in the ATP2C2 Gene Is Associated with Language Impairment and Related Measures. Hum. Mol. Genet. 2021, 30, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the Functional Impact of Protein Mutations: Application to Cancer Genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2018, 47, D886–D894. [Google Scholar] [CrossRef]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; Program, N.C.S.; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and Intensity of Constraint in Mammalian Genomic Sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and Integration of Deleteriousness Prediction Methods for Nonsynonymous SNVs in Whole Exome Sequencing Studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef] [Green Version]
- Andres, E.M.; Neely, H.L.; Hafeez, H.; Yasmin, T.; Kausar, F.; Basra, M.A.R.; Raza, M.H. Study of Rare Genetic Variants in TM4SF20, NFXL1, CNTNAP2, and ATP2C2 in Pakistani Probands and Families with Language Impairment. Meta Gene 2021, 30. [Google Scholar] [CrossRef]
- Hendam, A.; Al-Sadek, A.F.; Hefny, H.A. In Silico Deleterious Prediction of Nonsynonymous Single Nucleotide Polymorphisms in Neurexin1 Gene for Mental Disorders. Int. J. Bioinform. Res. Appl. 2020, 16, 1–24. [Google Scholar] [CrossRef]
- Ding, J.; Miao, Q.-F.; Zhang, J.-W.; Guo, Y.-X.; Zhang, Y.-X.; Zhai, Q.-X.; Chen, Z.-H. H258R Mutation in KCNAB3 Gene in a Family with Genetic Epilepsy and Febrile Seizures Plus. Brain Behav. 2020, 10, e01859. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; François, R.; Henry, L.; Müller, K. Dplyr: A Grammar of Data Manipulation. R Package Version 1.0.7. 2022. Available online: https://dplyr.tidyverse.org (accessed on 8 May 2023).
- Wickham, H.; Girlich, M. Tidyr: Tidy Messy Data. R Package Version 1.2.0. 2022. Available online: https://tidyr.tidyverse.org/ (accessed on 8 May 2023).
- R Core Team R: A Language and Environment for Statistical Computing. 2016. Available online: https://www.R-project.org/ (accessed on 8 May 2023).
- Mountford, H.S.; Villanueva, P.; Fernández, M.A.; De Barbieri, Z.; Cazier, J.B.; Newbury, D.F. Candidate Gene Variant Effects on Language Disorders in Robinson Crusoe Island. Ann. Hum. Biol. 2019, 46, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.; Cacabelos, R. Genomics of Speech and Language Disorders. J. Transl. Genet. Genom. 2019. [Google Scholar] [CrossRef]
- Lee, B.T.; Barber, G.P.; Benet-Pages, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, C.M.; et al. The UCSC Genome Browser Database: 2022 Update. Nucleic Acids Res. 2022, 50, D1115–D1122. [Google Scholar] [CrossRef]
- Adlof, S.M.; Hogan, T.P. If We Don’t Look, We Won’t See: Measuring Language Development to Inform Literacy Instruction. Policy Insights Behav. Brain Sci. 2019, 6, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Catts, H.W.; Adlof, S.M.; Ellis Weismer, S. Language Deficits in Poor Comprehenders: A Case for the Simple View of Reading. J. Speech Lang. Hear. Res. 2006, 49, 278–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [CrossRef]
- Jaiganesh, A.; Narui, Y.; Araya-Secchi, R.; Sotomayor, M. Beyond Cell–Cell Adhesion: Sensational Cadherins for Hearing and Balance. Cold Spring Harb. Perspect. Biol. 2018, 10, a029280. [Google Scholar] [CrossRef]
- Junghans, D.; Heidenreich, M.; Hack, I.; Taylor, V.; Frotscher, M.; Kemler, R. Postsynaptic and Differential Localization to Neuronal Subtypes of Protocadherin Beta16 in the Mammalian Central Nervous System. Eur. J. Neurosci. 2008, 27, 559–571. [Google Scholar] [CrossRef]
- Speevak, M.D.; Farrell, S.A. Non-Syndromic Language Delay in a Child with Disruption in the Protocadherin11X/Y Gene Pair. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 484–489. [Google Scholar] [CrossRef]
- Nardello, R.; Antona, V.; Mangano, G.D.; Salpietro, V.; Mangano, S.; Fontana, A. A Paradigmatic Autistic Phenotype Associated with Loss of PCDH11Y and NLGN4Y Genes. BMC Med. Genom. 2021, 14, 98. [Google Scholar] [CrossRef]
- Naskar, T.; Faruq, M.; Banerjee, P.; Khan, M.; Midha, R.; Kumari, R.; Devasenapathy, S.; Prajapati, B.; Sengupta, S.; Jain, D.; et al. Ancestral Variations of the PCDHG Gene Cluster Predispose to Dyslexia in a Multiplex Family. EBioMedicine 2018, 28, 168–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priddle, T.H.; Crow, T.J. Protocadherin 11X/Y a Human-Specific Gene Pair: An Immunohistochemical Survey of Fetal and Adult Brains. Cereb. Cortex 2013, 23, 1933–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastidar, S.G.; Nair, D. A Ribosomal Perspective on Neuronal Local Protein Synthesis. Front. Mol. Neurosci. 2022, 15, 823135. [Google Scholar] [CrossRef] [PubMed]
- Hetman, M.; Slomnicki, L.P. Ribosomal Biogenesis as an Emerging Target of Neurodevelopmental Pathologies. J. Neurochem. 2019, 148, 325–347. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.L.; Abel, L. Whole-Genome Sequencing Is More Powerful than Whole-Exome Sequencing for Detecting Exome Variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [Green Version]
- Rice, M.L.; Hoffman, L.; Wexler, K. Judgments of Omitted BE and DO in Questions as Extended Finiteness Clinical Markers of Specific Language Impairment (SLI) to 15 Years: A Study of Growth and Asymptote. J. Speech Lang. Hear. Res. 2009, 52, 1417–1433. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Ding, S.L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional Landscape of the Prenatal Human Brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Crain, S. Language Acquisition in the Absence of Experience. Behav. Brain Sci. 1991, 14, 597–612. [Google Scholar] [CrossRef]
- Ambridge, B.; Lieven, E.V.M. Child Language Acquisition: Contrasting Theoretical Approaches; Cambridge University Press: Cambridge, UK, 2011; ISBN 978-1-139-50051-7. [Google Scholar]
- SLI Consortium A Genomewide Scan Identifies Two Novel Loci Involved in Specific Language Impairment**Members of the Consortium Are Listed in the Appendix. Am. J. Hum. Genet. 2002, 70, 384–398. [CrossRef]
- SLI Consortium Highly Significant Linkage to the SLI1 Locus in an Expanded Sample of Individuals Affected by Specific Language Impairment. Am. J. Hum. Genet. 2004, 74, 1225–1238. [CrossRef] [Green Version]
- Hoff, E.; Tian, C. Socioeconomic Status and Cultural Influences on Language. J. Commun. Disord. 2005, 38, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Hoff, E. The Specificity of Environmental Influence: Socioeconomic Status Affects Early Vocabulary Development Via Maternal Speech. Child Dev. 2003, 74, 1368–1378. [Google Scholar] [CrossRef] [Green Version]
- Schuele, C.M. Socioeconomic Influences on Children’s Language Acquisition. J. Speech-Lang. Pathol. Audiol. 2001, 25, 77–88. [Google Scholar]
- Stromswold, K. Why Aren’t Identical Twins Linguistically Identical? Genetic, Prenatal and Postnatal Factors. Cognition 2006, 101, 333–384. [Google Scholar] [CrossRef] [PubMed]
- Piot, L.; Havron, N.; Cristia, A. Socioeconomic Status Correlates with Measures of Language Environment Analysis (LENA) System: A Meta-Analysis. J. Child Lang. 2022, 49, 1037–1051. [Google Scholar] [CrossRef]
- Rice, M.L.; Hoffman, L. Predicting Vocabulary Growth in Children with and without Specific Language Impairment: A Longitudinal Study from 2;6 to 21 Years of Age. J. Speech Lang. Hear. Res. 2015, 58, 345–359. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Discovery Pedigree (s) | Additional Probands Carrying Variant (s) | Fisher’s Test p-Value 2 | rsID | AA Change | MAF | # of Damaging In Silico Scores 3 | AA Change (HOPE) | Causality Prediction | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Affected | Unaffected | Size | Charge | ||||||||
| PDHA2 | 5463, 5886 | 2 ^ | 0 | 0.3590 | rs147966234 | pArg286Pro | 0.0089 1a | 5/5 | ∨ | POS to neu | P |
| PCDHB3 | 4093, 4130 | 1 | 0 | 0.5112 | rs61739886 | p.Thr81Ile | 0.0064 1a | 3/5 | ∧ | NCC | P |
| FURIN | 4075, 5886 | 0 | 0 | 0.06445 | rs150925934 | p.Arg462Trp | 0.0017 1a | 4/5 | ∧ | POS to neu | P |
| NOL6 | 4130 | 1 | 0 | <0.0001 *,1,4 | rs114465306 | p.Pro134Leu | 0.00008 1 | 4/5 | ∧ | NCR | P |
| 5931 | 0 | 0 | NA | rs114110943 | p.His366Tyr | 0.006 1b | 2/5 | ∧ | NCR | Likely P | |
| IQGAP3 | 4093 | 0 | 0 | NA | rs147754283 | p.Arg630Trp | 0.00005 1a | 3/5 | ∧ | POS to neu | Likely P |
| 5886 | 2 | 0 | 0.1094 | rs112144116 | p.Ala562Thr | 0.0034 1a | 4/5 | ∧ | NCC | P | |
| BAHCC1 | 4093 | 0 | 0 | NA | rs369588790 | p.Arg2199Gln | 0.00006 1a | 2/5 | NCR | NEG to neu | Likely P |
| 5931 | 0 | 0 | NA | rs200719992 | p.Gln2463Glu | 0.0066 1b | 4/5 | ∨ | POS to neu | Likely P | |
| GLI3 | 4093, 4130, 4132 | 6 ^ | 4 | 0.7946 | rs35364414 | p.Arg1537Cys | 0.0536 1a | 4/5 | ∨ | POS to neu | B |
| FLNB | 4132 | 2 + 1 ! | 0 | 0.3398 | rs116826041 | p.Ile2319Thr | 0.0093 1a | 3/5 | ∨ | NCR | B |
| KMT2D | 5463 | 0 | 0 | NA | rs146044282 | p.Asp3419Gly | 0.0015 1a | 3/5 | ∨ | NCR | Likely P |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andres, E.M.; Earnest, K.K.; Xuan, H.; Zhong, C.; Rice, M.L.; Raza, M.H. Innovative Family-Based Genetically Informed Series of Analyses of Whole-Exome Data Supports Likely Inheritance for Grammar in Children with Specific Language Impairment. Children 2023, 10, 1119. https://doi.org/10.3390/children10071119
Andres EM, Earnest KK, Xuan H, Zhong C, Rice ML, Raza MH. Innovative Family-Based Genetically Informed Series of Analyses of Whole-Exome Data Supports Likely Inheritance for Grammar in Children with Specific Language Impairment. Children. 2023; 10(7):1119. https://doi.org/10.3390/children10071119
Chicago/Turabian StyleAndres, Erin M., Kathleen Kelsey Earnest, Hao Xuan, Cuncong Zhong, Mabel L. Rice, and Muhammad Hashim Raza. 2023. "Innovative Family-Based Genetically Informed Series of Analyses of Whole-Exome Data Supports Likely Inheritance for Grammar in Children with Specific Language Impairment" Children 10, no. 7: 1119. https://doi.org/10.3390/children10071119