Recombinant Human Soluble Thrombomodulin Suppresses Monocyte Adhesion by Reducing Lipopolysaccharide-Induced Endothelial Cellular Stiffening

 ,
,  , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Determination of Cellular Stiffness
2.3. Fluorescent Imaging of F-Actin and Vinculin in HUVECs
2.4. Dye Transfer Assay
2.5. THP-1 Cell Adhesion to Substrates
2.6. THP-1 Cell Adhesion to HUVECs
2.7. Statistical Analyses
3. Results
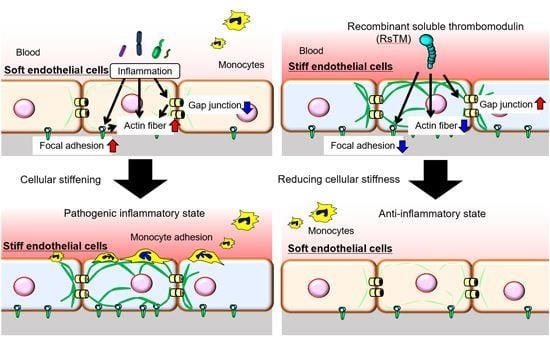
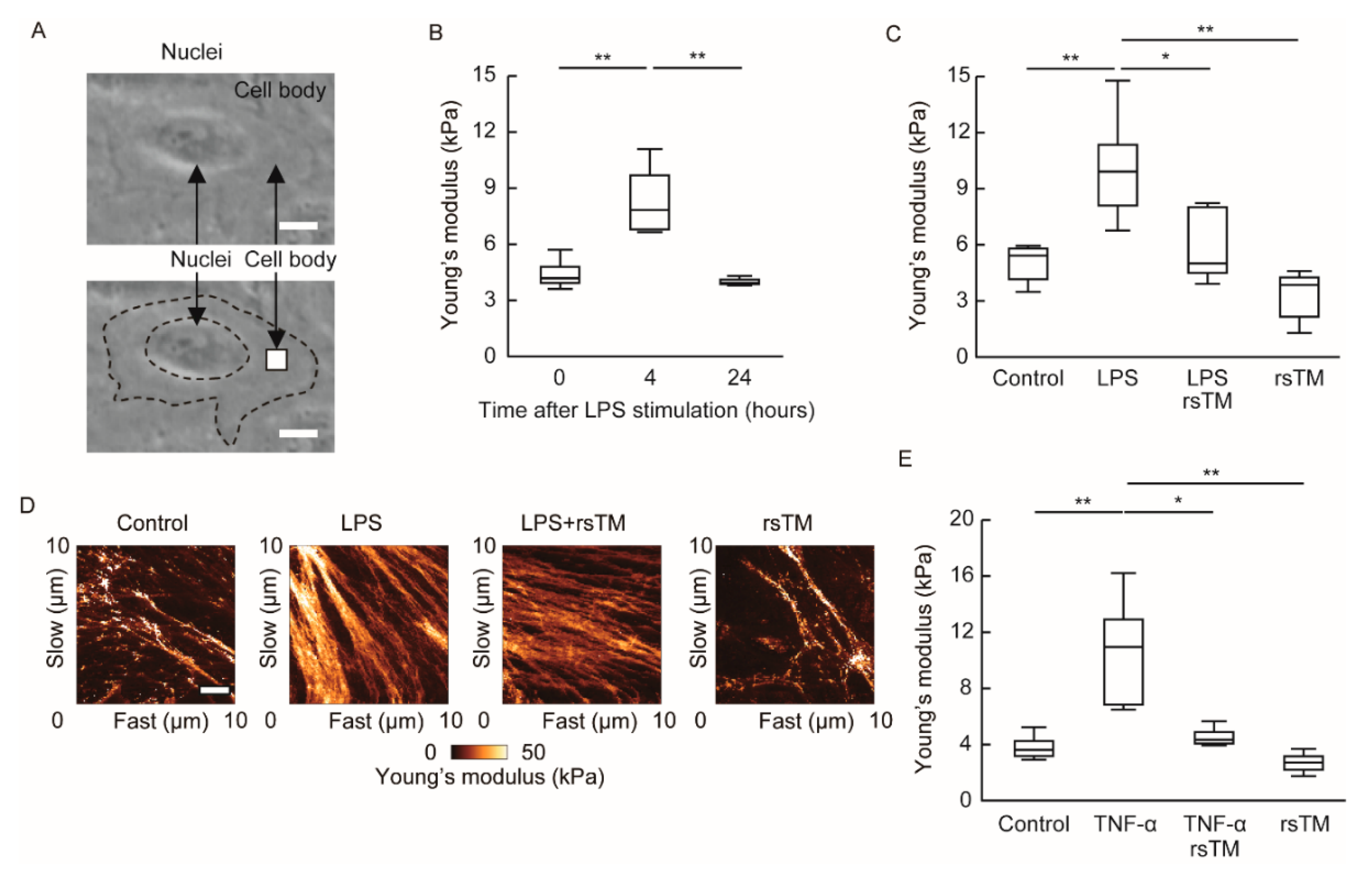
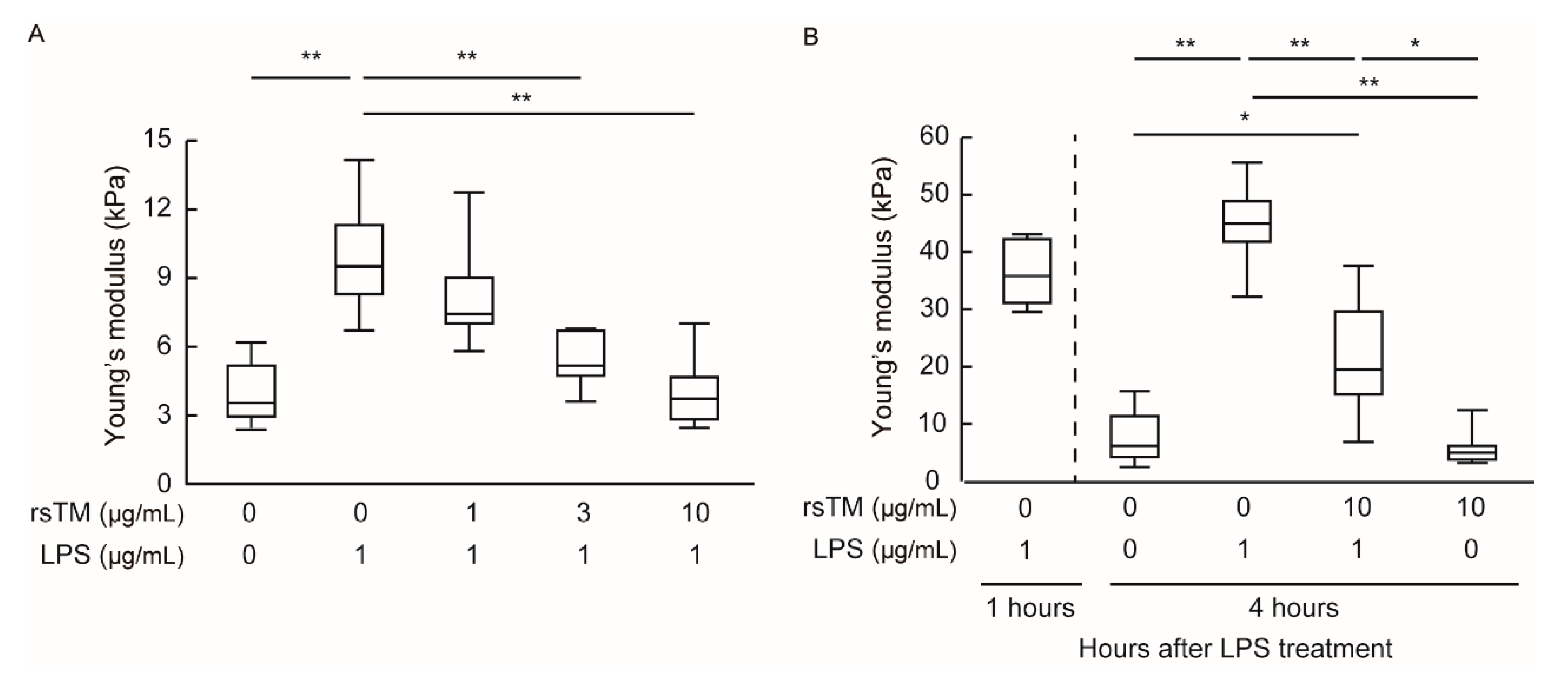
3.1. RsTM Reduces LPS-Induced Endothelial Cellular Stiffening
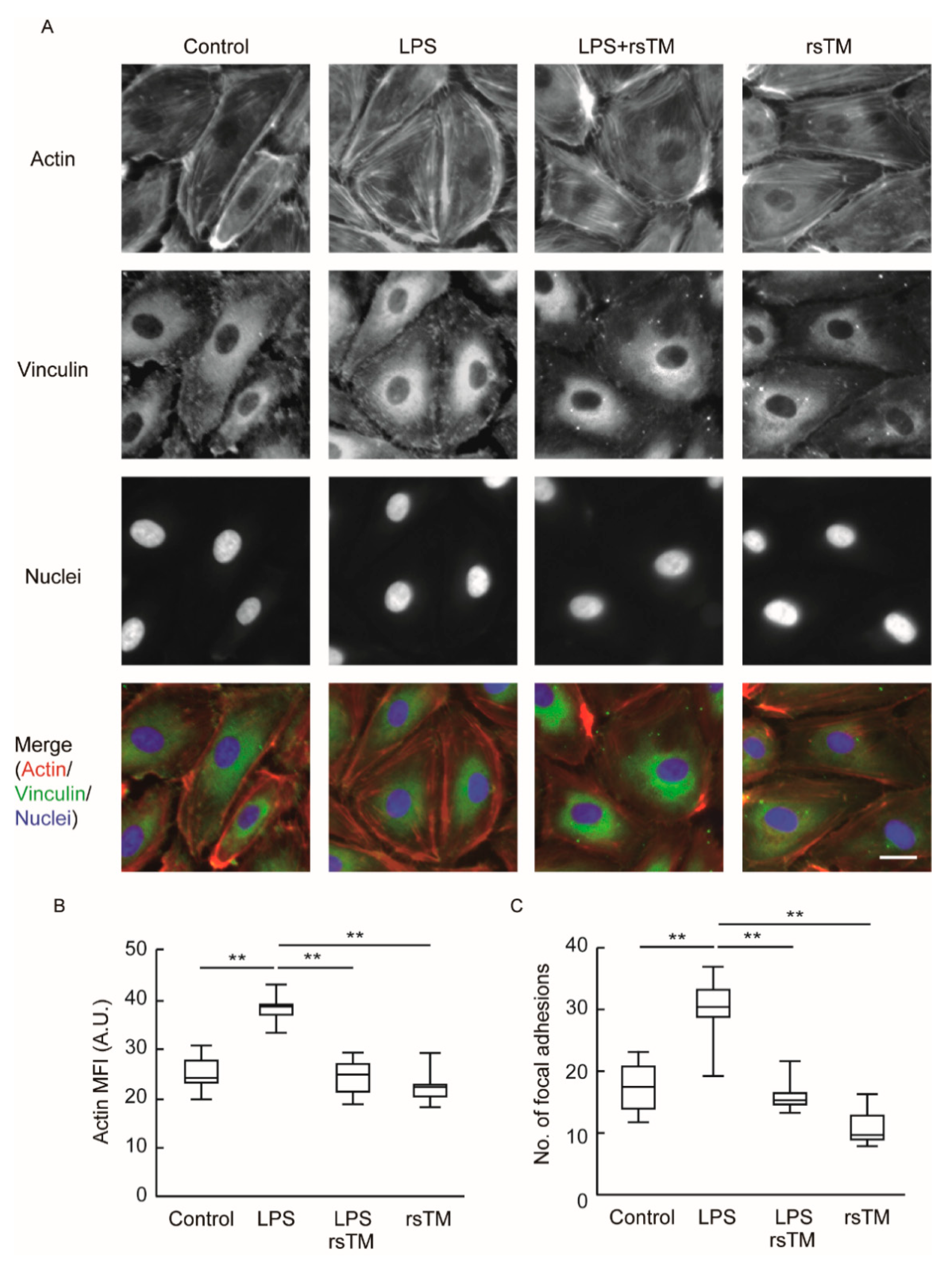
3.2. RsTM Improves Cytoskeletal Rearrangement and Focal Adhesion Formation
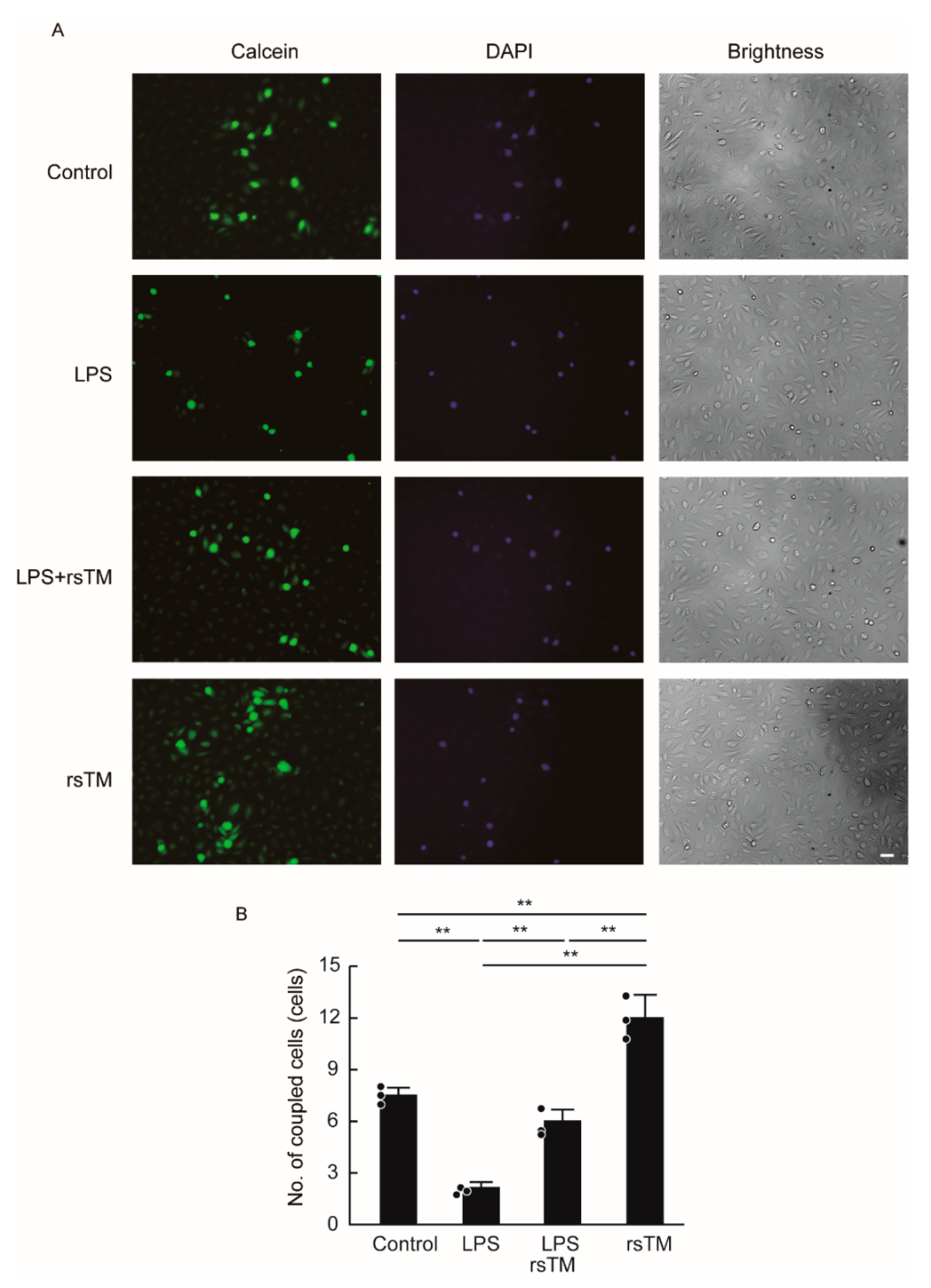
3.3. RsTM Enhances the Gap Junction Functionality of Endothelial Cells
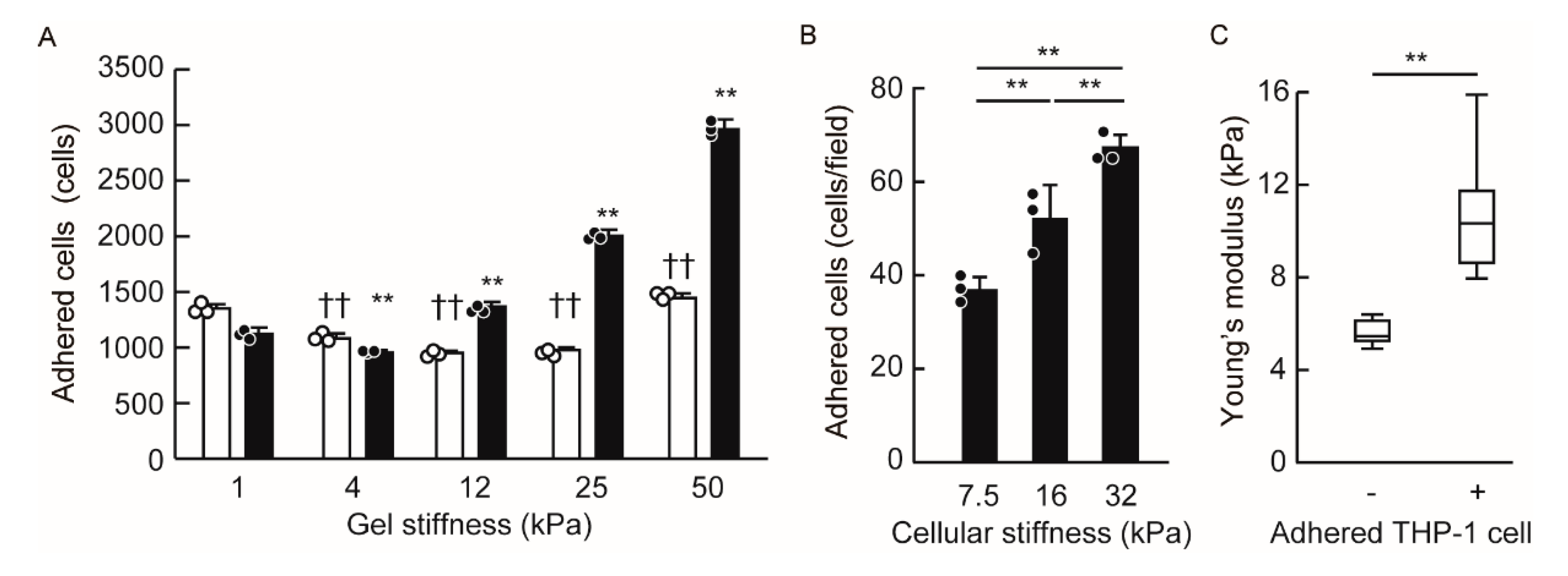
3.4. Endothelial Cellular Stiffening Promotes Monocyte Adhesion
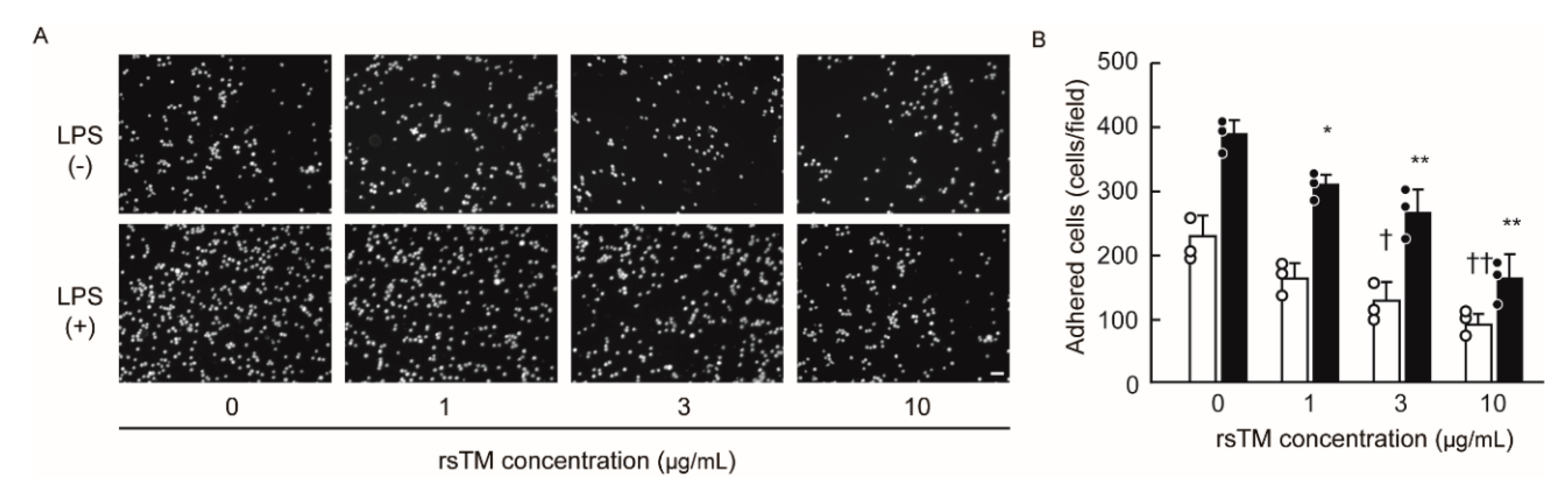
3.5. RsTM Suppresses Monocyte Adhesion by Reducing Endothelial Cellular Stiffness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Esmon, C.T.; Owen, W.G. Identification of an endothelial cell cofactor for thrombin-catalyzed activation of protein C. Proc. Natl. Acad. Sci. USA 1981, 78, 2249–2252. [Google Scholar] [CrossRef] [Green Version]
- Esmon, N.L.; Owen, W.G.; Esmon, C.T. Isolation of a membrane-bound cofactor for thrombin-catalyzed activation of protein C. J. Biol. Chem. 1982, 257, 859–864. [Google Scholar]
- Takehara, K.; Murakami, T.; Kuwahara-Arai, K.; Iba, T.; Nagaoka, I.; Sakamoto, K. Evaluation of the effect of recombinant thrombomodulin on a lipopolysaccharide-induced murine sepsis model. Exp. Ther. Med. 2017, 13, 2969–2974. [Google Scholar] [CrossRef]
- Suzuki, K.; Kusumoto, H.; Deyashiki, Y.; Nishioka, J.; Maruyama, I.; Zushi, M.; Kawahara, S.; Honda, G.; Yamamoto, S.; Horiguchi, S. Structure and expression of human thrombomodulin, a thrombin receptor on endothelium acting as a cofactor for protein C activation. EMBO J. 1987, 6, 1891–1897. [Google Scholar] [CrossRef]
- Uchiba, M.; Okajima, K.; Murakami, K.; Nawa, K.; Okabe, H.; Takatsuki, K. Recombinant human soluble thrombomodulin reduces endotoxin-induced pulmonary vascular injury via protein C activation in rats. Thromb. Haemost. 1995, 74, 1265–1270. [Google Scholar] [CrossRef] [PubMed]
- Abeyama, K.; Stern, D.M.; Ito, Y.; Kawahara, K.; Yoshimoto, Y.; Tanaka, M.; Uchimura, T.; Ida, N.; Yamazaki, Y.; Yamada, S.P.; et al. The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J. Clin. Investig. 2005, 115, 1267–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Hayashi, T.; Nishioka, J.; Kosaka, Y.; Zushi, M.; Honda, G.; Yamamoto, S. A domain composed of epidermal growth factor-like structures of human thrombomodulin is essential for thrombin binding and for protein C activation. J. Biol. Chem. 1989, 264, 4872–4876. [Google Scholar] [PubMed]
- Ito, T.; Kawahara, K.; Okamoto, K.; Yamada, S.; Yasuda, M.; Imaizumi, H.; Nawa, Y.; Meng, X.; Shrestha, B.; Hashiguchi, T.; et al. Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arter. Thromb. Vasc. Biol. 2008, 28, 1825–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.Y.; Chang, W.E.; Shi, G.Y.; Chang, B.Y.; Cheng, S.E.; Shih, Y.T.; Wu, H.L. Recombinant thrombomodulin inhibits lipopolysaccharide-induced inflammatory response by blocking the functions of CD14. J. Immunol. 2015, 194, 1905–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, E.M.; Van de Wouwer, M.; Pollefeyt, S.; Jurk, K.; Van Aken, H.; De Vriese, A.; Weitz, J.I.; Weiler, H.; Hellings, P.W.; Schaeffer, P.; et al. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor kappaB and mitogen-activated protein kinase pathways. J. Exp. Med. 2002, 196, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.F.; Hwang, S.J.; Vasan, R.S.; Larson, M.G.; Pencina, M.J.; Hamburg, N.M.; Vita, J.A.; Levy, D.; Benjamin, E.J. Arterial stiffness and cardiovascular events: The Framingham Heart Study. Circulation 2010, 121, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, A.; Montezano, A.C.; Lopes, R.A.; Rios, F.; Touyz, R.M. Vascular Fibrosis in Aging and Hypertension: Molecular Mechanisms and Clinical Implications. Can. J. Cardiol. 2016, 32, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kothapalli, D.; Liu, S.L.; Bae, Y.H.; Monslow, J.; Xu, T.; Hawthorne, E.A.; Byfield, F.J.; Castagnino, P.; Rao, S.; Rader, D.J.; et al. Cardiovascular protection by ApoE and ApoE-HDL linked to suppression of ECM gene expression and arterial stiffening. Cell Rep. 2012, 2, 1259–1271. [Google Scholar] [CrossRef] [Green Version]
- Hays, T.T.; Ma, B.; Zhou, N.; Stoll, S.; Pearce, W.J.; Qiu, H. Vascular smooth muscle cells direct extracellular dysregulation in aortic stiffening of hypertensive rats. Aging Cell 2018, 17, e12748. [Google Scholar] [CrossRef]
- Okamoto, T.; Kawamoto, E.; Takagi, Y.; Akita, N.; Hayashi, T.; Park, E.J.; Suzuki, K.; Shimaoka, M. Gap junction-mediated regulation of endothelial cellular stiffness. Sci. Rep. 2017, 7, 6134. [Google Scholar] [CrossRef] [Green Version]
- Huveneers, S.; Daemen, M.J.; Hordijk, P.L. Between Rho(k) and a hard place: The relation between vessel wall stiffness, endothelial contractility, and cardiovascular disease. Circ. Res. 2015, 116, 895–908. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Brown, A.C.; Myers, D.R.; Sakurai, Y.; Mannino, R.G.; Tran, R.; Ahn, B.; Hardy, E.T.; Kee, M.F.; Kumar, S.; et al. Platelet mechanosensing of substrate stiffness during clot formation mediates adhesion, spreading, and activation. Proc. Natl. Acad. Sci. USA 2014, 111, 14430–14435. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.M.; Gilula, N.B. The gap junction communication channel. Cell 1996, 84, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Giepmans, B.N. Gap junctions and connexin-interacting proteins. Cardiovasc. Res. 2004, 62, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Suzuki, K. The Role of Gap Junction-Mediated Endothelial Cell-Cell Interaction in the Crosstalk between Inflammation and Blood Coagulation. Int. J. Mol. Sci. 2017, 18, 2254. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Izawa, H.; Ichihara, S.; Takatsu, F.; Ishihara, H.; Hirayama, H.; Sone, T.; Tanaka, M.; Yokota, M. Prediction of the risk of myocardial infarction from polymorphisms in candidate genes. N. Engl. J. Med. 2002, 347, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Firouzi, M.; Kok, B.; Spiering, W.; Busjahn, A.; Bezzina, C.R.; Ruijter, J.M.; Koeleman, B.P.; Schipper, M.; Groenewegen, W.A.; Jongsma, H.J.; et al. Polymorphisms in human connexin40 gene promoter are associated with increased risk of hypertension in men. J. Hypertens. 2006, 24, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.W.; Christen, T.; Roth, I.; Chadjichristos, C.E.; Derouette, J.P.; Foglia, B.F.; Chanson, M.; Goodenough, D.A.; Kwak, B.R. Connexin37 protects against atherosclerosis by regulating monocyte adhesion. Nat. Med. 2006, 12, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Chadjichristos, C.E.; Scheckenbach, K.E.; van Veen, T.A.; Richani Sarieddine, M.Z.; de Wit, C.; Yang, Z.; Roth, I.; Bacchetta, M.; Viswambharan, H.; Foglia, B.; et al. Endothelial-specific deletion of connexin40 promotes atherosclerosis by increasing CD73-dependent leukocyte adhesion. Circulation 2010, 121, 123–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horimizu, M.; Kawase, T.; Tanaka, T.; Okuda, K.; Nagata, M.; Burns, D.M.; Yoshie, H. Biomechanical evaluation by AFM of cultured human cell-multilayered periosteal sheets. Micron 2013, 48, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Akiyama, M.; Takeda, M.; Gabazza, E.C.; Hayashi, T.; Suzuki, K. Connexin32 is expressed in vascular endothelial cells and participates in gap-junction intercellular communication. Biochem. Biophys. Res. Commun. 2009, 382, 264–268. [Google Scholar] [CrossRef]
- Stroka, K.M.; Aranda-Espinoza, H. Effects of Morphology vs. Cell-Cell Interactions on Endothelial Cell Stiffness. Cellar Mol. Bioeng. 2011, 4, 9–27. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, A.; Te Riet, J.; Ritz, K.; Hoogenboezem, M.; Anthony, E.C.; Mul, F.P.; de Vries, C.J.; Daemen, M.J.; Figdor, C.G.; van Buul, J.D.; et al. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J. Cell Sci. 2014, 127, 4470–4482. [Google Scholar] [CrossRef] [Green Version]
- Previtera, M.L.; Sengupta, A. Substrate Stiffness Regulates Proinflammatory Mediator Production through TLR4 Activity in Macrophages. PLoS ONE 2015, 10, e0145813. [Google Scholar] [CrossRef]
- Bashour, K.T.; Gondarenko, A.; Chen, H.; Shen, K.; Liu, X.; Huse, M.; Hone, J.C.; Kam, L.C. CD28 and CD3 have complementary roles in T-cell traction forces. Proc. Natl. Acad. Sci. USA 2014, 111, 2241–2246. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, T.; Takagi, Y.; Kawamoto, E.; Park, E.J.; Usuda, H.; Wada, K.; Shimaoka, M. Reduced substrate stiffness promotes M2-like macrophage activation and enhances peroxisome proliferator-activated receptor gamma expression. Exp. Cell Res. 2018, 367, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Kuebler, W.M.; Borges, J.; Sckell, A.; Kuhnle, G.E.; Bergh, K.; Messmer, K.; Goetz, A.E. Role of L-selectin in leukocyte sequestration in lung capillaries in a rabbit model of endotoxemia. Am. J. Respir. Crit. Care Med. 2000, 161, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Gavara, N.; Chadwick, R.S. Relationship between cell stiffness and stress fiber amount, assessed by simultaneous atomic force microscopy and live-cell fluorescence imaging. Biomech. Model. Mechanobiol. 2016, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Wu, M.H.; Yuan, S.Y. Nonmuscle myosin light-chain kinase deficiency attenuates atherosclerosis in apolipoprotein E-deficient mice via reduced endothelial barrier dysfunction and monocyte migration. Circulation 2011, 124, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essler, M.; Staddon, J.M.; Weber, P.C.; Aepfelbacher, M. Cyclic AMP blocks bacterial lipopolysaccharide-induced myosin light chain phosphorylation in endothelial cells through inhibition of Rho/Rho kinase signaling. J. Immunol. 2000, 164, 6543–6549. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Huang, X.; Guo, F.; Zhou, Z.; Chang, M.; Tang, J.; Huan, J. Lipopolysaccharide Induces Human Pulmonary Micro-Vascular Endothelial Apoptosis via the YAP Signaling Pathway. Front. Celluar Infect. Microbiol. 2016, 6, 133. [Google Scholar] [CrossRef] [Green Version]
- Nardone, G.; Oliver-De La Cruz, J.; Vrbsky, J.; Martini, C.; Pribyl, J.; Skladal, P.; Pesl, M.; Caluori, G.; Pagliari, S.; Martino, F.; et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat. Commun. 2017, 8, 15321. [Google Scholar] [CrossRef]
- Schaefer, A.; Hordijk, P.L. Cell-stiffness-induced mechanosignaling—A key driver of leukocyte transendothelial migration. J. Cell Sci. 2015, 128, 2221–2230. [Google Scholar] [CrossRef] [Green Version]
- Giri, H.; Cai, X.; Panicker, S.R.; Biswas, I.; Rezaie, A.R. Thrombomodulin Regulation of Mitogen-Activated Protein Kinases. Int. J. Mol. Sci. 2019, 20, 1851. [Google Scholar] [CrossRef] [Green Version]
- Stroka, K.M.; Aranda-Espinoza, H. Neutrophils display biphasic relationship between migration and substrate stiffness. Cell Motil. Cytoskelet. 2009, 66, 328–341. [Google Scholar] [CrossRef]
- Huynh, J.; Nishimura, N.; Rana, K.; Peloquin, J.M.; Califano, J.P.; Montague, C.R.; King, M.R.; Schaffer, C.B.; Reinhart-King, C.A. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 2011, 3, 112ra122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmel, L.; van der Stoel, M.; Rianna, C.; van Stalborch, A.M.; de Ligt, A.; Hoogenboezem, M.; Tol, S.; van Rijssel, J.; Szulcek, R.; Bogaard, H.J.; et al. Stiffness-Induced Endothelial DLC-1 Expression Forces Leukocyte Spreading through Stabilization of the ICAM-1 Adhesome. Cell Rep. 2018, 24, 3115–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, M.; Ito, T.; Kawahara, K.; Yamamoto, M.; Nagasato, T.; Shrestha, B.; Yamada, S.; Miyauchi, T.; Higuchi, K.; Takenaka, T.; et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS ONE 2013, 8, e75961. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.L.; Chang, C.F.; Shi, C.S.; Shi, G.Y.; Wu, H.L. Recombinant lectin-like domain of thrombomodulin suppresses vascular inflammation by reducing leukocyte recruitment via interacting with Lewis Y on endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2366–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, E.; Nago, N.; Okamoto, T.; Gaowa, A.; Masui-Ito, A.; Sakakura, Y.; Akama, Y.; Soe, Z.Y.; Prajuabjinda, O.; Darkwah, S.; et al. Anti-adhesive effects of human soluble thrombomodulin and its domains. Biochem. Biophys. Res. Commun. 2019, 511, 312–317. [Google Scholar] [CrossRef]
- Kawamoto, E.; Okamoto, T.; Takagi, Y.; Honda, G.; Suzuki, K.; Imai, H.; Shimaoka, M. LFA-1 and Mac-1 integrins bind to the serine/threonine-rich domain of thrombomodulin. Biochem. Biophys. Res. Commun. 2016, 473, 1005–1012. [Google Scholar] [CrossRef]
- Kasa, A.; Csortos, C.; Verin, A.D. Cytoskeletal mechanisms regulating vascular endothelial barrier function in response to acute lung injury. Tissue Barriers 2015, 3, e974448. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Zhang, W.G.; Sun, J.; Zhang, Y.; Lu, J.F.; Han, H.B.; Zhou, C.M.; Yan, J.H. Protective effects of thrombomodulin on microvascular permeability after subarachnoid hemorrhage in mouse model. Neuroscience 2015, 299, 18–27. [Google Scholar] [CrossRef]
- Hun Lee, J.; Won, S.; Stein, D.G. Progesterone attenuates thrombin-induced endothelial barrier disruption in the brain endothelial cell line bEnd.3: The role of tight junction proteins and the endothelial protein C receptor. Brain Res. 2015, 1613, 73–80. [Google Scholar] [CrossRef]
- Chen, C.H.; Mayo, J.N.; Gourdie, R.G.; Johnstone, S.R.; Isakson, B.E.; Bearden, S.E. The connexin 43/ZO-1 complex regulates cerebral endothelial F-actin architecture and migration. Am. J. Physiol. Cell. Physiol. 2015, 309, C600–C607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, H.S.; Iacobas, I.; Hotchkiss, K.; Hirst-Jensen, B.J.; Bosco, A.; Dandachi, N.; Dermietzel, R.; Sorgen, P.L.; Spray, D.C. The gap junction protein connexin32 interacts with the Src homology 3/hook domain of discs large homolog 1. J. Biol. Chem. 2007, 282, 9789–9796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talhouk, R.S.; Mroue, R.; Mokalled, M.; Abi-Mosleh, L.; Nehme, R.; Ismail, A.; Khalil, A.; Zaatari, M.; El-Sabban, M.E. Heterocellular interaction enhances recruitment of alpha and beta-catenins and ZO-2 into functional gap-junction complexes and induces gap junction-dependant differentiation of mammary epithelial cells. Exp. Cell Res. 2008, 314, 3275–3291. [Google Scholar] [CrossRef] [PubMed]
- Herve, J.C.; Bourmeyster, N.; Sarrouilhe, D. Diversity in protein-protein interactions of connexins: Emerging roles. Biochim. Biophys. Acta 2004, 1662, 22–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derouette, J.P.; Wong, C.; Burnier, L.; Morel, S.; Sutter, E.; Galan, K.; Brisset, A.C.; Roth, I.; Chadjichristos, C.E.; Kwak, B.R. Molecular role of Cx37 in advanced atherosclerosis: A micro-array study. Atherosclerosis 2009, 206, 69–76. [Google Scholar] [CrossRef]
- Vincent, J.L.; Francois, B.; Zabolotskikh, I.; Daga, M.K.; Lascarrou, J.B.; Kirov, M.Y.; Pettila, V.; Wittebole, X.; Meziani, F.; Mercier, E.; et al. Effect of a Recombinant Human Soluble Thrombomodulin on Mortality in Patients With Sepsis-Associated Coagulopathy: The SCARLET Randomized Clinical Trial. JAMA 2019, 321, 1993–2002. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okamoto, T.; Kawamoto, E.; Usuda, H.; Tanaka, T.; Nikai, T.; Asanuma, K.; Suzuki, K.; Shimaoka, M.; Wada, K. Recombinant Human Soluble Thrombomodulin Suppresses Monocyte Adhesion by Reducing Lipopolysaccharide-Induced Endothelial Cellular Stiffening. Cells 2020, 9, 1811. https://doi.org/10.3390/cells9081811
Okamoto T, Kawamoto E, Usuda H, Tanaka T, Nikai T, Asanuma K, Suzuki K, Shimaoka M, Wada K. Recombinant Human Soluble Thrombomodulin Suppresses Monocyte Adhesion by Reducing Lipopolysaccharide-Induced Endothelial Cellular Stiffening. Cells. 2020; 9(8):1811. https://doi.org/10.3390/cells9081811
Chicago/Turabian StyleOkamoto, Takayuki, Eiji Kawamoto, Haruki Usuda, Tetsuya Tanaka, Tetsuro Nikai, Kunihiro Asanuma, Koji Suzuki, Motomu Shimaoka, and Koichiro Wada. 2020. "Recombinant Human Soluble Thrombomodulin Suppresses Monocyte Adhesion by Reducing Lipopolysaccharide-Induced Endothelial Cellular Stiffening" Cells 9, no. 8: 1811. https://doi.org/10.3390/cells9081811