Multifaceted Aspects of Metabolic Plasticity in Human Cholangiocarcinoma: An Overview of Current Perspectives

 , , , and
, , , and
Abstract
:1. Introduction
2. Glucose Metabolism and CCA
3. Lipid Metabolism in CCA
4. Protein Metabolism and CCA
5. CCA and Iron
6. Molecular Aspects Underlying CCA Metabolism
6.1. PI3K-AKT-mTOR Signaling Pathway
6.2. SIRT2/cMYC Signaling Pathway
6.3. Uncoupling Protein 2 (UCP2)
6.4. Transcription Factor FOXO1
6.5. Nuclear Receptors: Farnesoid X Receptor (FXR) and Peroxisome Proliferator Activated Receptor-α (PPAR-α)
6.6. Isocitrate Dehidrogenase (IDH)
6.7. Tumor Suppressor p53
6.8. Hif-1α Signaling Pathway
7. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Steiner, P.E.; Higginson, J. Cholangiolocellular carcinoma of the liver. Cancer 1959, 12, 753–759. [Google Scholar] [CrossRef]
- Bragazzi, M.C.; Ridola, L.; Safarikia, S.; Matteo, S.D.; Costantini, D.; Nevi, L.; Cardinale, V. New insights into cholangiocarcinoma: Multiple stems and related cell lineages of origin. Ann. Gastroenterol. 2018, 31, 42–55. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Kakuda, Y. Pathologic classification of cholangiocarcinoma: New concepts. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 277–293. [Google Scholar] [CrossRef]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [Green Version]
- Krasinskas, A.M. Cholangiocarcinoma. Surg. Pathol. Clin. 2018, 11, 403–429. [Google Scholar] [CrossRef]
- Oliveira, I.S.; Kilcoyne, A.; Everett, J.M.; Mino-Kenudson, M.; Harisinghani, M.G.; Ganesan, K. Cholangiocarcinoma: Classification, diagnosis, staging, imaging features, and management. Abdom. Radiol. 2017, 42, 1637–1649. [Google Scholar] [CrossRef]
- Nakanuma, Y.; Sato, Y.; Harada, K.; Sasaki, M.; Xu, J.; Ikeda, H. Pathological classification of intrahepatic cholangiocarcinoma based on a new concept. World J. Hepatol. 2010, 2, 419–427. [Google Scholar] [CrossRef]
- Deoliveira, M.L.; Schulick, R.D.; Nimura, Y.; Rosen, C.; Gores, G.; Neuhaus, P.; Clavien, P.A. New staging system and a registry for perihilar cholangiocarcinoma. Hepatology 2011, 53, 1363–1371. [Google Scholar] [CrossRef]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef] [Green Version]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, S.; Gores, G.J. Emerging molecular therapeutic targets for cholangiocarcinoma. J. Hepatol. 2017, 67, 632–644. [Google Scholar] [CrossRef]
- Forner, A.; Vidili, G.; Rengo, M.; Bujanda, L.; Ponz-Sarvisé, M.; Lamarca, A. Clinical presentation, diagnosis and staging of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. 1), 98–107. [Google Scholar] [CrossRef] [Green Version]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Patel, T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer 2002, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef]
- Alvaro, D.; Crocetti, E.; Ferretti, S.; Bragazzi, M.C.; Capocaccia, R.; committee, A.C. Descriptive epidemiology of cholangiocarcinoma in Italy. Dig. Liver Dis. 2010, 42, 490–495. [Google Scholar] [CrossRef]
- Rizvi, S.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma-Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef] [Green Version]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Hepatol. 2019, 72, 95–103. [Google Scholar] [CrossRef]
- Lendvai, G.; Szekerczés, T.; Illyés, I.; Dóra, R.; Kontsek, E.; Gógl, A.; Kiss, A.; Werling, K.; Kovalszky, I.; Schaff, Z.; et al. Cholangiocarcinoma: Classification, Histopathology and Molecular Carcinogenesis. Pathol. Oncol. Res. 2018, 1–13. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Wolpaw, A.J.; Dang, C.V. Exploiting Metabolic Vulnerabilities of Cancer with Precision and Accuracy. Trends Cell Biol. 2018, 28, 201–212. [Google Scholar] [CrossRef]
- Vergara, D.; Stanca, E.; Guerra, F.; Priore, P.; Gaballo, A.; Franck, J.; Simeone, P.; Trerotola, M.; De Domenico, S.; Fournier, I.; et al. β-Catenin Knockdown Affects Mitochondrial Biogenesis and Lipid Metabolism in Breast Cancer Cells. Front. Physiol. 2017, 8, 544. [Google Scholar] [CrossRef] [Green Version]
- Agostinelli, C.; Carloni, S.; Limarzi, F.; Righi, S.; Laginestra, M.A.; Musuraca, G.; Fiorentino, M.; Napolitano, R.; Cuneo, A.; Vergara, D.; et al. The emerging role of GSK-3β in the pathobiology of classical Hodgkin lymphoma. Histopathology 2017, 71, 72–80. [Google Scholar] [CrossRef]
- Sharma, A.; Boise, L.H.; Shanmugam, M. Cancer Metabolism and the Evasion of Apoptotic Cell Death. Cancers 2019, 11, 1144. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Chen, X.; Wang, L.; Chen, S. The sweet trap in tumors: Aerobic glycolysis and potential targets for therapy. Oncotarget 2016, 7, 38908–38926. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Liu, S.; Yin, S.; Niu, W.; Xiong, W.; Tan, M.; Li, G.; Zhou, M. The reverse Warburg effect is likely to be an Achilles’ heel of cancer that can be exploited for cancer therapy. Oncotarget 2017, 8, 57813–57825. [Google Scholar] [CrossRef] [Green Version]
- Wilde, L.; Roche, M.; Domingo-Vidal, M.; Tanson, K.; Philp, N.; Curry, J.; Martinez-Outschoorn, U. Metabolic coupling and the Reverse Warburg Effect in cancer: Implications for novel biomarker and anticancer agent development. Semin. Oncol. 2017, 44, 198–203. [Google Scholar] [CrossRef]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Chiarugi, P.; Cirri, P. Metabolic exchanges within tumor microenvironment. Cancer Lett. 2016, 380, 272–280. [Google Scholar] [CrossRef]
- Kubo, Y.; Aishima, S.; Tanaka, Y.; Shindo, K.; Mizuuchi, Y.; Abe, K.; Shirabe, K.; Maehara, Y.; Honda, H.; Oda, Y. Different expression of glucose transporters in the progression of intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1610–1617. [Google Scholar] [CrossRef]
- Ikeno, Y.; Seo, S.; Iwaisako, K.; Yoh, T.; Nakamoto, Y.; Fuji, H.; Taura, K.; Okajima, H.; Kaido, T.; Sakaguchi, S.; et al. Preoperative metabolic tumor volume of intrahepatic cholangiocarcinoma measured by. J. Transl. Med. 2018, 16, 95. [Google Scholar] [CrossRef]
- Suzuki, H.; Komuta, M.; Bolog, A.; Yokobori, T.; Wada, S.; Araki, K.; Kubo, N.; Watanabe, A.; Tsukagoshi, M.; Kuwano, H. Relationship between 18-F-fluoro-deoxy-D-glucose uptake and expression of glucose transporter 1 and pyruvate kinase M2 in intrahepatic cholangiocarcinoma. Dig. Liver Dis. 2015, 47, 590–596. [Google Scholar] [CrossRef]
- Qu, X.; Sheng, J.; Shen, L.; Su, J.; Xu, Y.; Xie, Q.; Wu, Y.; Zhang, X.; Sun, L. Autophagy inhibitor chloroquine increases sensitivity to cisplatin in QBC939 cholangiocarcinoma cells by mitochondrial ROS. PLoS ONE 2017, 12, e0173712. [Google Scholar] [CrossRef]
- Sanmai, S.; Proungvitaya, T.; Limpaiboon, T.; Chua-On, D.; Seubwai, W.; Roytrakul, S.; Wongkham, S.; Wongkham, C.; Somintara, O.; Sangkhamanon, S.; et al. Serum pyruvate dehydrogenase kinase as a prognostic marker for cholangiocarcinoma. Oncol. Lett. 2019, 17, 5275–5282. [Google Scholar] [CrossRef]
- Xu, L.; Li, Y.; Zhou, L.; Dorfman, R.G.; Liu, L.; Cai, R.; Jiang, C.; Tang, D.; Wang, Y.; Zou, X.; et al. SIRT3 elicited an anti-Warburg effect through HIF1α/PDK1/PDHA1 to inhibit cholangiocarcinoma tumorigenesis. Cancer Med. 2019, 8, 2380–2391. [Google Scholar] [CrossRef] [Green Version]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Moon, A.; Chin, S.; Kim, H.K.; Kwak, J.J.; Koh, E.S.; Kim, Y.W.; Jang, K.T. EGFR, COX2, p-AKT expression and PIK3CA mutation in distal extrahepatic bile duct carcinoma. Pathology 2016, 48, 35–40. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, T.; Wu, Q.; Wang, J.; Wu, C. Immunohistochemical analysis of the mTOR pathway in intrahepatic cholangiocarcinoma. Neoplasma 2012, 59, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.Y.; Hong, S.M.; Choi, B.Y.; Cho, H.; Yu, E.; Hewitt, S.M. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin. Cancer Res. 2009, 15, 660–667. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Wang, L.; Zhou, L.; Dorfman, R.G.; Pan, Y.; Tang, D.; Wang, Y.; Yin, Y.; Jiang, C.; Zou, X.; et al. The SIRT2/cMYC Pathway Inhibits Peroxidation-Related Apoptosis in Cholangiocarcinoma through Metabolic Reprogramming. Neoplasia 2019, 21, 429–441. [Google Scholar] [CrossRef]
- Yu, J.; Shi, L.; Shen, X.; Zhao, Y. UCP2 regulates cholangiocarcinoma cell plasticity via mitochondria-to-AMPK signals. Biochem. Pharmacol. 2019, 166, 174–184. [Google Scholar] [CrossRef]
- Erice, O.; Labiano, I.; Arbelaiz, A.; Santos-Laso, A.; Munoz-Garrido, P.; Jimenez-Agüero, R.; Olaizola, P.; Caro-Maldonado, A.; Martín-Martín, N.; Carracedo, A.; et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1335–1344. [Google Scholar] [CrossRef]
- Xu, F.; Zhao, Y.; Qin, G.; Huan, Y.; Li, L.; Gao, W. Comprehensive analysis of competing endogenous RNA networks associated with cholangiocarcinoma. Exp. Ther. Med. 2019, 18, 4103–4112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Xu, C.; Zhang, J.G.; Zeng, C.P.; Wang, X.F.; Zhou, R.; Lin, X.; Ao, Z.X.; Lu, J.M.; Shen, J.; et al. Multivariate analysis of genomics data to identify potential pleiotropic genes for type 2 diabetes, obesity and dyslipidemia using Meta-CCA and gene-based approach. PLoS ONE 2018, 13, e0201173. [Google Scholar] [CrossRef]
- Fujiwara, H.; Tateishi, K.; Misumi, K.; Hayashi, A.; Igarashi, K.; Kato, H.; Nakatsuka, T.; Suzuki, N.; Yamamoto, K.; Kudo, Y.; et al. Mutant IDH1 confers resistance to energy stress in normal biliary cells through PFKP-induced aerobic glycolysis and AMPK activation. Sci. Rep. 2019, 9, 18859. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, C.; Ma, P.; Yu, Q.; Gu, M.; Dong, L.; Jiang, W.; Pan, S.; Xie, C.; Han, J.; et al. PGC1α promotes cholangiocarcinoma metastasis by upregulating PDHA1 and MPC1 expression to reverse the Warburg effect. Cell Death Dis. 2018, 9, 466. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, A.; Qi, L.; Na, C.; Jiang, R.; Fan, Z.; Chen, J. FOXO1, a Potential Therapeutic Target, Regulates Autophagic Flux, Oxidative Stress, Mitochondrial Dysfunction, and Apoptosis in Human Cholangiocarcinoma QBC939 Cells. Cell Physiol. Biochem. 2018, 45, 1506–1514. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Che, L.; Tharp, K.M.; Park, H.M.; Pilo, M.G.; Cao, D.; Cigliano, A.; Latte, G.; Xu, Z.; Ribback, S.; et al. Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 2016, 63, 1900–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, R.; Hiep, N.C.; Ouchi, H.; Sato, Y.; Harada, K. Expression of fatty-acid-binding protein 5 in intrahepatic and extrahepatic cholangiocarcinoma: The possibility of different energy metabolisms in anatomical location. Med. Mol. Morphol. 2019, 53, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Yoon, B.I.; Pairojkul, C.; Demetris, A.J.; Sirica, A.E. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology 2002, 36, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Yamamoto, H.; Hiraoka, N.; Dono, K.; Ito, Y.; Okami, J.; Kondo, M.; Nagano, H.; Umeshita, K.; Sakon, M.; et al. Differential expression of cyclooxygenase-2 (COX-2) in human bile duct epithelial cells and bile duct neoplasm. Hepatology 2001, 34, 638–650. [Google Scholar] [CrossRef]
- Chariyalertsak, S.; Sirikulchayanonta, V.; Mayer, D.; Kopp-Schneider, A.; Fürstenberger, G.; Marks, F.; Müller-Decker, K. Aberrant cyclooxygenase isozyme expression in human intrahepatic cholangiocarcinoma. Gut 2001, 48, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirica, A.E.; Lai, G.H.; Zhang, Z. Biliary cancer growth factor pathways, cyclo-oxygenase-2 and potential therapeutic strategies. J. Gastroenterol. Hepatol. 2001, 16, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirica, A.E.; Lai, G.H.; Endo, K.; Zhang, Z.; Yoon, B.I. Cyclooxygenase-2 and ERBB-2 in cholangiocarcinoma: Potential therapeutic targets. Semin. Liver Dis. 2002, 22, 303–313. [Google Scholar] [CrossRef]
- Wappler, J.; Arts, M.; Röth, A.; Heeren, R.M.A.; Peter Neumann, U.; Olde Damink, S.W.; Soons, Z.; Cramer, T. Glutamine deprivation counteracts hypoxia-induced chemoresistance. Neoplasia 2020, 22, 22–32. [Google Scholar] [CrossRef]
- Satriano, L.; Lewinska, M.; Rodrigues, P.M.; Banales, J.M.; Andersen, J.B. Metabolic rearrangements in primary liver cancers: Cause and consequences. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 748–766. [Google Scholar] [CrossRef]
- Kaira, K.; Sunose, Y.; Ohshima, Y.; Ishioka, N.S.; Arakawa, K.; Ogawa, T.; Sunaga, N.; Shimizu, K.; Tominaga, H.; Oriuchi, N.; et al. Clinical significance of L-type amino acid transporter 1 expression as a prognostic marker and potential of new targeting therapy in biliary tract cancer. BMC Cancer 2013, 13, 482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janpipatkul, K.; Suksen, K.; Borwornpinyo, S.; Jearawiriyapaisarn, N.; Hongeng, S.; Piyachaturawat, P.; Chairoungdua, A. Downregulation of LAT1 expression suppresses cholangiocarcinoma cell invasion and migration. Cell Signal. 2014, 26, 1668–1679. [Google Scholar] [CrossRef] [PubMed]
- Yothaisong, S.; Dokduang, H.; Anzai, N.; Hayashi, K.; Namwat, N.; Yongvanit, P.; Sangkhamanon, S.; Jutabha, P.; Endou, H.; Loilome, W. Inhibition of l-type amino acid transporter 1 activity as a new therapeutic target for cholangiocarcinoma treatment. Tumour. Biol. 2017, 39, 1010428317694545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamnongkan, W.; Thanan, R.; Techasen, A.; Namwat, N.; Loilome, W.; Intarawichian, P.; Titapun, A.; Yongvanit, P. Upregulation of transferrin receptor-1 induces cholangiocarcinoma progression via induction of labile iron pool. Tumour Biol. 2017, 39, 1010428317717655. [Google Scholar] [CrossRef] [Green Version]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef] [Green Version]
- Saengboonmee, C.; Seubwai, W.; Pairojkul, C.; Wongkham, S. High glucose enhances progression of cholangiocarcinoma cells via STAT3 activation. Sci. Rep. 2016, 6, 18995. [Google Scholar] [CrossRef]
- Phoomak, C.; Vaeteewoottacharn, K.; Silsirivanit, A.; Saengboonmee, C.; Seubwai, W.; Sawanyawisuth, K.; Wongkham, C.; Wongkham, S. High glucose levels boost the aggressiveness of highly metastatic cholangiocarcinoma cells via O-GlcNAcylation. Sci. Rep. 2017, 7, 43842. [Google Scholar] [CrossRef] [Green Version]
- Thonsri, U.; Seubwai, W.; Waraasawapati, S.; Sawanyawisuth, K.; Vaeteewoottacharn, K.; Boonmars, T.; Cha’on, U. Overexpression of lactate dehydrogenase A in cholangiocarcinoma is correlated with poor prognosis. Histol. Histopathol. 2017, 32, 503–510. [Google Scholar] [CrossRef]
- Yu, Y.; Liao, M.; Liu, R.; Chen, J.; Feng, H.; Fu, Z. Overexpression of lactate dehydrogenase-A in human intrahepatic cholangiocarcinoma: Its implication for treatment. World J. Surg. Oncol. 2014, 12, 78. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.A.; Chao, Y.; Shiah, S.G.; Lin, W.W. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim. Biophys. Acta 2013, 1833, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Chaiteerakij, R.; Yang, J.D.; Harmsen, W.S.; Slettedahl, S.W.; Mettler, T.A.; Fredericksen, Z.S.; Kim, W.R.; Gores, G.J.; Roberts, R.O.; Olson, J.E.; et al. Risk factors for intrahepatic cholangiocarcinoma: Association between metformin use and reduced cancer risk. Hepatology 2013, 57, 648–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, S.; Feng, T.; Ke, Q.; Fan, N.; Li, L.; Li, Z.; Dong, C.; Wang, C.; Xu, F.; Li, Y.; et al. Metformin inhibits proliferation and enhances chemosensitivity of intrahepatic cholangiocarcinoma cell lines. Oncol. Rep. 2014, 31, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Ma, N.; Wang, D.; Li, F.; He, R.; Li, D.; Zhao, R.; Zhou, Q.; Wang, Y.; Zhang, F.; et al. Metformin inhibits tumor growth by regulating multiple miRNAs in human cholangiocarcinoma. Oncotarget 2015, 6, 3178–3194. [Google Scholar] [CrossRef] [Green Version]
- Anastasiou, D.; Yu, Y.; Israelsen, W.J.; Jiang, J.K.; Boxer, M.B.; Hong, B.S.; Tempel, W.; Dimov, S.; Shen, M.; Jha, A.; et al. Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat. Chem. Biol. 2012, 8, 839–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Zhang, X.; Roberts, R.O.; Roberts, L.R.; Chaiteerakij, R. Metformin does not improve survival of cholangiocarcinoma patients with diabetes. Hepatology 2016, 63, 667–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Li, J.N.; Mahmoud, M.A.; Han, W.F.; Ripple, M.; Pizer, E.S. Sterol regulatory element-binding protein-1 participates in the regulation of fatty acid synthase expression in colorectal neoplasia. Exp. Cell Res. 2000, 261, 159–165. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Vanderhoydonc, F.; Elgamal, A.A.; Eelen, M.; Vercaeren, I.; Joniau, S.; Van Poppel, H.; Baert, L.; Goossens, K.; Heyns, W.; et al. Selective activation of the fatty acid synthesis pathway in human prostate cancer. Int. J. Cancer 2000, 88, 176–179. [Google Scholar] [CrossRef]
- Yoon, S.; Lee, M.Y.; Park, S.W.; Moon, J.S.; Koh, Y.K.; Ahn, Y.H.; Park, B.W.; Kim, K.S. Up-regulation of acetyl-CoA carboxylase alpha and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem. 2007, 282, 26122–26131. [Google Scholar] [CrossRef] [Green Version]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat: The role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Pilo, G.M.; Li, X.; Cigliano, A.; Latte, G.; Che, L.; Joseph, C.; Mela, M.; Wang, C.; Jiang, L.; et al. Inactivation of fatty acid synthase impairs hepatocarcinogenesis driven by AKT in mice and humans. J. Hepatol. 2016, 64, 333–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, C.; Wang, C.; Mattu, S.; Destefanis, G.; Ladu, S.; Delogu, S.; Armbruster, J.; Fan, L.; Lee, S.A.; Jiang, L.; et al. AKT (v-akt murine thymoma viral oncogene homolog 1) and N-Ras (neuroblastoma ras viral oncogene homolog) coactivation in the mouse liver promotes rapid carcinogenesis by way of mTOR (mammalian target of rapamycin complex 1), FOXM1 (forkhead box M1)/SKP2, and c-Myc pathways. Hepatology 2012, 55, 833–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amemori, S.; Ootani, A.; Aoki, S.; Fujise, T.; Shimoda, R.; Kakimoto, T.; Shiraishi, R.; Sakata, Y.; Tsunada, S.; Iwakiri, R.; et al. Adipocytes and preadipocytes promote the proliferation of colon cancer cells in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G923–G929. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Buache, E.; Chenard, M.P.; Dali-Youcef, N.; Rio, M.C. Adipocyte is a non-trivial, dynamic partner of breast cancer cells. Int. J. Dev. Biol. 2011, 55, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, S.J.; Sacca, P.A.; Pistone-Creydt, M.; Coló, F.A.; Serra, M.F.; Santino, F.E.; Sasso, C.V.; Lopez-Fontana, C.M.; Carón, R.W.; Calvo, J.C.; et al. Human breast adipose tissue: Characterization of factors that change during tumor progression in human breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 26. [Google Scholar] [CrossRef] [Green Version]
- Ito, R.; Narita, S.; Huang, M.; Nara, T.; Numakura, K.; Takayama, K.; Tsuruta, H.; Maeno, A.; Saito, M.; Inoue, T.; et al. The impact of obesity and adiponectin signaling in patients with renal cell carcinoma: A potential mechanism for the “obesity paradox”. PLoS ONE 2017, 12, e0171615. [Google Scholar] [CrossRef]
- Zi, X.; Lusch, A.; Blair, C.A.; Okhunov, Z.; Yokoyama, N.N.; Liu, S.; Baker, M.; Huynh, V.; Landman, J. Effect of perineoplasm perinephric adipose tissues on migration of clear cell renal cell carcinoma cells: A potential role of WNT signaling. Oncotarget 2016, 7, 53277–53288. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Zhang, J.; Wang, L.; Lu, L.; Yuan, Q.; An, F.; Zhang, S.; Jiao, Y. Adipocytes promote cholangiocarcinoma metastasis through fatty acid binding protein 4. J. Exp. Clin. Cancer Res. 2017, 36, 183. [Google Scholar] [CrossRef] [Green Version]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar] [CrossRef]
- Han, C.; Leng, J.; Demetris, A.J.; Wu, T. Cyclooxygenase-2 promotes human cholangiocarcinoma growth: Evidence for cyclooxygenase-2-independent mechanism in celecoxib-mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004, 64, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Leng, J.; Han, C.; Demetris, A.J. The cyclooxygenase-2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol. Cancer Ther. 2004, 3, 299–307. [Google Scholar] [PubMed]
- Wu, T. Cyclooxygenase-2 and prostaglandin signaling in cholangiocarcinoma. Biochim. Biophys. Acta 2005, 1755, 135–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lai, G.H.; Sirica, A.E. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology 2004, 39, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Lai, G.H.; Zhang, Z.; Sirica, A.E. Celecoxib acts in a cyclooxygenase-2-independent manner and in synergy with emodin to suppress rat cholangiocarcinoma growth in vitro through a mechanism involving enhanced Akt inactivation and increased activation of caspases-9 and -3. Mol. Cancer Ther. 2003, 2, 265–271. [Google Scholar] [PubMed]
- Poitout, V.; Robertson, R.P. Glucolipotoxicity: Fuel excess and beta-cell dysfunction. Endocr. Rev. 2008, 29, 351–366. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef]
- Shida, D.; Takabe, K.; Kapitonov, D.; Milstien, S.; Spiegel, S. Targeting SphK1 as a new strategy against cancer. Curr. Drug Targets 2008, 9, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; El Buri, A.; Adams, D.R.; Pyne, S. Sphingosine 1-phosphate and cancer. Adv. Biol. Regul. 2018, 68, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.H.; Yen, C.C.; Cheng, C.T.; Wu, R.C.; Huang, S.C.; Yu, C.S.; Chung, Y.H.; Liu, C.Y.; Chang, P.M.; Chao, Y.; et al. Identification of SPHK1 as a therapeutic target and marker of poor prognosis in cholangiocarcinoma. Oncotarget 2015, 6, 23594–23608. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Peterson, Y.K.; Smith, R.A.; Smith, C.D. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS ONE 2012, 7, e44543. [Google Scholar] [CrossRef]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Chaiteerakij, R.; Moser, C.D.; Shaleh, H.; Boakye, J.; Chen, G.; Ndzengue, A.; Li, Y.; Zhou, Y.; Huang, S.; et al. Antitumor effect of the novel sphingosine kinase 2 inhibitor ABC294640 is enhanced by inhibition of autophagy and by sorafenib in human cholangiocarcinoma cells. Oncotarget 2016, 7, 20080–20092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A Phase I Study of ABC294640, a First-in-Class Sphingosine Kinase-2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzoni, E.; Pusceddu, S.; Buzzoni, R.; Garanzini, E.; Damato, A.; Biondani, P.; Testa, I.; Grassi, P.; Bajetta, E.; de Braud, F.; et al. Safety profile and treatment response of everolimus in different solid tumors: An observational study. Future Oncol. 2014, 10, 1611–1617. [Google Scholar] [CrossRef]
- Lau, D.K.; Tay, R.Y.; Yeung, Y.H.; Chionh, F.; Mooi, J.; Murone, C.; Skrinos, E.; Price, T.J.; Mariadason, J.M.; Tebbutt, N.C. Phase II study of everolimus (RAD001) monotherapy as first-line treatment in advanced biliary tract cancer with biomarker exploration: The RADiChol Study. Br. J. Cancer 2018, 118, 966–971. [Google Scholar] [CrossRef] [Green Version]
- Buzzoni, R.; Pusceddu, S.; Bajetta, E.; De Braud, F.; Platania, M.; Iannacone, C.; Cantore, M.; Mambrini, A.; Bertolini, A.; Alabiso, O.; et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: A phase II ITMO study. Ann. Oncol. 2014, 25, 1597–1603. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Kang, W.K.; Lim, H.Y. Prospective phase II trial of everolimus in PIK3CA amplification/mutation and/or PTEN loss patients with advanced solid tumors refractory to standard therapy. BMC Cancer 2017, 17, 211. [Google Scholar] [CrossRef] [Green Version]
- Rizell, M.; Andersson, M.; Cahlin, C.; Hafström, L.; Olausson, M.; Lindnér, P. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int. J. Clin. Oncol. 2008, 13, 66–70. [Google Scholar] [CrossRef]
- Jung, K.S.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, S.T. Pilot study of sirolimus in patients with PIK3CA mutant/amplified refractory solid cancer. Mol. Clin. Oncol. 2017, 7, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Costello, B.A.; Borad, M.J.; Qi, Y.; Kim, G.P.; Northfelt, D.W.; Erlichman, C.; Alberts, S.R. Phase I trial of everolimus, gemcitabine and cisplatin in patients with solid tumors. Investig. New Drugs 2014, 32, 710–716. [Google Scholar] [CrossRef]
- Leelawat, K.; Leelawat, S.; Narong, S.; Hongeng, S. Roles of the MEK1/2 and AKT pathways in CXCL12/CXCR4 induced cholangiocarcinoma cell invasion. World J. Gastroenterol. 2007, 13, 1561–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Jin, M.H.; Nam, A.R.; Park, J.E.; Bang, J.H.; Oh, D.Y.; Bang, Y.J. Anti-tumor effects of NVP-BKM120 alone or in combination with MEK162 in biliary tract cancer. Cancer Lett. 2017, 411, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.V.; Boulter, L.; Dwyer, B.J.; Kendall, T.J.; Man, T.Y.; Minnis-Lyons, S.E.; Lu, W.Y.; Robson, A.J.; Gonzalez, S.F.; Raven, A.; et al. Notch3 drives development and progression of cholangiocarcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, 12250–12255. [Google Scholar] [CrossRef] [Green Version]
- McRee, A.J.; Sanoff, H.K.; Carlson, C.; Ivanova, A.; O’Neil, B.H. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Investig. New Drugs 2015, 33, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.D.; Alberts, S.R.; Peña, C.; Genvresse, I.; Ajavon-Hartmann, A.; Xia, C.; Kelly, A.; Grilley-Olson, J.E. Phase I dose-escalation study of copanlisib in combination with gemcitabine or cisplatin plus gemcitabine in patients with advanced cancer. Br. J. Cancer 2018, 118, 462–470. [Google Scholar] [CrossRef] [Green Version]
- Ahn, D.H.; Li, J.; Wei, L.; Doyle, A.; Marshall, J.L.; Schaaf, L.J.; Phelps, M.A.; Villalona-Calero, M.A.; Bekaii-Saab, T. Results of an abbreviated phase-II study with the Akt Inhibitor MK-2206 in Patients with Advanced Biliary Cancer. Sci. Rep. 2015, 5, 12122. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef] [Green Version]
- Mertens, J.C.; Rizvi, S.; Gores, G.J. Targeting cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1454–1460. [Google Scholar] [CrossRef]
- Buranrat, B.; Senggunprai, L.; Prawan, A.; Kukongviriyapan, V. Simvastatin and atorvastatin as inhibitors of proliferation and inducers of apoptosis in human cholangiocarcinoma cells. Life Sci. 2016, 153, 41–49. [Google Scholar] [CrossRef]
- Roeksomtawin, S.; Navasumrit, P.; Waraprasit, S.; Parnlob, V.; Sricharunrat, T.; Bhudhisawasdi, V.; Savaraj, N.; Ruchirawat, M. Decreased argininosuccinate synthetase expression in Thai patients with cholangiocarcinoma and the effects of ADI-PEG20 treatment in CCA cell lines. Oncol. Lett. 2018, 16, 1529–1538. [Google Scholar] [CrossRef] [Green Version]
- Abou-Alfa, G.K.; Qin, S.; Ryoo, B.Y.; Lu, S.N.; Yen, C.J.; Feng, Y.H.; Lim, H.Y.; Izzo, F.; Colombo, M.; Sarker, D.; et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2018, 29, 1402–1408. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cui, H. Targeting Glutamine Induces Apoptosis: A Cancer Therapy Approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Dong, H.; Robertson, K.; Liu, C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am. J. Pathol. 2011, 178, 652–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Kubo, S.; Tamori, A.; Itami, S.; Kawamura, E.; Iwaisako, K.; Ikeda, K.; Kawada, N.; Ochiya, T.; Taguchi, Y.H. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 16294. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxidative Med. Cell Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef] [Green Version]
- Recalcati, S.; Gammella, E.; Buratti, P.; Cairo, G. Molecular regulation of cellular iron balance. IUBMB Life 2017, 69, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Fonseca-Nunes, A.; Jakszyn, P.; Agudo, A. Iron and cancer risk—A systematic review and meta-analysis of the epidemiological evidence. Cancer Epidemiol. Biomark. Prev. 2014, 23, 12–31. [Google Scholar] [CrossRef] [Green Version]
- Niederau, C.; Fischer, R.; Sonnenberg, A.; Stremmel, W.; Trampisch, H.J.; Strohmeyer, G. Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis. N. Engl. J. Med. 1985, 313, 1256–1262. [Google Scholar] [CrossRef]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Ocana, A.; Vera-Badillo, F.; Al-Mubarak, M.; Templeton, A.J.; Corrales-Sanchez, V.; Diez-Gonzalez, L.; Cuenca-Lopez, M.D.; Seruga, B.; Pandiella, A.; Amir, E. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: Systematic review and meta-analysis. PLoS ONE 2014, 9, e95219. [Google Scholar] [CrossRef] [PubMed]
- Pignochino, Y.; Sarotto, I.; Peraldo-Neia, C.; Penachioni, J.Y.; Cavalloni, G.; Migliardi, G.; Casorzo, L.; Chiorino, G.; Risio, M.; Bardelli, A.; et al. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer 2010, 10, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treekitkarnmongkol, W.; Suthiphongchai, T. High expression of ErbB2 contributes to cholangiocarcinoma cell invasion and proliferation through AKT/p70S6K. World J. Gastroenterol. 2010, 16, 4047–4054. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Lorenz, J.; Mössner, J.; Wiedmann, M. Treatment of biliary tract cancer with NVP-AEW541: Mechanisms of action and resistance. World J. Gastroenterol. 2010, 16, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, H.; Adachi, Y.; Yamamoto, H.; Taniguchi, H.; Nosho, K.; Suzuki, H.; Arimura, Y.; Imai, K.; Carbone, D.P.; Shinomura, Y. Insulin-like growth factor receptor expression is associated with aggressive phenotypes and has therapeutic activity in biliary tract cancers. Cancer Sci. 2012, 103, 252–261. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kunnimalaiyaan, S.; Kunnimalaiyaan, M.; Gamblin, T.C. Inhibition of the AKT pathway in cholangiocarcinoma by MK2206 reduces cellular viability via induction of apoptosis. Cancer Cell Int. 2015, 15, 13. [Google Scholar] [CrossRef] [Green Version]
- Frampton, G.; Invernizzi, P.; Bernuzzi, F.; Pae, H.Y.; Quinn, M.; Horvat, D.; Galindo, C.; Huang, L.; McMillin, M.; Cooper, B.; et al. Interleukin-6-driven progranulin expression increases cholangiocarcinoma growth by an Akt-dependent mechanism. Gut 2012, 61, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Moolthiya, P.; Tohtong, R.; Keeratichamroen, S.; Leelawat, K. Role of mTOR inhibitor in cholangiocarcinoma cell progression. Oncol. Lett. 2014, 7, 854–860. [Google Scholar] [CrossRef]
- Zhang, M.; Pan, Y.; Tang, D.; Dorfman, R.G.; Xu, L.; Zhou, Q.; Zhou, L.; Wang, Y.; Li, Y.; Yin, Y.; et al. Low levels of pyruvate induced by a positive feedback loop protects cholangiocarcinoma cells from apoptosis. Cell Commun. Signal. 2019, 17, 23–37. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Bui, T.; Thompson, C.B. Cancer’s sweet tooth. Cancer Cell 2006, 9, 419–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. Energy deregulation: Licensing tumors to grow. Science 2006, 312, 1158–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zhu, L.; Sun, D.; Gan, F.; Gao, S.; Yin, Y.; Chen, L. Natural products as modulator of autophagy with potential clinical prospects. Apoptosis 2017, 22, 325–356. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef]
- Preidis, G.A.; Kim, K.H.; Moore, D.D. Nutrient-sensing nuclear receptors PPARα and FXR control liver energy balance. J. Clin. Investig. 2017, 127, 1193–1201. [Google Scholar] [CrossRef]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Zhu, C.; Fuchs, C.D.; Halilbasic, E.; Trauner, M. Bile acids in regulation of inflammation and immunity: Friend or foe? Clin. Exp. Rheumatol. 2016, 34, 25–31. [Google Scholar]
- Vaquero, J.; Briz, O.; Herraez, E.; Muntané, J.; Marin, J.J. Activation of the nuclear receptor FXR enhances hepatocyte chemoprotection and liver tumor chemoresistance against genotoxic compounds. Biochim. Biophys. Acta 2013, 1833, 2212–2219. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.D.; Chen, W.D.; Wang, X.; Lou, G.; Liu, N.; Lin, M.; Forman, B.M.; Huang, W. Promotion of liver regeneration/repair by farnesoid X receptor in both liver and intestine in mice. Hepatology 2012, 56, 2336–2343. [Google Scholar] [CrossRef] [Green Version]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Bleeker, F.E.; Atai, N.A.; Lamba, S.; Jonker, A.; Rijkeboer, D.; Bosch, K.S.; Tigchelaar, W.; Troost, D.; Vandertop, W.P.; Bardelli, A.; et al. The prognostic IDH1(R132) mutation is associated with reduced NADP+-dependent IDH activity in glioblastoma. Acta Neuropathol. 2010, 119, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Janin, M.; Mylonas, E.; Saada, V.; Micol, J.B.; Renneville, A.; Quivoron, C.; Koscielny, S.; Scourzic, L.; Forget, S.; Pautas, C.; et al. Serum 2-hydroxyglutarate production in IDH1- and IDH2-mutated de novo acute myeloid leukemia: A study by the Acute Leukemia French Association group. J. Clin. Oncol. 2014, 32, 297–305. [Google Scholar] [CrossRef] [Green Version]
- Poinsignon, V.; Mercier, L.; Nakabayashi, K.; David, M.D.; Lalli, A.; Penard-Lacronique, V.; Quivoron, C.; Saada, V.; De Botton, S.; Broutin, S.; et al. Quantitation of isocitrate dehydrogenase (IDH)-induced D and L enantiomers of 2-hydroxyglutaric acid in biological fluids by a fully validated liquid tandem mass spectrometry method, suitable for clinical applications. J. Chromatogr. B Anal. Technol. BioMed Life Sci. 2016, 1022, 290–297. [Google Scholar] [CrossRef]
- Delahousse, J.; Verlingue, L.; Broutin, S.; Legoupil, C.; Touat, M.; Doucet, L.; Ammari, S.; Lacroix, L.; Ducreux, M.; Scoazec, J.Y.; et al. Circulating oncometabolite D-2-hydroxyglutarate enantiomer is a surrogate marker of isocitrate dehydrogenase-mutated intrahepatic cholangiocarcinomas. Eur. J. Cancer 2018, 90, 83–91. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipp, B.R.; Voss, J.S.; Kerr, S.E.; Barr Fritcher, E.G.; Graham, R.P.; Zhang, L.; Highsmith, W.E.; Zhang, J.; Roberts, L.R.; Gores, G.J.; et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum. Pathol. 2012, 43, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Govindan, A.; Sheth, R.A.; Nardi, V.; Blaszkowsky, L.S.; Faris, J.E.; Clark, J.W.; Ryan, D.P.; Kwak, E.L.; Allen, J.N.; et al. Prognosis and Clinicopathologic Features of Patients With Advanced Stage Isocitrate Dehydrogenase (IDH) Mutant and IDH Wild-Type Intrahepatic Cholangiocarcinoma. Oncologist 2015, 20, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madala, H.R.; Punganuru, S.R.; Arutla, V.; Misra, S.; Thomas, T.J.; Srivenugopal, K.S. Beyond Brooding on Oncometabolic Havoc in IDH-Mutant Gliomas and AML: Current and Future Therapeutic Strategies. Cancers 2018, 10, 49. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Xiao, M.T.; Liu, L.X.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Clark, O.; Yen, K.; Mellinghoff, I.K. Molecular Pathways: Isocitrate Dehydrogenase Mutations in Cancer. Clin. Cancer Res. 2016, 22, 1837–1842. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Parker, S.J.; Metallo, C.M. Metabolic consequences of oncogenic IDH mutations. Pharmacol. Ther. 2015, 152, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Voss, J.S.; Holtegaard, L.M.; Kerr, S.E.; Fritcher, E.G.; Roberts, L.R.; Gores, G.J.; Zhang, J.; Highsmith, W.E.; Halling, K.C.; Kipp, B.R. Molecular profiling of cholangiocarcinoma shows potential for targeted therapy treatment decisions. Hum. Pathol. 2013, 44, 1216–1222. [Google Scholar] [CrossRef]
- Borger, D.R.; Goyal, L.; Yau, T.; Poon, R.T.; Ancukiewicz, M.; Deshpande, V.; Christiani, D.C.; Liebman, H.M.; Yang, H.; Kim, H.; et al. Circulating oncometabolite 2-hydroxyglutarate is a potential surrogate biomarker in patients with isocitrate dehydrogenase-mutant intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2014, 20, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Ong, C.K.; Subimerb, C.; Pairojkul, C.; Wongkham, S.; Cutcutache, I.; Yu, W.; McPherson, J.R.; Allen, G.E.; Ng, C.C.; Wong, B.H.; et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat. Genet. 2012, 44, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Drill, E.; Vakiani, E.; Pak, L.M.; Boerner, T.; Askan, G.; Schvartzman, J.M.; Simpson, A.L.; Jarnagin, W.R.; Sigel, C.S. Distinct histomorphological features are associated with IDH1 mutation in intrahepatic cholangiocarcinoma. Hum. Pathol. 2019, 91, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Nepal, C.; O’Rourke, C.J.; Oliveira, D.V.N.P.; Taranta, A.; Shema, S.; Gautam, P.; Calderaro, J.; Barbour, A.; Raggi, C.; Wennerberg, K.; et al. Genomic perturbations reveal distinct regulatory networks in intrahepatic cholangiocarcinoma. Hepatology 2018, 68, 949–963. [Google Scholar] [CrossRef] [Green Version]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef] [Green Version]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [Green Version]
- Isomoto, H.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Frank, D.A.; Gores, G.J. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 2005, 42, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Vicentini, C.; Ruzzenente, A.; Brunelli, M.; Conci, S.; Fassan, M.; Mafficini, A.; Rusev, B.; Corbo, V.; Capelli, P.; et al. Genetic alterations analysis in prognostic stratified groups identified TP53 and ARID1A as poor clinical performance markers in intrahepatic cholangiocarcinoma. Sci. Rep. 2018, 8, 7119. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Ochiai, T.; Nomura, F.; Group, J.A.R. Titration of serum p53 antibodies in 1,085 patients with various types of malignant tumors: A multiinstitutional analysis by the Japan p53 Antibody Research Group. Cancer 2003, 97, 682–689. [Google Scholar] [CrossRef]
- Okada, R.; Shimada, H.; Otsuka, Y.; Tsuchiya, M.; Ishii, J.; Katagiri, T.; Maeda, T.; Kubota, Y.; Nemoto, T.; Kaneko, H. Serum p53 antibody as a potential tumor marker in extrahepatic cholangiocarcinoma. Surg. Today 2017, 47, 1492–1499. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Alexander, W.B.; Guo, B.; Kato, Y.; Patra, K.; O’Dell, M.R.; McCall, M.N.; Whitney-Miller, C.L.; Bardeesy, N.; Hezel, A.F. Kras and Tp53 Mutations Cause Cholangiocyte- and Hepatocyte-Derived Cholangiocarcinoma. Cancer Res. 2018, 78, 4445–4451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.; Wu, J. Hypoxia inducible factor in hepatocellular carcinoma: A therapeutic target. World J. Gastroenterol. 2015, 21, 12171–12178. [Google Scholar] [CrossRef] [PubMed]
- Nagao, A.; Kobayashi, M.; Koyasu, S.; Chow, C.C.T.; Harada, H. HIF-1-Dependent Reprogramming of Glucose Metabolic Pathway of Cancer Cells and Its Therapeutic Significance. Int. J. Mol. Sci. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Morine, Y.; Shimada, M.; Utsunomiya, T.; Imura, S.; Ikemoto, T.; Mori, H.; Hanaoka, J.; Kanamoto, M.; Iwahashi, S.; Miyake, H. Hypoxia inducible factor expression in intrahepatic cholangiocarcinoma. Hepatogastroenterology 2011, 58, 1439–1444. [Google Scholar] [CrossRef]
- Thongchot, S.; Yongvanit, P.; Loilome, W.; Seubwai, W.; Phunicom, K.; Tassaneeyakul, W.; Pairojkul, C.; Promkotra, W.; Techasen, A.; Namwat, N. High expression of HIF-1α, BNIP3 and PI3KC3: Hypoxia-induced autophagy predicts cholangiocarcinoma survival and metastasis. Asian Pac. J. Cancer Prev. 2014, 15, 5873–5878. [Google Scholar] [CrossRef] [Green Version]
- Vanichapol, T.; Leelawat, K.; Hongeng, S. Hypoxia enhances cholangiocarcinoma invasion through activation of hepatocyte growth factor receptor and the extracellular signal-regulated kinase signaling pathway. Mol. Med. Rep. 2015, 12, 3265–3272. [Google Scholar] [CrossRef] [Green Version]
- Thongchot, S.; Loilome, W.; Yongvanit, P.; Dokduang, H.; Thanan, R.; Techasen, A.; Namwat, N. Chloroquine exerts anti-metastatic activities under hypoxic conditions in cholangiocarcinoma cells. Asian Pac. J. Cancer Prev. 2015, 16, 2031–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, R.; Li, Q.; Ge, X.; Nie, J.; Wang, F.; Xu, B.; Pan, Z.; Miao, L. NK4 suppresses cholangiocarcinoma angiogenesis and invasion through targeting HIF-1α pathway. Int. J. Clin. Exp. Med. 2017, 10, 2345–2352. [Google Scholar]
- Thomas, J.D.; Zhang, Y.J.; Wei, Y.H.; Cho, J.H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F.S. Rab1A Is an mTORC1 Activator and a Colorectal Oncogene. Cancer Cell 2016, 30, 181–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, P.; Kang, Y.; Luo, J. Hypoxia-mediated miR-212-3p downregulation enhances progression of intrahepatic cholangiocarcinoma through upregulation of Rab1a. Cancer Biol. Ther. 2018, 19, 984–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef]
- Rankin, E.B.; Rha, J.; Selak, M.A.; Unger, T.L.; Keith, B.; Liu, Q.; Haase, V.H. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell Biol. 2009, 29, 4527–4538. [Google Scholar] [CrossRef] [Green Version]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [Green Version]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V.; Kim, J.W. Convergence of Cancer Metabolism and Immunity: An Overview. Biomol. Ther. 2018, 26, 4–9. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Tavakoli, S.; Zamora, D.; Ullevig, S.; Asmis, R. Bioenergetic profiles diverge during macrophage polarization: Implications for the interpretation of 18F-FDG PET imaging of atherosclerosis. J. Nucl. Med. 2013, 54, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehla, K.; Singh, P.K. Metabolic Regulation of Macrophage Polarization in Cancer. Trends Cancer 2019, 5, 822–834. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; In’t Veld, P.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Z.; Shi, Q.; Zhang, W.; Shu, Y.; Yang, N.; Chen, B.; Wang, Q.; Zhao, X.; Chen, J.; Cheng, N.; et al. Caspase-1 cleaves PPARγ for potentiating the pro-tumor action of TAMs. Nat. Commun. 2017, 8, 766. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.C.; Bihuniak, J.D.; Macintyre, A.N.; Staron, M.; Liu, X.; Amezquita, R.; Tsui, Y.C.; Cui, G.; Micevic, G.; Perales, J.C.; et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 2015, 162, 1217–1228. [Google Scholar] [CrossRef] [Green Version]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [Green Version]


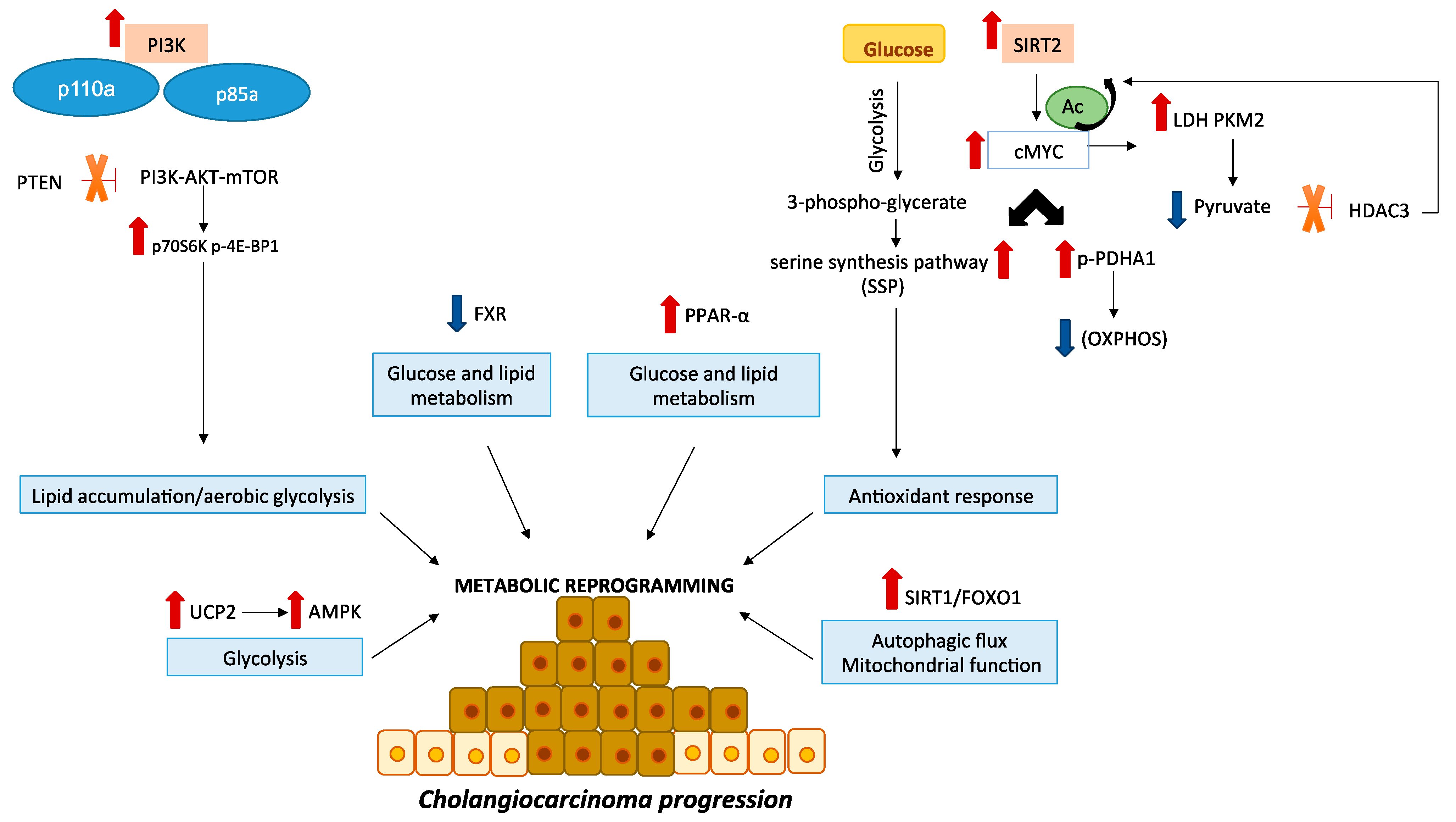{kind=link}
| Metabolic Pathways | Metabolic Target | Effects on Cholangiocarcinoma (CCA) | Reference |
|---|---|---|---|
| Glucose metabolism | GLUT-1 upregulation | Correlation with in vitro CCA cell invasion. Correlation with larger tumor size, poor differentiation, metastasis and poor prognosis of CCA. | [33,34,35] |
| PPP upstimulation | Sustainment of both the antioxidant capacity of CCA cells and cisplatin resistance. | [36] | |
| PDK overexpression | High serum PDK3 levels correlate with short survival of patients with CCA. PDK1 expression promotes glycolysis and CCA cell proliferation. | [37] | |
| SIRT3 effects mediated by HIF1α/ PDK1/PDHA1 pathway | Decrease of SIRT3 expression induced the glycolytic flux through the hypoxia inducible factor α (HIF1α)/PDK1/ PDHA1 axis, promoting CCA progression. | [38] | |
| Deregulation of PI3K-AKT-mTOR signaling | Protein overexpression and activation of PI3K have been associated with tumor progression, differentiation, nodal involvement and reduced OS. Upregulation of activated form of AKT have been reported in neoplastic cells compared to the surrounding normal tissue; Increased mTOR gene copy number and elevated phospho-mTOR levels have been described in biliary cancer specimens in comparison to the adjacent normal or dysplastic epithelium. PTEN loss has been related to poor tumor differentiation, nodal involvement and shorter survival in CCA. | [39,40,41,42] | |
| SIRT2 overexpression | SIRT2 and its downstream target cMYC, were overexpressed both in human CCA cell lines and in 48 CCA samples compared to adjacent tissues, The SIRT2/cMYC pathway is able to reprogram CCA metabolism through inhibition of OXPHOS and activation of SSP to counteract ROS production, thus protecting CCA cells from oxidative stress-induced apoptosis. | [43] | |
| UCP2 overexpression | Up-regulation of UCP2 sustains the EMT and cell invasion of CCA cells. | [44] | |
| FXR downregulation | Initiation and progression of CCA, and the downregulation of FXR expression could promote cancer development, modulating the energy metabolism of CCA cells. | [45] | |
| PPAR-α upregulation | Tumor occurrence and progression in CCA patients with. | [46,47] | |
| IDH1 and IDH2 mutations | Increase of glucose uptake and glucose metabolism as well as upregulation of some metabolites in TCA cycle. Upregulation of PFKP. | [48] | |
| Mitochondrial metabolism | PGC1α upregulation | Promotion of CCA metastasis both in vitro and in vivo. | [49] |
| Sirt1/FOXO1 stimulation | Involvement in autophagy and mitochondrial dysfunction in CCA cells. | [50] | |
| Lipid metabolism | FASN down-regulation | In human and mouse iCCA tissues FASN expression was down- regulated respect to non-tumor adjacent tissues. | [51] |
| FA transporter (SLC27A1) overexpression | SCL27A1 silencing in CCA cell lines led to a decrease of cells growth. | [52] | |
| FA transporter (FABP5) over expression | Correlation with worse prognosis in eCCA. | [53] | |
| COX-2 upregulation | Promotion of CCA growth and invasion. | [54,55,56,57,58] | |
| Protein metabolism | Glutamine depletion | Strong depletion: induction a cessation of proliferation or cell death (in vitro addiction to glutamine). Gradual reduction: eCCA cells (EGI-1 and TFK-1) could proliferate under long-term glutamine withdrawal overcoming their addiction to glutamine. | [59] |
| ASS deficiency | Reduction of arginine in the surrounding tumor cells could lead to a reduction in CCA cell proliferation. | [60] | |
| LAT1 overexpression | Activation of mTOR pathway thus affecting cell proliferation and viability. | [61,62,63] | |
| Iron Metabolism | TfR1 high expression | Contribution to CCA progression and poorer clinical outcomes. | [64] |
| Ferritin high expression | Negative prognostic index for CCA patients. | [65] | |
| Fpn reduced mRNA levels | Reduction of iron release, in tumor cells of CCA patients sample compared to matched surrounding liver, suggests that elevated iron content is a negative prognostic factor. | [65] |
| Target | Glucose Metabolism | Reference |
|---|---|---|
| mTOR | Everolimus | [104,105,106,107] |
| Sirolimus | [108,109] | |
| Everolimus + CisGem | [110] | |
| PI3K | LY294002 | [111] |
| Buparlisib | [112] | |
| PI-103 | [113] | |
| Buparlisib + mFOLFOX6 | [114] | |
| Copanlisib ± GemorCisGem | [115] | |
| AKT | MK2206 | [116] |
| IDH1 | AG-120 | [117] |
| AG-221 | [118] | |
| AGI-5198 | [48] | |
| Lipid Metabolism | ||
| SPHK2 | ABC294640 | [100,101,102,103] |
| HMGCR | Simvastatin, Atorvastatin | [119] |
| Protein Metabolism | ||
| LAT1 | 2-aminobicyclo-(2,2,1)-heptane-2-carboxylic acid | [62] |
| JPH203 | [63] | |
| ASS | ADI-PEG 20 | [120,121] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastore, M.; Lori, G.; Gentilini, A.; Taddei, M.L.; Di Maira, G.; Campani, C.; Recalcati, S.; Invernizzi, P.; Marra, F.; Raggi, C. Multifaceted Aspects of Metabolic Plasticity in Human Cholangiocarcinoma: An Overview of Current Perspectives. Cells 2020, 9, 596. https://doi.org/10.3390/cells9030596
Pastore M, Lori G, Gentilini A, Taddei ML, Di Maira G, Campani C, Recalcati S, Invernizzi P, Marra F, Raggi C. Multifaceted Aspects of Metabolic Plasticity in Human Cholangiocarcinoma: An Overview of Current Perspectives. Cells. 2020; 9(3):596. https://doi.org/10.3390/cells9030596
Chicago/Turabian StylePastore, Mirella, Giulia Lori, Alessandra Gentilini, Maria Letizia Taddei, Giovanni Di Maira, Claudia Campani, Stefania Recalcati, Pietro Invernizzi, Fabio Marra, and Chiara Raggi. 2020. "Multifaceted Aspects of Metabolic Plasticity in Human Cholangiocarcinoma: An Overview of Current Perspectives" Cells 9, no. 3: 596. https://doi.org/10.3390/cells9030596