
Phosphorylations and Acetylations of Cytochrome c Control Mitochondrial Respiration, Mitochondrial Membrane Potential, Energy, ROS, and Apoptosis
 ,
,  , , and
, , and
Abstract
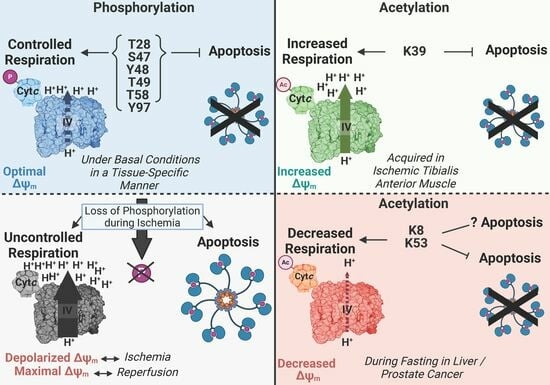
:
1. Introduction
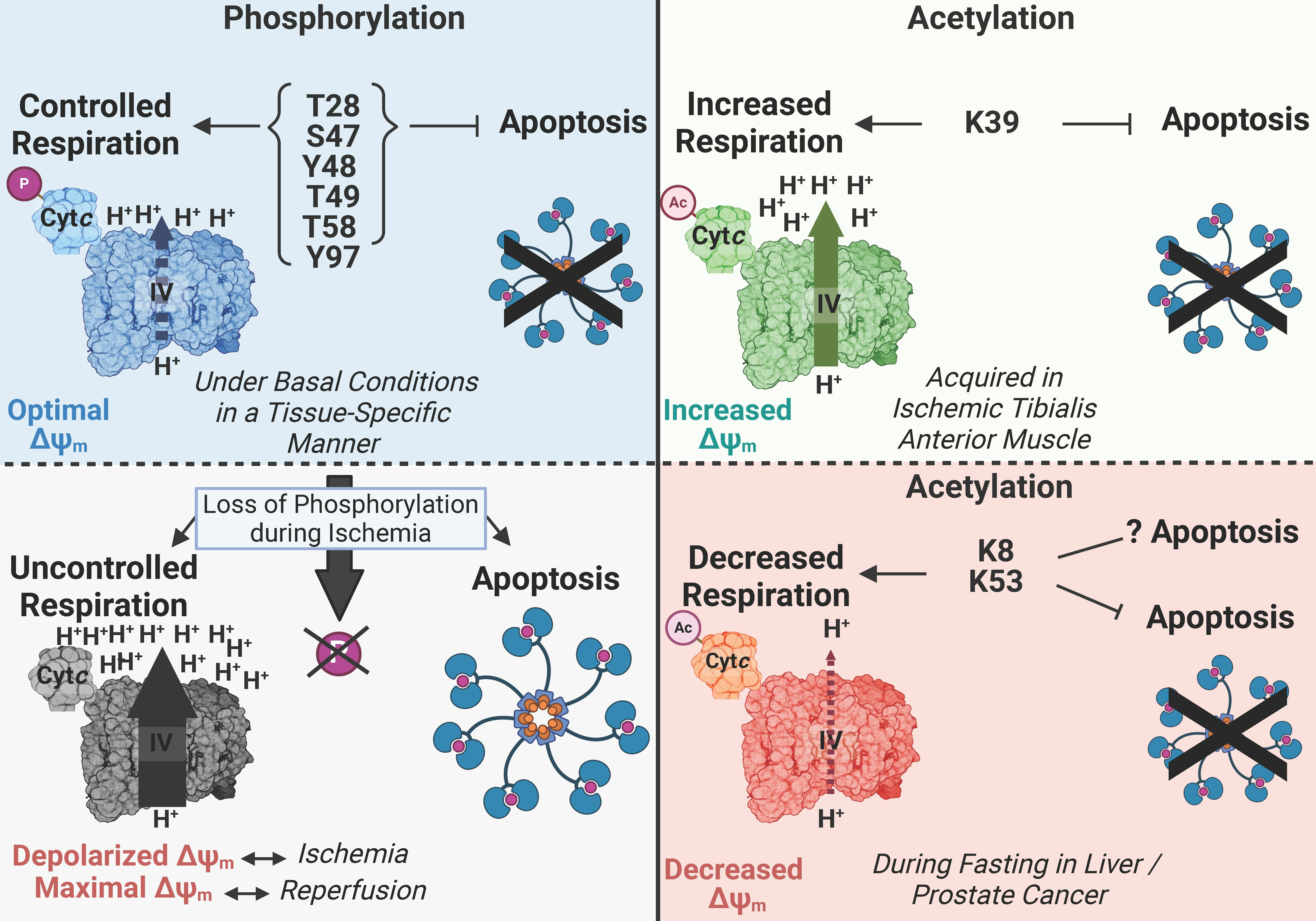
2. Various Functions of Cytc
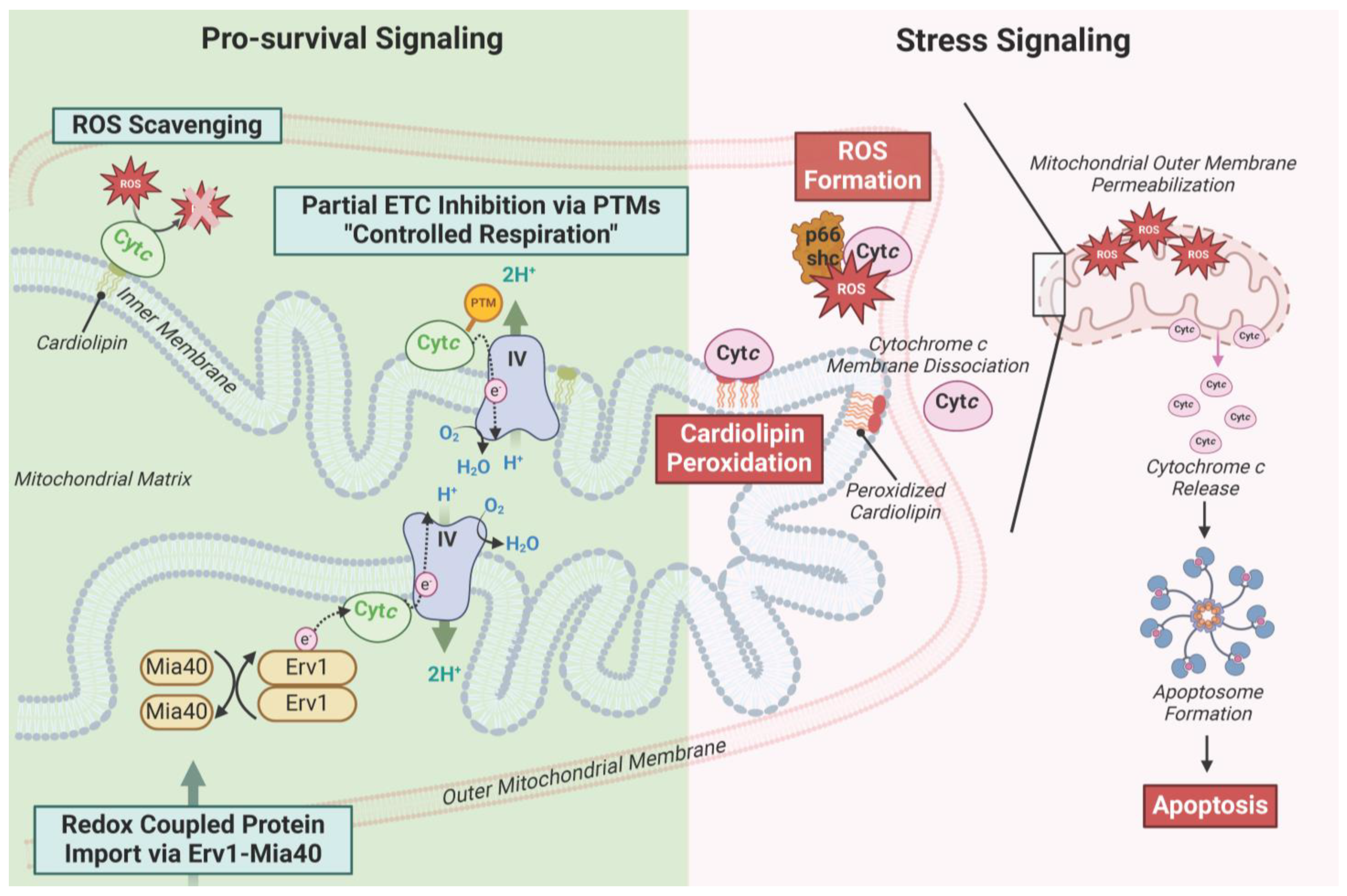
3. Characterized Phosphorylation Sites of Cytc
4. Most Phosphorylations of Cytc Are Protective through Partial Inhibition of ETC Flux and Decreased Apoptotic Activity
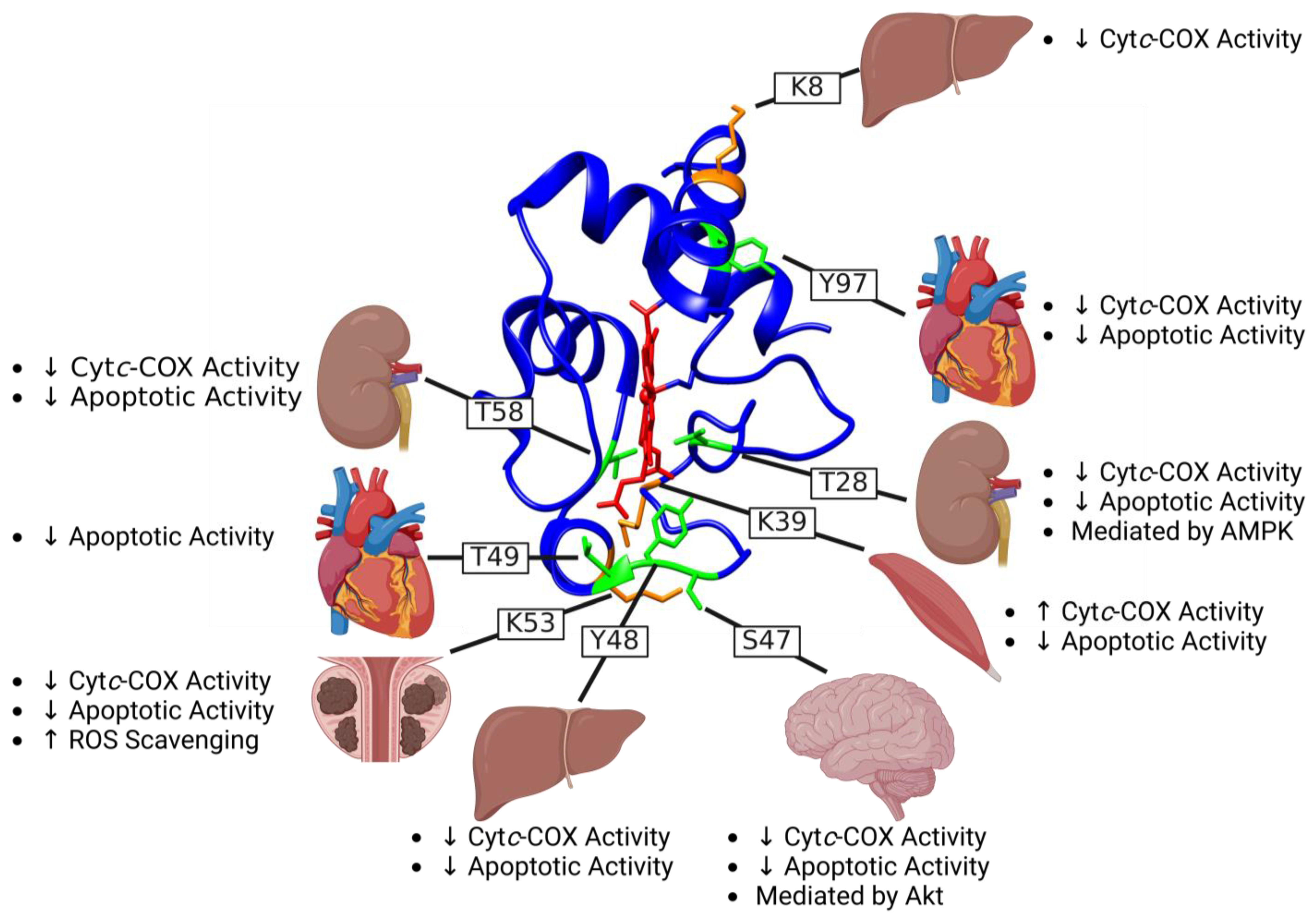
4.1. Phosphorylation of Threonine 28 (T28)
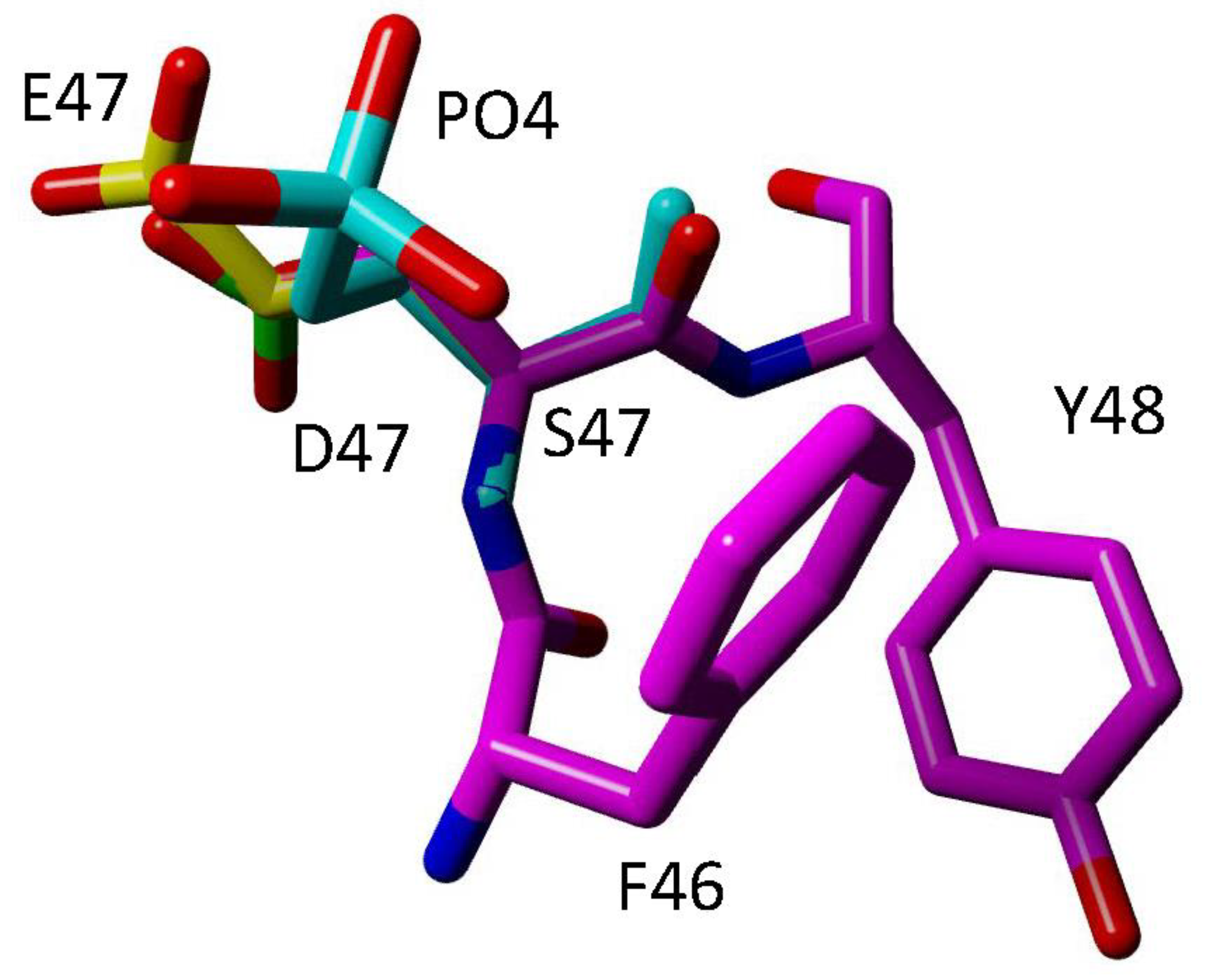
4.2. Phosphorylation of Serine 47 (S47)
4.3. Phosphorylation of Tyrosine 48 (Y48)
4.4. Phosphorylation of Threonine 49 (T49)
4.5. Phosphorylation of Threonine 58 (T58)
4.6. Phosphorylation of Tyrosine 97 (Y97)
5. Characterized Acetylation Sites of Cytc

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | Tissue of Origin | Experimental Models | Findings |
|---|---|---|---|
| Lysine 8 | Fasted Mouse Liver | Recombinant acetylmimetic K8Q Cytc | Decreased Cytc-COX activity, decreased KD to cytochrome c1 [98] |
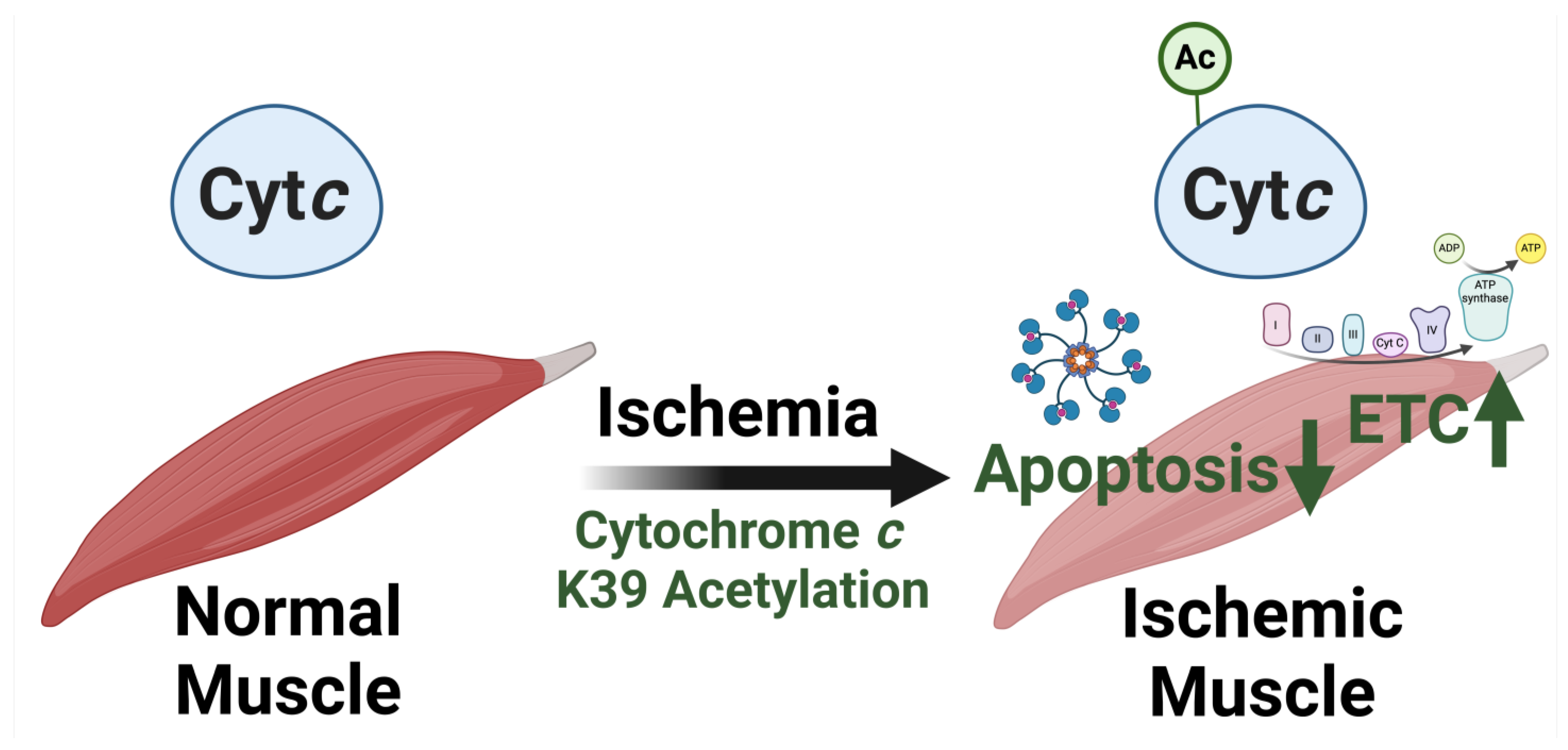
| Lysine 39 | Ischemic Porcine Tibialis Anterior Muscle | In vivo acetylated Cytc purified from ischemic porcine muscle | Increased Cytc-COX Vmax, decreased caspase-3 activity [93] |
| Recombinant acetylmimetic K39Q Cytc | Increased Cytc-COX Vmax, decreased caspase-3 activity, increased rate of oxidation, decreased rate of reduction, decreased cardiolipin peroxidase activity [93] | ||
| Cytc double knockout mouse lung fibroblasts expressing K39Q Cytc | Increased respiration, increased ΔΨm, increased mitochondrial ROS production, increased ATP levels, decreased cell death, reduced responsiveness to oxygen-glucose deprivation followed by reoxygenation [93] | ||
| Lysine 53 | Human Prostate Cancer | Recombinant acetylmimetic K53Q Cytc | Decreased Cytc-COX Vmax, decreased caspase-3 activity, increased rate of oxidation, increased rate of reduction, increased heme degradation, decreased cardiolipin peroxidase activity [92,98] |
5.1. Acetylation of Lysine 8 (K8)
5.2. Acetylation of Lysine 39 (K39)
5.3. Acetylation of Lysine 53 (K53)
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Apaf-1 | apoptosis protease-activating factor-1 |
| Cytc | cytochrome c |
| COX | cytochrome c oxidase (complex IV) |
| ETC | electron transport chain |
| IMS | intermembrane space |
| ΔΨm | mitochondrial membrane potential |
| K8 | lysine 8 (of Cytc) |
| K39 | lysine 39 (of Cytc) |
| K53 | lysine 53 (of Cytc) |
| OGD/R | oxygen-glucose deprivation followed by reoxygenation |
| OxPhos | oxidative phosphorylation |
| pCMF | p-carboxymethyl-L-phenylalanine |
| ROS | reactive oxygen species |
| S47 | serine 47 (of Cytc) |
| TMPD | tetramethyl-p-phenylenediamine |
| T28 | threonine 28 (of Cytc) |
| T49 | threonine 49 (of Cytc) |
| T58 | threonine 58 (of Cytc) |
| WT | wild-type (Cytc) |
| Y48 | tyrosine 48 (of Cytc) |
| Y97 | tyrosine 97 (of Cytc) |
References
- Rich, P. Chemiosmotic coupling: The cost of living. Nature 2003, 421, 583. [Google Scholar] [CrossRef]
- Moe, A.; Di Trani, J.; Rubinstein, J.L.; Brzezinski, P. Cryo-EM structure and kinetics reveal electron transfer by 2D diffusion of cytochrome c in the yeast III-IV respiratory supercomplex. Proc. Natl. Acad. Sci. USA 2021, 118, e2021157118. [Google Scholar] [CrossRef]
- Perez-Mejias, G.; Guerra-Castellano, A.; Diaz-Quintana, A.; De la Rosa, M.A.; Diaz-Moreno, I. Cytochrome c: Surfing Off of the Mitochondrial Membrane on the Tops of Complexes III and IV. Comput. Struct. Biotechnol. J. 2019, 17, 654–660. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Benard, G.; Bellance, N.; Jose, C.; Melser, S.; Nouette-Gaulain, K.; Rossignol, R. Multi-site control and regulation of mitochondrial energy production. Biochim. Biophys. Acta 2010, 1797, 698–709. [Google Scholar] [CrossRef]
- Cogliati, S.; Lorenzi, I.; Rigoni, G.; Caicci, F.; Soriano, M.E. Regulation of Mitochondrial Electron Transport Chain Assembly. J. Mol. Biol. 2018, 430, 4849–4873. [Google Scholar] [CrossRef]
- Hüttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [PubMed]
- Pieczenik, S.R.; Neustadt, J. Mitochondrial dysfunction and molecular pathways of disease. Exp. Mol. Pathol. 2007, 83, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Hüttemann, M.; Lee, I.; Pecinova, A.; Pecina, P.; Przyklenk, K.; Doan, J.W. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J. Bioenerg. Biomembr. 2008, 40, 445–456. [Google Scholar] [CrossRef]
- Ott, M.; Robertson, J.D.; Gogvadze, V.; Zhivotovsky, B.; Orrenius, S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc. Natl. Acad. Sci. USA 2002, 99, 1259–1263. [Google Scholar] [CrossRef]
- Kagan, V.E.; Bayir, H.A.; Belikova, N.A.; Kapralov, O.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Stoyanovsky, D.A.; Wipf, P.; Kochanek, P.M.; et al. Cytochrome c/cardiolipin relations in mitochondria: A kiss of death. Free Radic. Biol. Med. 2009, 46, 1439–1453. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Ashiya, M.; Buttle, K.; Weiler, S.; Oakes, S.A.; Mannella, C.A.; Korsmeyer, S.J. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 2002, 2, 55–67. [Google Scholar] [CrossRef]
- Petronilli, V.; Nicolli, A.; Costantini, P.; Colonna, R.; Bernardi, P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim. Biophys. Acta 1994, 1187, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar] [CrossRef]
- Bernardi, P.; Azzone, G.F. Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J. Biol. Chem. 1981, 256, 7187–7192. [Google Scholar] [CrossRef] [PubMed]
- Ripple, M.O.; Abajian, M.; Springett, R. Cytochrome c is rapidly reduced in the cytosol after mitochondrial outer membrane permeabilization. Apoptosis 2010, 15, 563–573. [Google Scholar] [CrossRef]
- Cheng, T.C.; Hong, C.; Akey, I.V.; Yuan, S.; Akey, C.W. A near atomic structure of the active human apoptosome. Elife 2016, 5, e17755. [Google Scholar] [CrossRef]
- Brown, G.C.; Borutaite, V. Regulation of apoptosis by the redox state of cytochrome c. Biochim. Biophys. Acta 2008, 1777, 877–881. [Google Scholar] [CrossRef]
- Skemiene, K.; Rakauskaite, G.; Trumbeckaite, S.; Liobikas, J.; Brown, G.C.; Borutaite, V. Anthocyanins block ischemia-induced apoptosis in the perfused heart and support mitochondrial respiration potentially by reducing cytosolic cytochrome c. Int. J. Biochem. Cell Biol. 2013, 45, 23–29. [Google Scholar] [CrossRef]
- Liobikas, J.; Skemiene, K.; Trumbeckaite, S.; Borutaite, V. Anthocyanins in cardioprotection: A path through mitochondria. Pharmacol. Res. 2016, 113, 808–815. [Google Scholar] [CrossRef]
- Kalkavan, H.; Chen, M.J.; Crawford, J.C.; Quarato, G.; Fitzgerald, P.; Tait, S.W.G.; Goding, C.R.; Green, D.R. Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 2022, 185, 3356–3374.e22. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Zurek, M.P.; Turner, A.A.; Khobeir, A.; Yazdi, N.; Hakim, L.; Liu, J.; Vaishnav, A.; et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int. J. Biochem. Cell Biol. 2020, 121, 105704. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Marquez, I.; Perez-Mejias, G.; Diaz-Quintana, A.; De la Rosa, M.A.; Diaz-Moreno, I. Post-Translational Modifications of Cytochrome c in Cell Life and Disease. Int. J. Mol. Sci. 2020, 21, 8483. [Google Scholar] [CrossRef] [PubMed]
- Pasdois, P.; Parker, J.E.; Griffiths, E.J.; Halestrap, A.P. The role of oxidized cytochrome c in regulating mitochondrial reactive oxygen species production and its perturbation in ischaemia. Biochem. J. 2011, 436, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.B.; Li, M.; Zhao, Y.; Xu, J.X. Cytochrome c is a hydrogen peroxide scavenger in mitochondria. Protein Pept. Lett. 2003, 10, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Pereverzev, M.O.; Vygodina, T.V.; Konstantinov, A.A.; Skulachev, V.P. Cytochrome c, an ideal antioxidant. Biochem. Soc. Trans. 2003, 31, 1312–1315. [Google Scholar] [CrossRef]
- Allen, S.; Balabanidou, V.; Sideris, D.P.; Lisowsky, T.; Tokatlidis, K. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J. Mol. Biol. 2005, 353, 937–944. [Google Scholar] [CrossRef]
- Gonzalez-Arzola, K.; Diaz-Quintana, A.; Bernardo-Garcia, N.; Martinez-Fabregas, J.; Rivero-Rodriguez, F.; Casado-Combreras, M.A.; Elena-Real, C.A.; Velazquez-Cruz, A.; Gil-Caballero, S.; Velazquez-Campoy, A.; et al. Nucleus-translocated mitochondrial cytochrome c liberates nucleophosmin-sequestered ARF tumor suppressor by changing nucleolar liquid-liquid phase separation. Nat. Struct. Mol. Biol. 2022, 29, 1024–1036. [Google Scholar] [CrossRef]
- Rivero-Rodriguez, F.; Diaz-Quintana, A.; Velazquez-Cruz, A.; Gonzalez-Arzola, K.; Gavilan, M.P.; Velazquez-Campoy, A.; Rios, R.M.; De la Rosa, M.A.; Diaz-Moreno, I. Inhibition of the PP2A activity by the histone chaperone ANP32B is long-range allosterically regulated by respiratory cytochrome c. Redox Biol. 2021, 43, 101967. [Google Scholar] [CrossRef]
- Gonzalez-Arzola, K.; Diaz-Moreno, I.; Cano-Gonzalez, A.; Diaz-Quintana, A.; Velazquez-Campoy, A.; Moreno-Beltran, B.; Lopez-Rivas, A.; De la Rosa, M.A. Structural basis for inhibition of the histone chaperone activity of SET/TAF-Ibeta by cytochrome c. Proc. Natl. Acad. Sci. USA 2015, 112, 9908–9913. [Google Scholar] [CrossRef]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66Shc in ischaemia reperfusion injury and other cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Steele, H.B.B.; Elmer-Dixon, M.M.; Rogan, J.T.; Ross, J.B.A.; Bowler, B.E. The Human Cytochrome c Domain-Swapped Dimer Facilitates Tight Regulation of Intrinsic Apoptosis. Biochemistry 2020, 59, 2055–2068. [Google Scholar] [CrossRef]
- Diaz-Quintana, A.; Perez-Mejias, G.; Guerra-Castellano, A.; De la Rosa, M.A.; Diaz-Moreno, I. Wheel and Deal in the Mitochondrial Inner Membranes: The Tale of Cytochrome c and Cardiolipin. Oxid. Med. Cell Longev. 2020, 2020, 6813405. [Google Scholar] [CrossRef]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef]
- Xu, Y.; Anjaneyulu, M.; Donelian, A.; Yu, W.; Greenberg, M.L.; Ren, M.; Owusu-Ansah, E.; Schlame, M. Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin. Proc. Natl. Acad. Sci. USA 2019, 116, 11235–11240. [Google Scholar] [CrossRef]
- Montero, J.; Mari, M.; Colell, A.; Morales, A.; Basanez, G.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Cholesterol and peroxidized cardiolipin in mitochondrial membrane properties, permeabilization and cell death. Biochim. Biophys. Acta 2010, 1797, 1217–1224. [Google Scholar] [CrossRef]
- Paradies, G.; Petrosillo, G.; Paradies, V.; Ruggiero, F.M. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic. Biol. Med. 2010, 48, 1286–1295. [Google Scholar] [CrossRef] [PubMed]
- Aluri, H.S.; Simpson, D.C.; Allegood, J.C.; Hu, Y.; Szczepanek, K.; Gronert, S.; Chen, Q.; Lesnefsky, E.J. Electron flow into cytochrome c coupled with reactive oxygen species from the electron transport chain converts cytochrome c to a cardiolipin peroxidase: Role during ischemia-reperfusion. Biochim. Biophys. Acta 2014, 1840, 3199–3207. [Google Scholar] [CrossRef]
- Lee, I.; Salomon, A.R.; Yu, K.; Doan, J.W.; Grossman, L.I.; Hüttemann, M. New prospects for an old enzyme: Mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry 2006, 45, 9121–9128. [Google Scholar] [CrossRef]
- Yu, H.; Lee, I.; Salomon, A.R.; Yu, K.; Hüttemann, M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim. Biophys. Acta 2008, 1777, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, G.; Varughese, A.; Ji, Q.; Lee, I.; Liu, J.; Vaishnav, A.; Sinkler, C.; Kapralov, A.A.; Moraes, C.T.; Sanderson, T.H.; et al. Phosphorylation of Cytochrome c Threonine 28 Regulates Electron Transport Chain Activity in Kidney: IMPLICATIONS FOR AMP KINASE. J. Biol. Chem. 2017, 292, 64–79. [Google Scholar] [CrossRef]
- Wan, J.; Kalpage, H.A.; Vaishnav, A.; Liu, J.; Lee, I.; Mahapatra, G.; Turner, A.A.; Zurek, M.P.; Ji, Q.; Moraes, C.T.; et al. Regulation of Respiration and Apoptosis by Cytochrome c Threonine 58 Phosphorylation. Sci. Rep. 2019, 9, 15815. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Vaishnav, A.; Liu, J.; Varughese, A.; Wan, J.; Turner, A.A.; Ji, Q.; Zurek, M.P.; Kapralov, A.A.; Kagan, V.E.; et al. Serine-47 phosphorylation of cytochrome c in the mammalian brain regulates cytochrome c oxidase and caspase-3 activity. FASEB J. 2019, 33, 13503–13514. [Google Scholar] [CrossRef]
- Li, F.; Sun, H.; Lin, X.; Li, Q.; Zhao, D.; Cheng, Z.; Liu, J.; Fan, Q. Increased cytochrome C threonine 50 phosphorylation in aging heart as a novel defensive signaling against hypoxia/reoxygenation induced apoptosis. Aging (Albany NY) 2022, 14, 5699–5709. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Diaz-Moreno, I.; Velazquez-Campoy, A.; De la Rosa, M.A.; Diaz-Quintana, A. Structural and functional characterization of phosphomimetic mutants of cytochrome c at threonine 28 and serine 47. Biochim. Biophys. Acta 2016, 1857, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Kalpage, H.A.; Wan, J.; Morse, P.T.; Lee, I.; Hüttemann, M. Brain-Specific Serine-47 Modification of Cytochrome c Regulates Cytochrome c Oxidase Activity Attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling. Cells 2020, 9, 1843. [Google Scholar] [CrossRef]
- Pecina, P.; Borisenko, G.G.; Belikova, N.A.; Tyurina, Y.Y.; Pecinova, A.; Lee, I.; Samhan-Arias, A.K.; Przyklenk, K.; Kagan, V.E.; Hüttemann, M. Phosphomimetic substitution of cytochrome c tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry 2010, 49, 6705–6714. [Google Scholar] [CrossRef]
- Garcia-Heredia, J.M.; Diaz-Quintana, A.; Salzano, M.; Orzaez, M.; Perez-Paya, E.; Teixeira, M.; De la Rosa, M.A.; Diaz-Moreno, I. Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti-apoptotic switch. J. Biol. Inorg. Chem. 2011, 16, 1155–1168. [Google Scholar] [CrossRef]
- Moreno-Beltran, B.; Guerra-Castellano, A.; Diaz-Quintana, A.; Del Conte, R.; Garcia-Maurino, S.M.; Diaz-Moreno, S.; Gonzalez-Arzola, K.; Santos-Ocana, C.; Velazquez-Campoy, A.; De la Rosa, M.A.; et al. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc. Natl. Acad. Sci. USA 2017, 114, E3041–E3050. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Diaz-Quintana, A.; Perez-Mejias, G.; Elena-Real, C.A.; Gonzalez-Arzola, K.; Garcia-Maurino, S.M.; De la Rosa, M.A.; Diaz-Moreno, I. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 7955–7960. [Google Scholar] [CrossRef]
- Roberts, V.A.; Pique, M.E. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J. Biol. Chem. 1999, 274, 38051–38060. [Google Scholar] [CrossRef]
- Schmidt, T.R.; Wildman, D.E.; Uddin, M.; Opazo, J.C.; Goodman, M.; Grossman, L.I. Rapid electrostatic evolution at the binding site for cytochrome c on cytochrome c oxidase in anthropoid primates. Proc. Natl. Acad. Sci. USA 2005, 102, 6379–6384. [Google Scholar] [CrossRef]
- Purring-Koch, C.; McLendon, G. Cytochrome c binding to Apaf-1: The effects of dATP and ionic strength. Proc. Natl. Acad. Sci. USA 2000, 97, 11928–11931. [Google Scholar] [CrossRef]
- Yu, T.; Wang, X.; Purring-Koch, C.; Wei, Y.; McLendon, G.L. A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J. Biol. Chem. 2001, 276, 13034–13038. [Google Scholar] [CrossRef]
- Zhou, M.; Li, Y.; Hu, Q.; Bai, X.C.; Huang, W.; Yan, C.; Scheres, S.H.; Shi, Y. Atomic structure of the apoptosome: Mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes. Dev. 2015, 29, 2349–2361. [Google Scholar] [CrossRef]
- Dorstyn, L.; Akey, C.W.; Kumar, S. New insights into apoptosome structure and function. Cell Death Differ. 2018, 25, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Elena-Real, C.A.; Diaz-Quintana, A.; Gonzalez-Arzola, K.; Velazquez-Campoy, A.; Orzaez, M.; Lopez-Rivas, A.; Gil-Caballero, S.; De la Rosa, M.A.; Diaz-Moreno, I. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3epsilon-dependent Apaf-1 inhibition. Cell Death Dis. 2018, 9, 365. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef]
- Kalpage, H.A.; Bazylianska, V.; Recanati, M.A.; Fite, A.; Liu, J.; Wan, J.; Mantena, N.; Malek, M.H.; Podgorski, I.; Heath, E.I.; et al. Tissue-specific regulation of cytochrome c by post-translational modifications: Respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 2019, 33, 1540–1553. [Google Scholar] [CrossRef] [PubMed]
- Kaim, G.; Dimroth, P. ATP synthesis by F-type ATP synthase is obligatorily dependent on the transmembrane voltage. EMBO J. 1999, 18, 4118–4127. [Google Scholar] [CrossRef]
- Korshunov, S.S.; Skulachev, V.P.; Starkov, A.A. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997, 416, 15–18. [Google Scholar] [CrossRef]
- Liu, S.S. Mitochondrial Q cycle-derived superoxide and chemiosmotic bioenergetics. Ann. N. Y Acad. Sci. 2010, 1201, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A.; Fiskum, G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J. Neurochem. 2003, 86, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijevic, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef]
- Rottenberg, H.; Covian, R.; Trumpower, B.L. Membrane potential greatly enhances superoxide generation by the cytochrome bc1 complex reconstituted into phospholipid vesicles. J. Biol. Chem. 2009, 284, 19203–19210. [Google Scholar] [CrossRef]
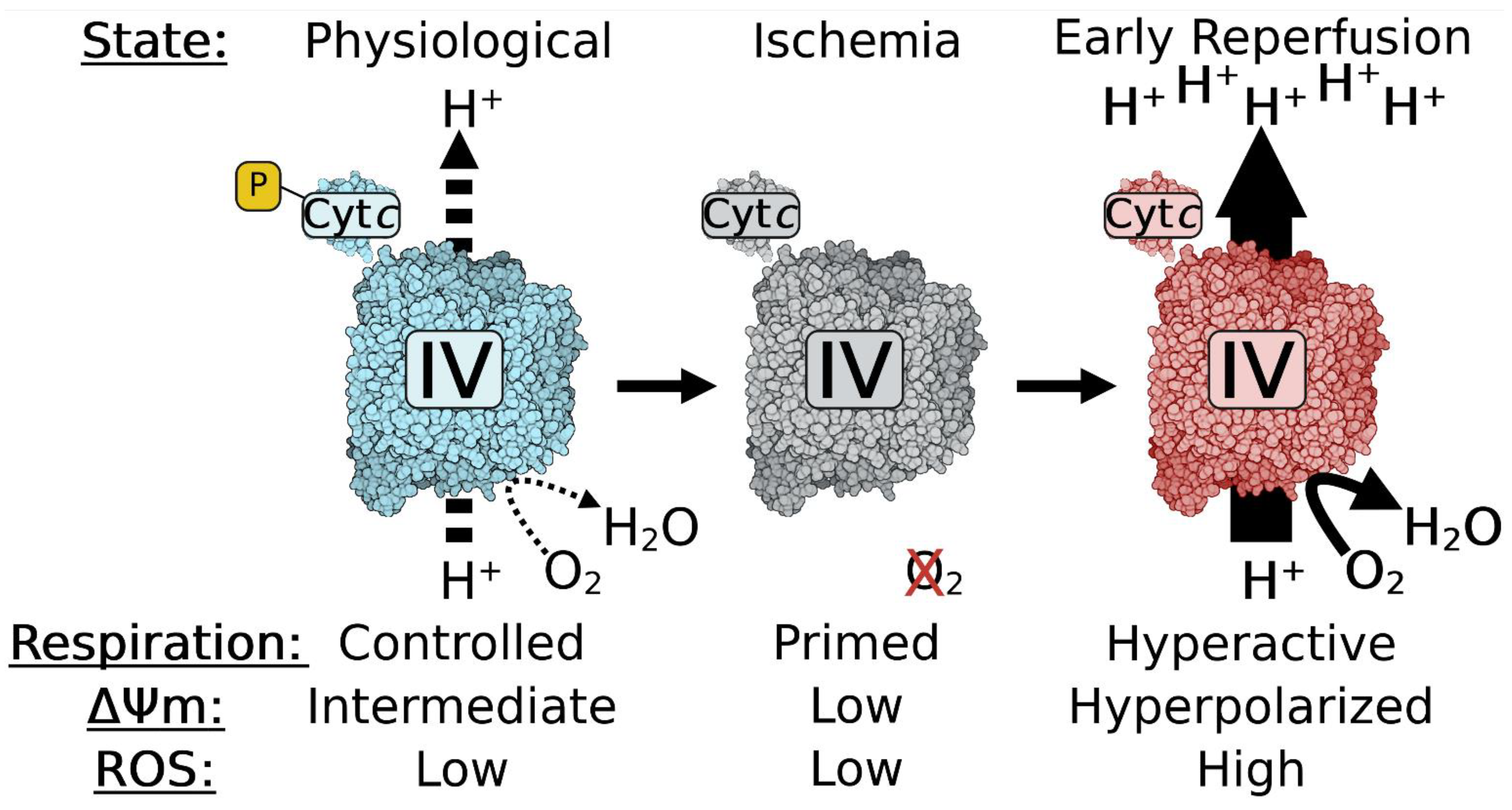
- Morse, P.T.; Wan, J.; Bell, J.; Lee, I.; Goebel, D.J.; Malek, M.H.; Sanderson, T.H.; Hüttemann, M. Sometimes less is more: Inhibitory infrared light during early reperfusion calms hyperactive mitochondria and suppresses reperfusion injury. Biochem. Soc. Trans. 2022, 50, 1377–1388. [Google Scholar] [CrossRef]
- Sanishvili, R.; Volz, K.W.; Westbrook, E.M.; Margoliash, E. The low ionic strength crystal structure of horse cytochrome c at 2.1 A resolution and comparison with its high ionic strength counterpart. Structure 1995, 3, 707–716. [Google Scholar] [CrossRef]
- Sugitani, R.; Stuchebrukhov, A.A. Molecular dynamics simulation of water in cytochrome c oxidase reveals two water exit pathways and the mechanism of transport. Biochim. Biophys. Acta 2009, 1787, 1140–1150. [Google Scholar] [CrossRef]
- Tsukihara, T.; Shimokata, K.; Katayama, Y.; Shimada, H.; Muramoto, K.; Aoyama, H.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Yao, M.; et al. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc. Natl. Acad. Sci. USA 2003, 100, 15304–15309. [Google Scholar] [CrossRef]
- Zaidi, S.; Hassan, M.I.; Islam, A.; Ahmad, F. The role of key residues in structure, function, and stability of cytochrome-c. Cell Mol. Life Sci. 2014, 71, 229–255. [Google Scholar] [CrossRef]
- Shimada, S.; Shinzawa-Itoh, K.; Baba, J.; Aoe, S.; Shimada, A.; Yamashita, E.; Kang, J.; Tateno, M.; Yoshikawa, S.; Tsukihara, T. Complex structure of cytochrome c-cytochrome c oxidase reveals a novel protein-protein interaction mode. EMBO J. 2017, 36, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Cammack, R. Redox States and Potentials. In Bioenergetics: A Practical Approach; Brown, G.C., Cooper, C.E., Eds.; IRL Press: Oxford, UK, 1995; pp. 85–109. [Google Scholar]
- Vempati, U.D.; Diaz, F.; Barrientos, A.; Narisawa, S.; Mian, A.M.; Millan, J.L.; Boise, L.H.; Moraes, C.T. Role of cytochrome c in apoptosis: Increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome c release. Mol. Cell Biol. 2007, 27, 1771–1783. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Juszczak, F.; Caron, N.; Mathew, A.V.; Decleves, A.E. Critical Role for AMPK in Metabolic Disease-Induced Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 7994. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, J.; Finckenberg, P.; Levijoki, J.; Mervaala, E. AMPK activator AICAR ameliorates ischaemia reperfusion injury in the rat kidney. Br. J. Pharmacol. 2012, 166, 1905–1915. [Google Scholar] [CrossRef]
- Hüttemann, M.; Pecina, P.; Rainbolt, M.; Sanderson, T.H.; Kagan, V.E.; Samavati, L.; Doan, J.W.; Lee, I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion 2011, 11, 369–381. [Google Scholar] [CrossRef]
- Bijur, G.N.; Jope, R.S. Rapid accumulation of Akt in mitochondria following phosphatidylinositol 3-kinase activation. J. Neurochem. 2003, 87, 1427–1435. [Google Scholar] [CrossRef]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M.; et al. Akt-mediated phosphorylation of MICU1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Xie, X.; Shu, R.; Yu, C.; Fu, Z.; Li, Z. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis. 2022, 13, 157–174. [Google Scholar] [CrossRef]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Cell Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.D.; Gellerfors, P. The redox properties of the cytochromes of purified complex 3. Biochim. Biophys. Acta 1974, 357, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Kokhan, O.; Wraight, C.A.; Tajkhorshid, E. The binding interface of cytochrome c and cytochrome c(1) in the bc(1) complex: Rationalizing the role of key residues. Biophys. J. 2010, 99, 2647–2656. [Google Scholar] [CrossRef]
- Guerra-Castellano, A.; Diaz-Quintana, A.; Moreno-Beltran, B.; Lopez-Prados, J.; Nieto, P.M.; Meister, W.; Staffa, J.; Teixeira, M.; Hildebrandt, P.; De la Rosa, M.A.; et al. Mimicking Tyrosine Phosphorylation in Human Cytochrome c by the Evolved tRNA Synthetase Technique. Chemistry 2015, 21, 15004–15012. [Google Scholar] [CrossRef]
- De Rocco, D.; Cerqua, C.; Goffrini, P.; Russo, G.; Pastore, A.; Meloni, F.; Nicchia, E.; Moraes, C.T.; Pecci, A.; Salviati, L.; et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim. Biophys. Acta 2014, 1842, 269–274. [Google Scholar] [CrossRef]
- Deacon, O.M.; Karsisiotis, A.I.; Moreno-Chicano, T.; Hough, M.A.; Macdonald, C.; Blumenschein, T.M.A.; Wilson, M.T.; Moore, G.R.; Worrall, J.A.R. Heightened Dynamics of the Oxidized Y48H Variant of Human Cytochrome c Increases Its Peroxidatic Activity. Biochemistry 2017, 56, 6111–6124. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.S.; Nucci, N.V.; Fuglestad, B.; Tommos, C.; Wand, A.J. Defining the Apoptotic Trigger: THE INTERACTION OF CYTOCHROME c AND CARDIOLIPIN. J. Biol. Chem. 2015, 290, 30879–30887. [Google Scholar] [CrossRef]
- Italia, J.S.; Peeler, J.C.; Hillenbrand, C.M.; Latour, C.; Weerapana, E.; Chatterjee, A. Genetically encoded protein sulfation in mammalian cells. Nat. Chem. Biol. 2020, 16, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, T.H.; Mahapatra, G.; Pecina, P.; Ji, Q.; Yu, K.; Sinkler, C.; Varughese, A.; Kumar, R.; Bukowski, M.J.; Tousignant, R.N.; et al. Cytochrome C is tyrosine 97 phosphorylated by neuroprotective insulin treatment. PLoS ONE 2013, 8, e78627. [Google Scholar] [CrossRef]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Bazylianska, V.; Kalpage, H.A.; Wan, J.; Vaishnav, A.; Mahapatra, G.; Turner, A.A.; Chowdhury, D.D.; Kim, K.; Morse, P.T.; Lee, I.; et al. Lysine 53 Acetylation of Cytochrome c in Prostate Cancer: Warburg Metabolism and Evasion of Apoptosis. Cells 2021, 10, 802. [Google Scholar] [CrossRef]
- Morse, P.T.; Perez-Mejias, G.; Wan, J.; Turner, A.A.; Marquez, I.; Kalpage, H.A.; Vaishnav, A.; Zurek, M.P.; Huettemann, P.P.; Kim, K.; et al. Cytochrome c lysine acetylation regulates cellular respiration and cell death in ischemic skeletal muscle. Nat. Commun. 2023, 14, 4166. [Google Scholar] [CrossRef]
- Wagner, G.R.; Payne, R.M. Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 2013, 288, 29036–29045. [Google Scholar] [CrossRef]
- Konig, A.C.; Hartl, M.; Boersema, P.J.; Mann, M.; Finkemeier, I. The mitochondrial lysine acetylome of Arabidopsis. Mitochondrion 2014, 19 Pt B, 252–260. [Google Scholar] [CrossRef]
- Hong, S.Y.; Ng, L.T.; Ng, L.F.; Inoue, T.; Tolwinski, N.S.; Hagen, T.; Gruber, J. The Role of Mitochondrial Non-Enzymatic Protein Acylation in Ageing. PLoS ONE 2016, 11, e0168752. [Google Scholar] [CrossRef]
- Kamieniarz, K.; Schneider, R. Tools to tackle protein acetylation. Chem. Biol. 2009, 16, 1027–1029. [Google Scholar] [CrossRef]
- Marquez, I.; Perez-Mejias, G.; Guerra-Castellano, A.; Olloqui-Sariego, J.L.; Andreu, R.; Calvente, J.J.; De la Rosa, M.A.; Diaz-Moreno, I. Structural and functional insights into lysine acetylation of cytochrome c using mimetic point mutants. FEBS Open Bio 2021, 11, 3304–3323. [Google Scholar] [CrossRef]
- Moreno-Beltran, B.; Diaz-Moreno, I.; Gonzalez-Arzola, K.; Guerra-Castellano, A.; Velazquez-Campoy, A.; De la Rosa, M.A.; Diaz-Quintana, A. Respiratory complexes III and IV can each bind two molecules of cytochrome c at low ionic strength. FEBS Lett. 2015, 589, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Rieder, R.; Bosshard, H.R. Comparison of the binding sites on cytochrome c for cytochrome c oxidase, cytochrome bc1, and cytochrome c1. Differential acetylation of lysyl residues in free and complexed cytochrome c. J. Biol. Chem. 1980, 255, 4732–4739. [Google Scholar] [CrossRef]
- Eckert, P.; Schnackerz, K. Ischemic tolerance of human skeletal muscle. Ann. Plast. Surg. 1991, 26, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Gillani, S.; Cao, J.; Suzuki, T.; Hak, D.J. The effect of ischemia reperfusion injury on skeletal muscle. Injury 2012, 43, 670–675. [Google Scholar] [CrossRef]
- Nakamura, Y.; Ogura, M.; Tanaka, D.; Inagaki, N. Localization of mouse mitochondrial SIRT proteins: Shift of SIRT3 to nucleus by co-expression with SIRT5. Biochem. Biophys. Res. Commun. 2008, 366, 174–179. [Google Scholar] [CrossRef]
- Schlicker, C.; Gertz, M.; Papatheodorou, P.; Kachholz, B.; Becker, C.F.; Steegborn, C. Substrates and regulation mechanisms for the human mitochondrial sirtuins Sirt3 and Sirt5. J. Mol. Biol. 2008, 382, 790–801. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, Y.M.; Chun, Y.S.; Chen, J.; Kim, J.E.; Park, J.W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Hitaoka, S.; Inoue, K.; Imai, M.; Saio, T.; Uchida, T.; Shinzawa-Itoh, K.; Yoshikawa, S.; Yoshizawa, K.; Ishimori, K. Energetic Mechanism of Cytochrome c-Cytochrome c Oxidase Electron Transfer Complex Formation under Turnover Conditions Revealed by Mutational Effects and Docking Simulation. J. Biol. Chem. 2016, 291, 15320–15331. [Google Scholar] [CrossRef]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.A.; Stebbins, C.L.; Bonigut, S.; Halliwell, B.; Longhurst, J.C. Production of hydroxyl radicals in contracting skeletal muscle of cats. J. Appl. Physiol. (1985) 1996, 81, 1197–1206. [Google Scholar] [CrossRef]
- Pearson, T.; Kabayo, T.; Ng, R.; Chamberlain, J.; McArdle, A.; Jackson, M.J. Skeletal muscle contractions induce acute changes in cytosolic superoxide, but slower responses in mitochondrial superoxide and cellular hydrogen peroxide. PLoS ONE 2014, 9, e96378. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Alonso, J.; Richardson, R.S.; Saltin, B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J. Physiol. 2001, 530, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, I.H.; Kemppainen, J.; Kaskinoro, K.; Peltonen, J.E.; Borra, R.; Lindroos, M.; Oikonen, V.; Nuutila, P.; Knuuti, J.; Boushel, R.; et al. Regulation of human skeletal muscle perfusion and its heterogeneity during exercise in moderate hypoxia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 299, R72–R79. [Google Scholar] [CrossRef] [PubMed]
- Kuhns, K.J.; Zhang, G.; Wang, Z.; Liu, W. ARD1/NAA10 acetylation in prostate cancer. Exp. Mol. Med. 2018, 50, 1–8. [Google Scholar] [CrossRef]
- Xie, X.; Xu, Z.; Wang, C.; Fang, C.; Zhao, J.; Xu, L.; Qian, X.; Dai, J.; Sun, F.; Xu, D.; et al. Tip60 is associated with resistance to X-ray irradiation in prostate cancer. FEBS Open Bio 2018, 8, 271–278. [Google Scholar] [CrossRef]
- Villani, G.; Attardi, G. In vivo control of respiration by cytochrome c oxidase in wild-type and mitochondrial DNA mutation-carrying human cells. Proc. Natl. Acad. Sci. USA 1997, 94, 1166–1171. [Google Scholar] [CrossRef]
- Villani, G.; Greco, M.; Papa, S.; Attardi, G. Low reserve of cytochrome c oxidase capacity in vivo in the respiratory chain of a variety of human cell types. J. Biol. Chem. 1998, 273, 31829–31836. [Google Scholar] [CrossRef] [PubMed]
- Kunz, W.S.; Kudin, A.; Vielhaber, S.; Elger, C.E.; Attardi, G.; Villani, G. Flux control of cytochrome c oxidase in human skeletal muscle. J. Biol. Chem. 2000, 275, 27741–27745. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Scrima, R.; Boffoli, D.; Capitanio, N. Control by cytochrome c oxidase of the cellular oxidative phosphorylation system depends on the mitochondrial energy state. Biochem. J. 2006, 396, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, R.; Malgat, M.; Mazat, J.P.; Letellier, T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 1999, 274, 33426–33432. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef]





| Residue | Tissue of Origin | Experimental Models | Findings |
|---|---|---|---|
| Threonine 28 | Bovine Kidney | In vivo phosphorylated Cytc purified from bovine kidney | Decreased Cytc-COX Vmax and Km, phosphorylated by AMPK [42] |
| Recombinant phosphomimetic T28E Cytc | Decreased Cytc-COX Vmax and Km, decreased redox potential, increased rate of reduction, decreased degradation by H2O2, decreased cardiolipin peroxidase activity [42] | ||
| Cytc double knockout mouse lung fibroblasts expressing T28E Cytc | Decreased respiration, decreased ΔΨm, decreased mitochondrial ROS production, decreased ATP levels, decreased cell death [42] | ||
| Recombinant phosphomimetic T28D Cytc | * Increased Cytc-COX activity, decreased redox potential, increased cardiolipin peroxidase activity [46] | ||
| Serine 47 | Porcine Brain, Rat Brain | In vivo phosphorylated Cytc purified from porcine brain | Decreased Cytc-COX activity, decreased caspase-3 activity, phosphorylated by Akt [44,47] |
| Recombinant phosphomimetic S47E Cytc | Decreased Cytc-COX activity, decreased caspase-3 activity, decreased heme degradation, decreased cardiolipin peroxidase activity [44] | ||
| Cytc double knockout mouse lung fibroblasts expressing S47E Cytc | Decreased respiration, decreased ΔΨm, decreased mitochondrial ROS production, decreased cell death, reduced responsiveness to oxygen-glucose deprivation followed by reoxygenation [47] | ||
| Recombinant phosphomimetic S47D Cytc | * Increased Cytc-COX activity, decreased caspase-3 activity, decreased cardiolipin peroxidase activity [46] | ||
| Tyrosine 48 | Bovine Liver | In vivo phosphorylated Cytc purified from bovine liver | Decreased Cytc-COX Vmax and Km [41] |
| Recombinant phosphomimetic Y48E Cytc | Decreased Cytc-COX Vmax, increased COX Km, decreased caspase-9 activity, decreased caspase-3 activity, decreased redox potential, decreased cardiolipin peroxidase activity [48,49] | ||
| Recombinant phosphomimetic Y48pCMF Cytc | * Overall decreased supercomplex activity (increased isolated Cytc-COX activity), decreased caspase-3 activity, increased cardiolipin peroxidase activity [50] | ||
| Threonine 49 | Mouse Heart | AC16 cardiomyocytes concurrently expressing WT Cytc and T49E Cytc | Decreased cell death, decreased caspase-9 activity, decreased caspase-3 activity [45]; methodological limitations are discussed in the text |
| Threonine 58 | Rat Kidney | Recombinant phosphomimetic T58E Cytc | Decreased Cytc-COX Vmax, decreased caspase-3 activity, decreased rate of oxidation, increased rate of reduction, decreased heme degradation, decreased cardiolipin peroxidase activity [43] |
| Cytc double knockout mouse lung fibroblasts expressing T58E Cytc | Decreased respiration, decreased ΔΨm, decreased mitochondrial ROS production, decreased ATP levels, decreased cell death [43] | ||
| Tyrosine 97 | Bovine Heart | In vivo phosphorylated Cytc purified from bovine heart | Decreased Cytc-COX Km, spectral shift of characteristic 695 nm peak to 687 nm (indicates changes to heme group) [40] |
| Recombinant phosphomimetic Y97E Cytc | Decreased melting temperature [49] | ||
| Recombinant phosphomimetic Y97pCMF Cytc | * Increased Cytc-COX activity, decreased caspase-3 activity [51] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morse, P.T.; Arroum, T.; Wan, J.; Pham, L.; Vaishnav, A.; Bell, J.; Pavelich, L.; Malek, M.H.; Sanderson, T.H.; Edwards, B.F.P.; et al. Phosphorylations and Acetylations of Cytochrome c Control Mitochondrial Respiration, Mitochondrial Membrane Potential, Energy, ROS, and Apoptosis. Cells 2024, 13, 493. https://doi.org/10.3390/cells13060493
Morse PT, Arroum T, Wan J, Pham L, Vaishnav A, Bell J, Pavelich L, Malek MH, Sanderson TH, Edwards BFP, et al. Phosphorylations and Acetylations of Cytochrome c Control Mitochondrial Respiration, Mitochondrial Membrane Potential, Energy, ROS, and Apoptosis. Cells. 2024; 13(6):493. https://doi.org/10.3390/cells13060493
Chicago/Turabian StyleMorse, Paul T., Tasnim Arroum, Junmei Wan, Lucynda Pham, Asmita Vaishnav, Jamie Bell, Lauren Pavelich, Moh H. Malek, Thomas H. Sanderson, Brian F.P. Edwards, and et al. 2024. "Phosphorylations and Acetylations of Cytochrome c Control Mitochondrial Respiration, Mitochondrial Membrane Potential, Energy, ROS, and Apoptosis" Cells 13, no. 6: 493. https://doi.org/10.3390/cells13060493