
Benefits and Pitfalls of a Glycosylation Inhibitor Tunicamycin in the Therapeutic Implication of Cancers
 , , , ,
, , , , 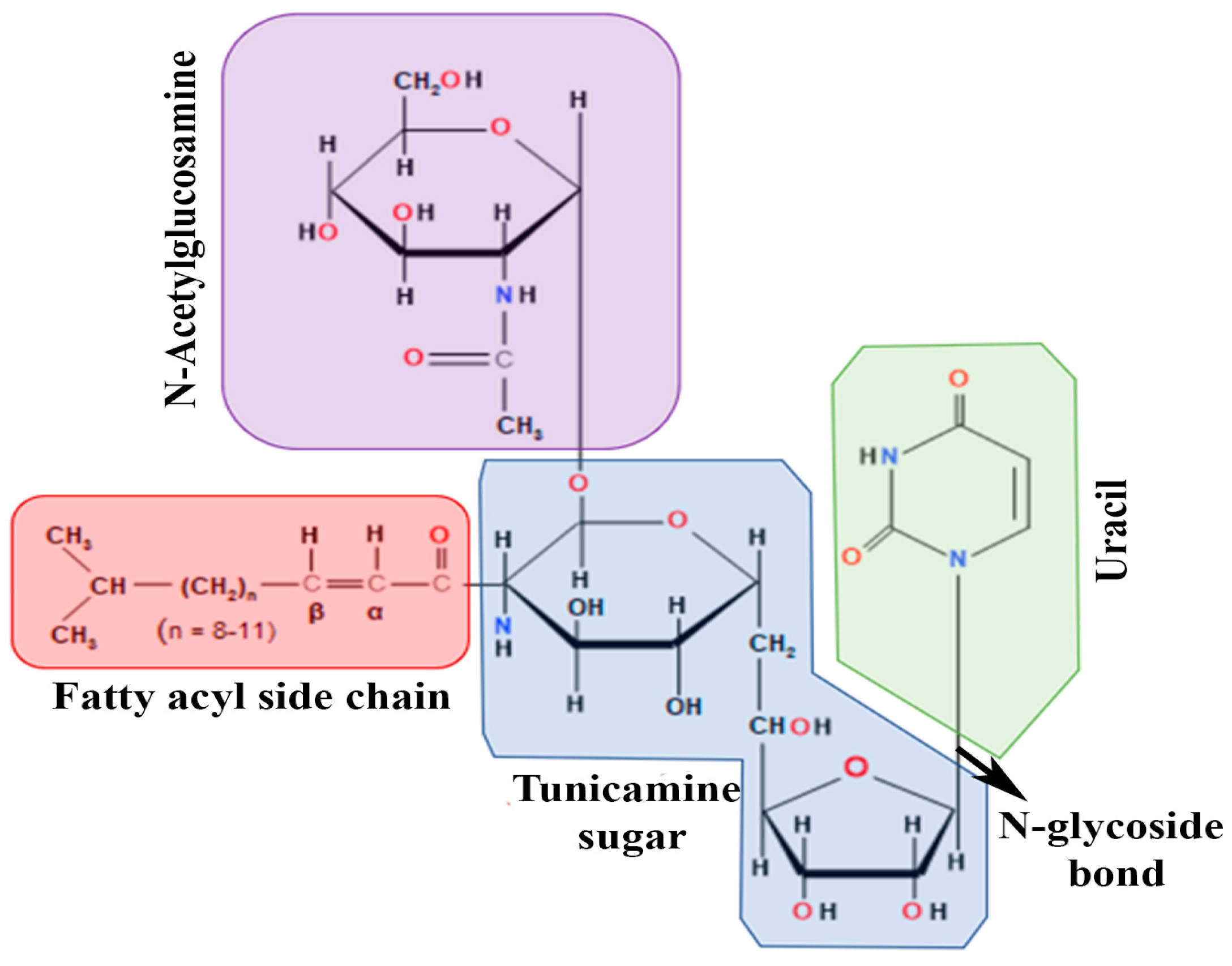{kind=link}
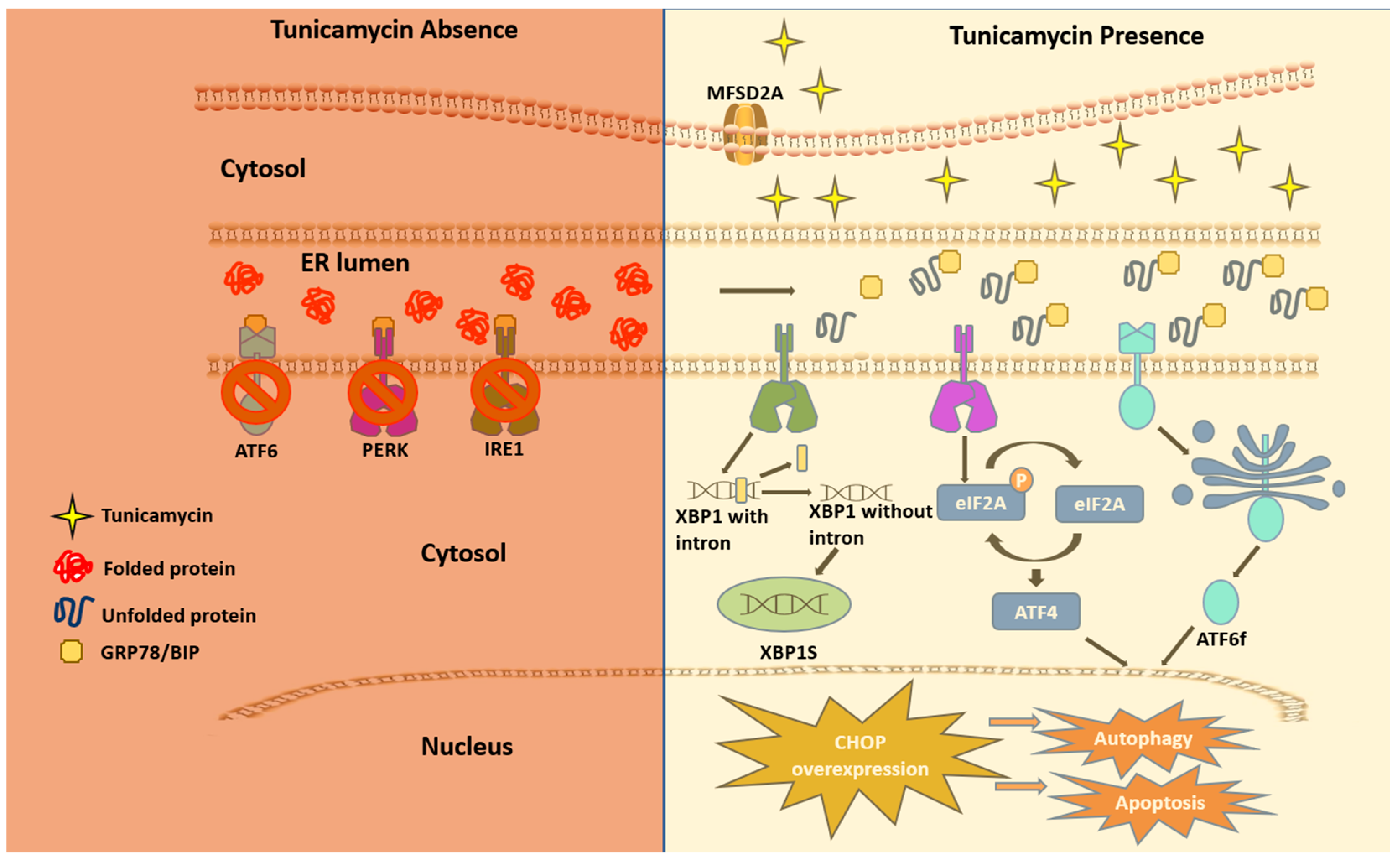{kind=link}
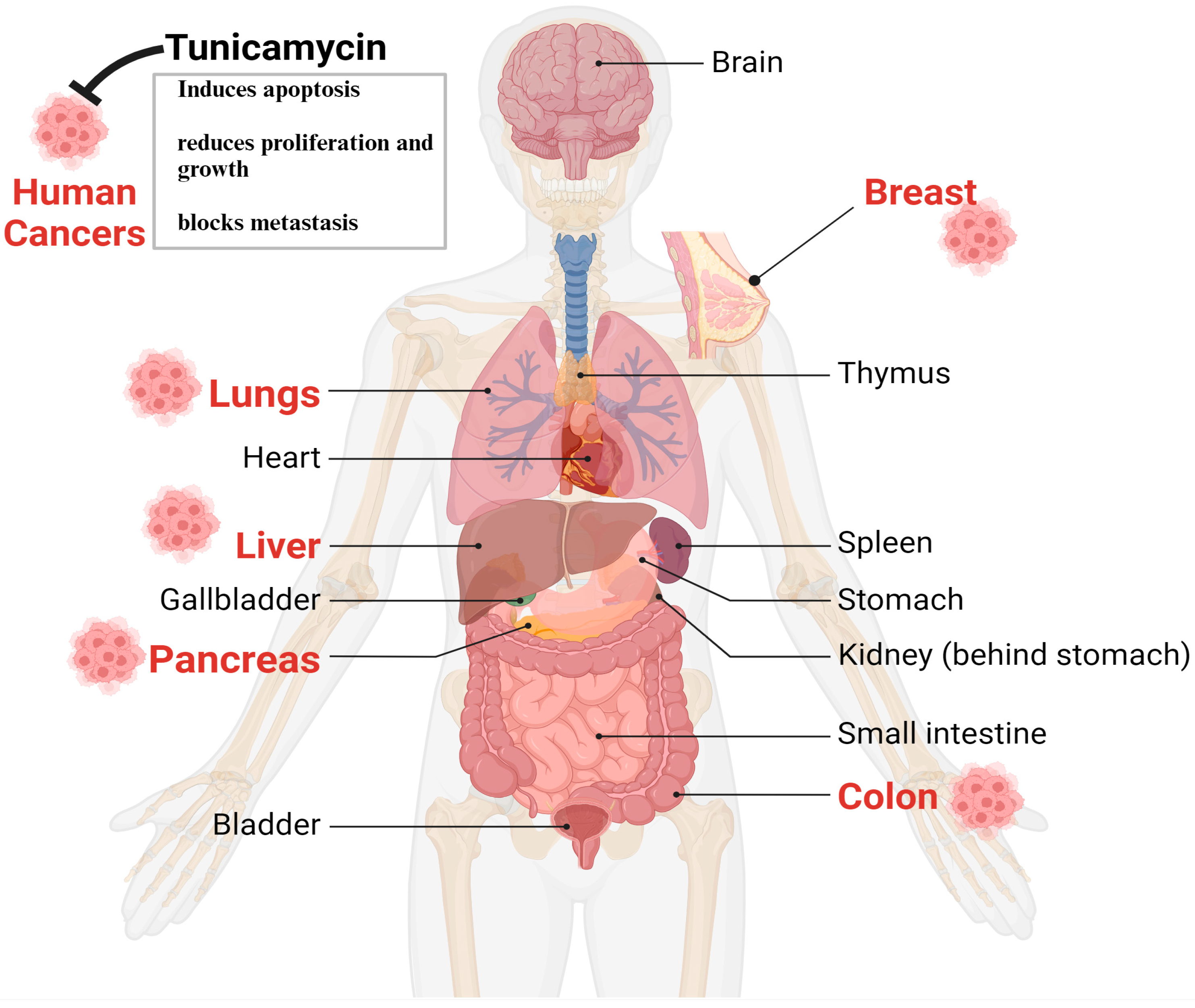{kind=link}
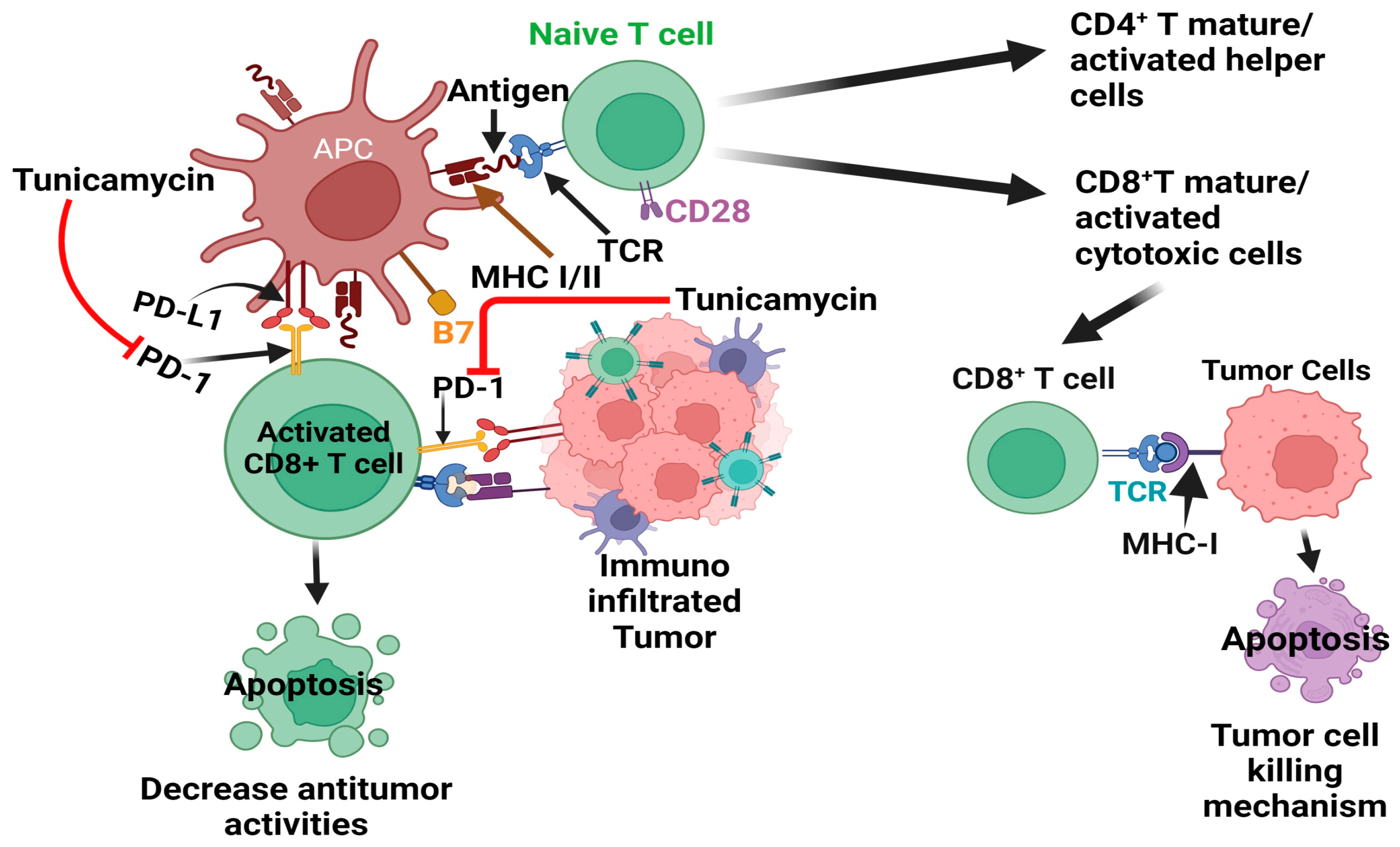{kind=link}
Abstract
:1. Introduction
2. Origin and Basic Biology of Tunicamycin
3. How Does Tunicamycin Work?
4. Tunicamycin in Cancer Therapy
5. Tunicamycin in Immunotherapy
6. The Pitfalls of the Use of Tunicamycin
7. Can Nanoencapsulation Overcome the Pitfall?
8. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bennett, E.P.; Mandel, U.; Clausen, H.; Gerken, T.A.; Fritz, T.A.; Tabak, L.A. Control of mucin-type O-glycosylation: A classification of the polypeptide GalNAc-transferase gene family. Glycobiology 2012, 22, 736–756. [Google Scholar] [CrossRef]
- Clausen, H.; Bennett, E.P. A family of UDP-GalNAc: Polypeptide N-acetylgalactosaminyl-transferases control the initiation of mucin-type O-linked glycosylation. Glycobiology 1996, 6, 635–646. [Google Scholar] [CrossRef]
- Schoberer, J.; Shin, Y.J.; Vavra, U.; Veit, C.; Strasser, R. Analysis of Protein Glycosylation in the ER. Methods Mol. Biol. 2018, 1691, 205–222. [Google Scholar] [CrossRef]
- Yang, G.; Zhang, H.; Yi, W.; Yan, S.; Cao, L. Editorial: Protein Glycosylation-Advances in Identification, Characterization and Biological Function Elucidation Using Mass Spectrometry. Front. Chem. 2022, 10, 847242. [Google Scholar] [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef]
- Rudd, P.M.; Elliott, T.; Cresswell, P.; Wilson, I.A.; Dwek, R.A. Glycosylation and the immune system. Science 2001, 291, 2370–2376. [Google Scholar] [CrossRef]
- Takahashi, K.; Raska, M.; Stuchlova Horynova, M.; Hall, S.D.; Poulsen, K.; Kilian, M.; Hiki, Y.; Yuzawa, Y.; Moldoveanu, Z.; Julian, B.A.; et al. Enzymatic sialylation of IgA1 O-glycans: Implications for studies of IgA nephropathy. PLoS ONE 2014, 9, e99026. [Google Scholar] [CrossRef]
- Hakomori, S. Tumor-associated carbohydrate antigens defining tumor malignancy: Basis for development of anti-cancer vaccines. Adv. Exp. Med. Biol. 2001, 491, 369–402. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Ju, T. Aberrant Glycosylation as Immune Therapeutic Targets for Solid Tumors. Cancers 2023, 15, 3536. [Google Scholar] [CrossRef]
- Veillon, L.; Fakih, C.; Abou-El-Hassan, H.; Kobeissy, F.; Mechref, Y. Glycosylation Changes in Brain Cancer. ACS Chem. Neurosci. 2018, 9, 51–72. [Google Scholar] [CrossRef]
- Hauselmann, I.; Borsig, L. Altered tumor-cell glycosylation promotes metastasis. Front. Oncol. 2014, 4, 28. [Google Scholar] [CrossRef]
- Munkley, J.; Elliott, D.J. Hallmarks of glycosylation in cancer. Oncotarget 2016, 7, 35478–35489. [Google Scholar] [CrossRef]
- Meany, D.L.; Chan, D.W. Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin. Proteom. 2011, 8, 7. [Google Scholar] [CrossRef]
- Arnal-Estape, A.; Nguyen, D.X. Sweets for a bitter end: Lung cancer cell-surface protein glycosylation mediates metastatic colonization. Cancer Discov. 2015, 5, 109–111. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis: Past, present and the near future. Carcinogenesis 2000, 21, 505–515. [Google Scholar] [CrossRef]
- MacDougall, J.R.; Matrisian, L.M. Contributions of tumor and stromal matrix metalloproteinases to tumor progression, invasion and metastasis. Cancer Metastasis Rev. 1995, 14, 351–362. [Google Scholar] [CrossRef]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Cancer Biol. 2000, 10, 415–433. [Google Scholar] [CrossRef]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6–16. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Liu, H.; Zhang, Z.; Ni, Z.; Chen, J.; Yang, Z.; Nie, Y.; Fan, D. Tunicamycin specifically aggravates ER stress and overcomes chemoresistance in multidrug-resistant gastric cancer cells by inhibiting N-glycosylation. J. Exp. Clin. Cancer Res. 2018, 37, 272. [Google Scholar] [CrossRef]
- Guha, P.; Kaptan, E.; Gade, P.; Kalvakolanu, D.V.; Ahmed, H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget 2017, 8, 68191–68207. [Google Scholar] [CrossRef]
- Jung, Y.H.; Lim, E.J.; Heo, J.; Kwon, T.K.; Kim, Y.H. Tunicamycin sensitizes human prostate cells to TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of cIAP2. Int. J. Oncol. 2012, 40, 1941–1948. [Google Scholar] [CrossRef] [PubMed]
- Akira, T.; Gakuzo, T. Tunicamycin, A New Antibiotic, I Isolation And Characterization of Tunicamycin. J. Antibiot. 1970, 24, 215–223. [Google Scholar]
- Yoo, J.; Mashalidis, E.H.; Kuk, A.C.Y.; Yamamoto, K.; Kaeser, B.; Ichikawa, S.; Lee, S.Y. GlcNAc-1-P-transferase-tunicamycin complex structure reveals basis for inhibition of N-glycosylation. Nat. Struct. Mol. Biol. 2018, 25, 217–224. [Google Scholar] [CrossRef]
- Duksin, D.; Bornstein, P. Changes in surface properties of normal and transformed cells caused by tunicamycin, an inhibitor of protein glycosylation. Proc. Natl. Acad. Sci. USA 1977, 74, 3433–3437. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Arey, B.J. The Role of Glycosylation in Receptor Signaling. Glycosylation 2012, 26, 50262. [Google Scholar] [CrossRef]
- Seales, E.C.; Jurado, G.A.; Singhal, A.; Bellis, S.L. Ras oncogene directs expression of a differentially sialylated, functionally altered beta1 integrin. Oncogene 2003, 22, 7137–7145. [Google Scholar] [CrossRef]
- Very, N.; Lefebvre, T.; El Yazidi-Belkoura, I. Drug resistance related to aberrant glycosylation in colorectal cancer. Oncotarget 2018, 9, 1380–1402. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; He, Z.; Deng, J.; Zhang, Z.; Liu, L.; Ye, W.; Liu, S. Tunicamycin induces ER stress and inhibits tumorigenesis of head and neck cancer cells by inhibiting N-glycosylation. Am. J. Transl. Res. 2020, 12, 541–550. [Google Scholar]
- Bassik, M.C.; Kampmann, M. Knocking out the door to tunicamycin entry. Proc. Natl. Acad. Sci. USA 2011, 108, 11731–11732. [Google Scholar] [CrossRef]
- Reiling, J.H.; Clish, C.B.; Carette, J.E.; Varadarajan, M.; Brummelkamp, T.R.; Sabatini, D.M. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl. Acad. Sci. USA 2011, 108, 11756–11765. [Google Scholar] [CrossRef]
- de-Freitas-Junior, J.C.; Bastos, L.G.; Freire-Neto, C.A.; Rocher, B.D.; Abdelhay, E.S.; Morgado-Diaz, J.A. N-glycan biosynthesis inhibitors induce in vitro anticancer activity in colorectal cancer cells. J. Cell. Biochem. 2012, 113, 2957–2966. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Scheuner, D.; Schroder, M.; Shen, X.; Lee, K.; Liu, C.Y.; Arnold, S.M. The unfolded protein response in nutrient sensing and differentiation. Nat. Rev. Mol. Cell Biol. 2002, 3, 411–421. [Google Scholar] [CrossRef]
- Read, A.; Schroder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Ron, D. Translational control in the endoplasmic reticulum stress response. J. Clin. Investig. 2002, 110, 1383–1388. [Google Scholar] [CrossRef]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef]
- Hasegawa, A.; Osuga, Y.; Hirota, Y.; Hamasaki, K.; Kodama, A.; Harada, M.; Tajima, T.; Takemura, Y.; Hirata, T.; Yoshino, O.; et al. Tunicamycin enhances the apoptosis induced by tumor necrosis factor-related apoptosis-inducing ligand in endometriotic stromal cells. Hum. Reprod. 2009, 24, 408–414. [Google Scholar] [CrossRef]
- Delom, F.; Emadali, A.; Cocolakis, E.; Lebrun, J.J.; Nantel, A.; Chevet, E. Calnexin-dependent regulation of tunicamycin-induced apoptosis in breast carcinoma MCF-7 cells. Cell Death Differ. 2007, 14, 586–596. [Google Scholar] [CrossRef]
- Banerjee, A.; Lang, J.Y.; Hung, M.C.; Sengupta, K.; Banerjee, S.K.; Baksi, K.; Banerjee, D.K. Unfolded protein response is required in nu/nu mice microvasculature for treating breast tumor with tunicamycin. J. Biol. Chem. 2011, 286, 29127–29138. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, Y.; Xue, J.; Li, J.; Yi, J.; Bu, J.; Zhang, Z.; Qiu, P.; Gu, X. Advances in immunotherapy for triple-negative breast cancer. Mol. Cancer 2023, 22, 145. [Google Scholar] [CrossRef]
- Wang, X.; Xiong, W.; Tang, Y. Tunicamycin suppresses breast cancer cell growth and metastasis via regulation of the protein kinase B/nuclear factor-kappaB signaling pathway. Oncol. Lett. 2018, 15, 4137–4142. [Google Scholar] [CrossRef]
- Han, X.; Zhang, X.; Li, H.; Huang, S.; Zhang, S.; Wang, F.; Shi, Y. Tunicamycin enhances the antitumor activity of trastuzumab on breast cancer in vitro and in vivo. Oncotarget 2015, 6, 38912–38925. [Google Scholar] [CrossRef]
- You, Z.; He, L.; Yan, N. Tunicamycin-Induced Endoplasmic Reticulum Stress Promotes Breast Cancer Cell MDA-MB-231 Apoptosis through Inhibiting Wnt/beta-Catenin Signaling Pathway. J. Healthc. Eng. 2021, 2021, 6394514. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Nami, B.; Donmez, H.; Kocak, N. Tunicamycin-induced endoplasmic reticulum stress reduces in vitro subpopulation and invasion of CD44+/CD24- phenotype breast cancer stem cells. Exp. Toxicol. Pathol. 2016, 68, 419–426. [Google Scholar] [CrossRef]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- You, S.; Li, W.; Guan, Y. Tunicamycin inhibits colon carcinoma growth and aggressiveness via modulation of the ERK-JNK-mediated AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 4203–4212. [Google Scholar] [CrossRef]
- Guo, X.; Meng, Y.; Sheng, X.; Guan, Y.; Zhang, F.; Han, Z.; Kang, Y.; Tai, G.; Zhou, Y.; Cheng, H. Tunicamycin enhances human colon cancer cells to TRAIL-induced apoptosis by JNK-CHOP-mediated DR5 upregulation and the inhibition of the EGFR pathway. Anti-Cancer Drugs 2017, 28, 66–74. [Google Scholar] [CrossRef]
- de Freitas Junior, J.C.; Silva Bdu, R.; de Souza, W.F.; de Araujo, W.M.; Abdelhay, E.S.; Morgado-Diaz, J.A. Inhibition of N-linked glycosylation by tunicamycin induces E-cadherin-mediated cell-cell adhesion and inhibits cell proliferation in undifferentiated human colon cancer cells. Cancer Chemother. Pharmacol. 2011, 68, 227–238. [Google Scholar] [CrossRef]
- Chern, Y.J.; Wong, J.C.T.; Cheng, G.S.W.; Yu, A.; Yin, Y.; Schaeffer, D.F.; Kennecke, H.F.; Morin, G.; Tai, I.T. The interaction between SPARC and GRP78 interferes with ER stress signaling and potentiates apoptosis via PERK/eIF2alpha and IRE1alpha/XBP-1 in colorectal cancer. Cell Death Dis. 2019, 10, 504. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Hsu, C.C.; Huang, T.T.; Lee, C.H.; Chen, J.L.; Yang, S.H.; Jiang, J.K.; Chen, W.S.; Lee, K.D.; Teng, H.W. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 2018, 12, 1706–1717. [Google Scholar] [CrossRef]
- Massion, P.P.; Carbone, D.P. The molecular basis of lung cancer: Molecular abnormalities and therapeutic implications. Respir. Res. 2003, 4, 12. [Google Scholar] [CrossRef]
- Ahmmed, B.; Khan, M.N.; Nisar, M.A.; Kampo, S.; Zheng, Q.; Li, Y.; Yan, Q. Tunicamycin enhances the suppressive effects of cisplatin on lung cancer growth through PTX3 glycosylation via AKT/NF-kappaB signaling pathway. Int. J. Oncol. 2019, 54, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Jehn, C.F.; Boulikas, T.; Kourvetaris, A.; Possinger, K.; Luftner, D. Pharmacokinetics of liposomal cisplatin (lipoplatin) in combination with 5-FU in patients with advanced head and neck cancer: First results of a phase III study. Anticancer. Res. 2007, 27, 471–475. [Google Scholar] [PubMed]
- Diamandis, E.P.; Goodglick, L.; Planque, C.; Thornquist, M.D. Pentraxin-3 is a novel biomarker of lung carcinoma. Clin. Cancer Res. 2011, 17, 2395–2399. [Google Scholar] [CrossRef] [PubMed]
- Bottazzi, B.; Inforzato, A.; Messa, M.; Barbagallo, M.; Magrini, E.; Garlanda, C.; Mantovani, A. The pentraxins PTX3 and SAP in innate immunity, regulation of inflammation and tissue remodelling. J. Hepatol. 2016, 64, 1416–1427. [Google Scholar] [CrossRef]
- Liu, L.; Li, S.; Qu, Y.; Bai, H.; Pan, X.; Wang, J.; Wang, Z.; Duan, J.; Zhong, J.; Wan, R.; et al. Ablation of ERO1A induces lethal endoplasmic reticulum stress responses and immunogenic cell death to activate anti-tumor immunity. Cell Rep. Med. 2023, 4, 101206. [Google Scholar] [CrossRef]
- Shin, S.; Solorzano, J.; Liauzun, M.; Pyronnet, S.; Bousquet, C.; Martineau, Y. Translational alterations in pancreatic cancer: A central role for the integrated stress response. NAR Cancer 2022, 4, zcac031. [Google Scholar] [CrossRef]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Wehner, M.R.; Matrisian, L.M.; Nead, K.T. Estimated Projection of US Cancer Incidence and Death to 2040. JAMA Netw. Open 2021, 4, e214708. [Google Scholar] [CrossRef] [PubMed]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Maity, G.; Haque, I.; Ghosh, A.; Sarkar, S.; Gupta, V.; Campbell, D.R.; Von Hoff, D.; Banerjee, S. Human pancreatic cancer progression: An anarchy among CCN-siblings. J. Cell Commun. Signal. 2016, 10, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.K.; Lee, S.; Salotti, J.; Basu, S.; Sakchaisri, K.; Xiao, Z.; Walia, V.; Westlake, C.J.; Morrison, D.K.; Johnson, P.F. Oncogenic RAS-Induced Perinuclear Signaling Complexes Requiring KSR1 Regulate Signal Transmission to Downstream Targets. Cancer Res. 2018, 78, 891–908. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.K.; Makdisi, W.F.; Weston, A.P.; Campbell, D.R. A two-step enriched-nested PCR technique enhances sensitivity for detection of codon 12 K-ras mutations in pancreatic adenocarcinoma. Pancreas 1997, 15, 16–24. [Google Scholar] [CrossRef]
- Brembeck, F.H.; Schreiber, F.S.; Deramaudt, T.B.; Craig, L.; Rhoades, B.; Swain, G.; Grippo, P.; Stoffers, D.A.; Silberg, D.G.; Rustgi, A.K. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer Res. 2003, 63, 2005–2009. [Google Scholar]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Birkeness, L.B.; Banerjee, S.; Quadir, M.; Banerjee, S.K. The role of CCNs in controlling cellular communication in the tumor microenvironment. J. Cell Commun. Signal. 2022, 17, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Dhar, G.; Mehta, S.; Banerjee, S.; Gardner, A.; McCarty, B.M.; Mathur, S.C.; Campbell, D.R.; Kambhampati, S.; Banerjee, S.K. Loss of WISP-2/CCN5 signaling in human pancreatic cancer: A potential mechanism for epithelial-mesenchymal-transition. Cancer Lett. 2007, 254, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Bannoura, S.F.; Khan, H.Y.; Azmi, A.S. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front. Oncol. 2022, 12, 1013902. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, S.; Saber-Ayad, M.; Abdel-Rahman, W.M. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 10219. [Google Scholar] [CrossRef] [PubMed]
- Dias Carvalho, P.; Guimaraes, C.F.; Cardoso, A.P.; Mendonca, S.; Costa, A.M.; Oliveira, M.J.; Velho, S. KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 2018, 78, 7–14. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Gross, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Cheng, N.C.; Vonderheide, R.H. Immune vulnerabilities of mutant KRAS in pancreatic cancer. Trends Cancer 2023, 9, 928–936. [Google Scholar] [CrossRef]
- Vonderheide, R.H. The Immune Revolution: A Case for Priming, Not Checkpoint. Cancer Cell 2018, 33, 563–569. [Google Scholar] [CrossRef]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in pancreatic cancer treatment: A new frontier. Therap Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef]
- Jacenik, D.; Karagiannidis, I.; Beswick, E.J. Th2 cells inhibit growth of colon and pancreas cancers by promoting anti-tumorigenic responses from macrophages and eosinophils. Br. J. Cancer 2023, 128, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Baylot, V.; Andrieu, C.; Katsogiannou, M.; Taieb, D.; Garcia, S.; Giusiano, S.; Acunzo, J.; Iovanna, J.; Gleave, M.; Garrido, C.; et al. OGX-427 inhibits tumor progression and enhances gemcitabine chemotherapy in pancreatic cancer. Cell Death Dis. 2011, 2, e221. [Google Scholar] [CrossRef]
- Topisirovic, I.; Ruiz-Gutierrez, M.; Borden, K.L. Phosphorylation of the eukaryotic translation initiation factor eIF4E contributes to its transformation and mRNA transport activities. Cancer Res. 2004, 64, 8639–8642. [Google Scholar] [CrossRef]
- Wendel, H.G.; Silva, R.L.; Malina, A.; Mills, J.R.; Zhu, H.; Ueda, T.; Watanabe-Fukunaga, R.; Fukunaga, R.; Teruya-Feldstein, J.; Pelletier, J.; et al. Dissecting eIF4E action in tumorigenesis. Genes Dev. 2007, 21, 3232–3237. [Google Scholar] [CrossRef]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Ge, Y.; Liu, C.; Zhang, X.; Jiang, J.; Wei, Y. ER stress inducer tunicamycin suppresses the self-renewal of glioma-initiating cell partly through inhibiting Sox2 translation. Oncotarget 2016, 7, 36395–36406. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.S.F.; Kwok, I.; Tan, L.; Shi, C.; Cerezo-Wallis, D.; Tan, Y.; Leong, K.; Calvo, G.F.; Yang, K.; Zhang, Y.; et al. Deterministic reprogramming of neutrophils within tumors. Science 2024, 383, eadf6493. [Google Scholar] [CrossRef]
- Awad, R.M.; De Vlaeminck, Y.; Maebe, J.; Goyvaerts, C.; Breckpot, K. Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression. Front. Immunol. 2018, 9, 1977. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Shiravand, Y.; Khodadadi, F.; Kashani, S.M.A.; Hosseini-Fard, S.R.; Hosseini, S.; Sadeghirad, H.; Ladwa, R.; O’Byrne, K.; Kulasinghe, A. Immune Checkpoint Inhibitors in Cancer Therapy. Curr. Oncol. 2022, 29, 3044–3060. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gogenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hammerling, G.J.; et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccin. Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune Checkpoint Blockade in Cancer Therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef]
- Postow, M.A.; Manuel, M.; Wong, P.; Yuan, J.; Dong, Z.; Liu, C.; Perez, S.; Tanneau, I.; Noel, M.; Courtier, A.; et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer 2015, 3, 23. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e487. [Google Scholar] [CrossRef]
- Cheong, J.E.; Sun, L. Targeting the IDO1/TDO2-KYN-AhR Pathway for Cancer Immunotherapy—Challenges and Opportunities. Trends Pharmacol. Sci. 2018, 39, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Chan, L.C.; Li, C.W.; Hsu, J.L.; Hung, M.C. Mechanisms Controlling PD-L1 Expression in Cancer. Mol. Cell 2019, 76, 359–370. [Google Scholar] [CrossRef]
- Sun, L.; Li, C.W.; Chung, E.M.; Yang, R.; Kim, Y.S.; Park, A.H.; Lai, Y.J.; Yang, Y.; Wang, Y.H.; Liu, J.; et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020, 80, 2298–2310. [Google Scholar] [CrossRef]
- Salatino, M.; Girotti, M.R.; Rabinovich, G.A. Glycans Pave the Way for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell 2018, 33, 155–157. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Chung, E.M.; Kim, Y.S.; Park, A.H.; Yao, J.; Cha, J.H.; Xia, W.; Chan, L.C.; Kim, T.; et al. Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 2018, 33, 187–201.e110. [Google Scholar] [CrossRef]
- Wang, Y.N.; Lee, H.H.; Hsu, J.L.; Yu, D.; Hung, M.C. The impact of PD-L1 N-linked glycosylation on cancer therapy and clinical diagnosis. J. Biomed. Sci. 2020, 27, 77. [Google Scholar] [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell 2019, 36, 168–178.e164. [Google Scholar] [CrossRef]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef]
- Yaghoubi, N.; Soltani, A.; Ghazvini, K.; Hassanian, S.M.; Hashemy, S.I. PD-1/ PD-L1 blockade as a novel treatment for colorectal cancer. Biomed. Pharmacother. 2019, 110, 312–318. [Google Scholar] [CrossRef]
- Samadi, M.; Kamrani, A.; Nasiri, H.; Shomali, N.; Heris, J.A.; Shahabi, P.; Ghahremanzadeh, K.; Mohammadinasab, R.; Sadeghi, M.; Sadeghvand, S.; et al. Cancer immunotherapy focusing on the role of interleukins: A comprehensive and updated study. Pathol. Res. Pract. 2023, 249, 154732. [Google Scholar] [CrossRef]
- Blomberg, O.S.; Spagnuolo, L.; Garner, H.; Voorwerk, L.; Isaeva, O.I.; van Dyk, E.; Bakker, N.; Chalabi, M.; Klaver, C.; Duijst, M.; et al. IL-5-producing CD4+ T cells and eosinophils cooperate to enhance response to immune checkpoint blockade in breast cancer. Cancer Cell 2023, 41, 106–123.e110. [Google Scholar] [CrossRef]
- Bohnacker, S.; Hildenbrand, K.; Aschenbrenner, I.; Muller, S.I.; Bieren, J.E.; Feige, M.J. Influence of glycosylation on IL-12 family cytokine biogenesis and function. Mol. Immunol. 2020, 126, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Takatsu, K. Interleukin-5 and IL-5 receptor in health and diseases. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2011, 87, 463–485. [Google Scholar] [CrossRef] [PubMed]
- Grisaru-Tal, S.; Munitz, A. T cell-eosinophil crosstalk-A new road for effective immune checkpoint blockade in breast cancer? Cancer Cell 2023, 41, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Tsujimoto, M.; Tsuruoka, N.; Sugo, T.; Endo, T.; Kobata, A. Role of sugar chains in the in-vitro activity of recombinant human interleukin 5. Eur. J. Biochem. 1993, 211, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Kunimoto, D.Y.; Allison, K.C.; Watson, C.; Fuerst, T.; Armstrong, G.D.; Paul, W.; Strober, W. High-level production of murine interleukin-5 (IL-5) utilizing recombinant baculovirus expression. Purification of the rIL-5 and its use in assessing the biologic role of IL-5 glycosylation. Cytokine 1991, 3, 224–230. [Google Scholar] [CrossRef]
- Greenfeder, S.; Umland, S.P.; Cuss, F.M.; Chapman, R.W.; Egan, R.W. Th2 cytokines and asthma. The role of interleukin-5 in allergic eosinophilic disease. Respir. Res. 2001, 2, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Hirai, K.; Yamaguchi, M.; Misaki, Y.; Takaishi, T.; Ohta, K.; Morita, Y.; Ito, K.; Miyamoto, T. Enhancement of human basophil histamine release by interleukin 5. J. Exp. Med. 1990, 172, 1525–1528. [Google Scholar] [CrossRef] [PubMed]
- Resnick, M.B.; Weller, P.F. Mechanisms of eosinophil recruitment. Am. J. Respir. Cell Mol. Biol. 1993, 8, 349–355. [Google Scholar] [CrossRef]
- Alam, A.; Levanduski, E.; Denz, P.; Villavicencio, H.S.; Bhatta, M.; Alhorebi, L.; Zhang, Y.; Gomez, E.C.; Morreale, B.; Senchanthisai, S.; et al. Fungal mycobiome drives IL-33 secretion and type 2 immunity in pancreatic cancer. Cancer Cell 2022, 40, 153–167.e111. [Google Scholar] [CrossRef]
- Davis, B.P.; Rothenberg, M.E. Eosinophils and cancer. Cancer Immunol. Res. 2014, 2, 1–8. [Google Scholar] [CrossRef]
- Gitto, S.B.; Beardsley, J.M.; Nakkina, S.P.; Oyer, J.L.; Cline, K.A.; Litherland, S.A.; Copik, A.J.; Khaled, A.S.; Fanaian, N.; Arnoletti, J.P.; et al. Identification of a novel IL-5 signaling pathway in chronic pancreatitis and crosstalk with pancreatic tumor cells. Cell Commun. Signal. 2020, 18, 95. [Google Scholar] [CrossRef]
- Li, A.; Song, N.J.; Riesenberg, B.P.; Li, Z. The Emerging Roles of Endoplasmic Reticulum Stress in Balancing Immunity and Tolerance in Health and Diseases: Mechanisms and Opportunities. Front. Immunol. 2019, 10, 3154. [Google Scholar] [CrossRef] [PubMed]
- Foufelle, F.; Fromenty, B. Role of endoplasmic reticulum stress in drug-induced toxicity. Pharmacol. Res. Perspect. 2016, 4, e00211. [Google Scholar] [CrossRef] [PubMed]
- Vogel, P.; Stynes, B.A.; Coackley, W.; Yeoh, G.T.; Petterson, D.S. Glycolipid toxins from parasitised annual ryegrass: A comparison with tunicamycin. Biochem. Biophys. Res. Commun. 1982, 105, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.Y.; Korolev, V.V. Specific toxicity of tunicamycin in induction of programmed cell death of sympathetic neurons. Exp. Neurol. 1996, 137, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Price, N.P.; Hartman, T.M.; Li, J.; Velpula, K.K.; Naumann, T.A.; Guda, M.R.; Yu, B.; Bischoff, K.M. Modified tunicamycins with reduced eukaryotic toxicity that enhance the antibacterial activity of beta-lactams. J. Antibiot. 2017, 70, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Raj, S.; Jose, S.; Sumod, U.S.; Sabitha, M. Nanotechnology in cosmetics: Opportunities and challenges. J. Pharm. Bioallied Sci. 2012, 4, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Rajput, P.; Kumar, P.; Priya, A.K.; Kumari, S.; Shiade, S.R.G.; Rajput, V.D.; Fathi, A.; Pradhan, A.; Sarfraz, R.; Sushkova, S.; et al. Nanomaterials and biochar mediated remediation of emerging contaminants. Sci. Total Environ. 2024, 916, 170064. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles:applications and hazards. Int. J. Nanomed. 2008, 3, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Evers, M.J.W.; van de Wakker, S.I.; de Groot, E.M.; de Jong, O.G.; Gitz-Francois, J.J.J.; Seinen, C.S.; Sluijter, J.P.G.; Schiffelers, R.M.; Vader, P. Functional siRNA Delivery by Extracellular Vesicle-Liposome Hybrid Nanoparticles. Adv. Healthc. Mater. 2022, 11, e2101202. [Google Scholar] [CrossRef] [PubMed]
- Gargioni, E.; Schulz, F.; Raabe, A.; Burdak-Rothkamm, S.; Rieckmann, T.; Rothkamm, K. Targeted nanoparticles for tumour radiotherapy enhancement-the long dawn of a golden era? Ann. Transl. Med. 2016, 4, 523. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Zhao, W.; Li, B.; Zhang, X.; Zhang, C.; Bratasz, A.; Deng, B.; McComb, D.W.; Dong, Y. Co-delivery of mRNA and SPIONs through amino-ester nanomaterials. Nano Res. 2018, 11, 5596–5603. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Gao, J.; Karp, J.M.; Langer, R.; Joshi, N. The Future of Drug Delivery. Chem. Mater. 2023, 35, 359–363. [Google Scholar] [CrossRef]
- Mura, S.; Nicolas, J.; Couvreur, P. Stimuli-responsive nanocarriers for drug delivery. Nat. Mater. 2013, 12, 991–1003. [Google Scholar] [CrossRef]
- Cheng, R.; Meng, F.; Deng, C.; Klok, H.A.; Zhong, Z. Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials 2013, 34, 3647–3657. [Google Scholar] [CrossRef]
- Han, Y.; Wen, P.; Li, J.; Kataoka, K. Targeted nanomedicine in cisplatin-based cancer therapeutics. J. Control. Release 2022, 345, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Din, F.U.; Choi, J.Y.; Kim, D.W.; Mustapha, O.; Kim, D.S.; Thapa, R.K.; Ku, S.K.; Youn, Y.S.; Oh, K.T.; Yong, C.S.; et al. Irinotecan-encapsulated double-reverse thermosensitive nanocarrier system for rectal administration. Drug Deliv. 2017, 24, 502–510. [Google Scholar] [CrossRef]
- Roy, B.; Murugesan, K.; Mathanmohun, M.; Sathian, B. Impact of nanoparticles on human health and disease. Nepal J. Epidemiol. 2023, 13, 1294–1297. [Google Scholar] [CrossRef]
- Chen, W.; Wang, W.; Xie, Z.; Centurion, F.; Sun, B.; Paterson, D.J.; Tsao, S.C.; Chu, D.; Shen, Y.; Mao, G.; et al. Size-Dependent Penetration of Nanoparticles in Tumor Spheroids: A Multidimensional and Quantitative Study of Transcellular and Paracellular Pathways. Small 2023, e2304693. [Google Scholar] [CrossRef]
- Banerjee, A.; Johnson, K.T.; Banerjee, I.A.; Banerjee, D.K. Nanoformulation enhances anti-angiogenic efficacy of tunicamycin. Transl. Cancer Res. 2013, 2, 240–255. [Google Scholar]
- Ray, P.; Dutta, D.; Haque, I.; Nair, G.; Mohammed, J.; Parmer, M.; Kale, N.; Orr, M.; Jain, P.; Banerjee, S.; et al. pH-Sensitive Nanodrug Carriers for Codelivery of ERK Inhibitor and Gemcitabine Enhance the Inhibition of Tumor Growth in Pancreatic Cancer. Mol. Pharm. 2021, 18, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.; Haideri, N.; Haque, I.; Mohammed, O.; Chakrabarty, S.; Banerjee, S.; Quadir, M.; Brinker, A.E.; Banerjee, S.K. The Impact of Nanoparticles on the Immune System: A Gray Zone of Nanomedicine. J. Immunol. Sci. 2021, 5, 14. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng. Transl. Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, Z.; Gu, Z. Bioinspired and Biomimetic Nanomedicines. Acc. Chem. Res. 2019, 52, 1255–1264. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, P.D.; Tripathi, A.; Ansari, K.M.; Shanker, R.; Das, M. Impact of nanoparticles on the immune system. J. Biomed. Nanotechnol. 2011, 7, 193–194. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, S.; Ansari, A.A.; Upadhyay, S.P.; Mettman, D.J.; Hibdon, J.R.; Quadir, M.; Ghosh, P.; Kambhampati, A.; Banerjee, S.K. Benefits and Pitfalls of a Glycosylation Inhibitor Tunicamycin in the Therapeutic Implication of Cancers. Cells 2024, 13, 395. https://doi.org/10.3390/cells13050395
Banerjee S, Ansari AA, Upadhyay SP, Mettman DJ, Hibdon JR, Quadir M, Ghosh P, Kambhampati A, Banerjee SK. Benefits and Pitfalls of a Glycosylation Inhibitor Tunicamycin in the Therapeutic Implication of Cancers. Cells. 2024; 13(5):395. https://doi.org/10.3390/cells13050395
Chicago/Turabian StyleBanerjee, Snigdha, Affan A. Ansari, Sunil P. Upadhyay, Daniel J. Mettman, Jamie R. Hibdon, Mohiuddin Quadir, Pratyusha Ghosh, Anjali Kambhampati, and Sushanta K. Banerjee. 2024. "Benefits and Pitfalls of a Glycosylation Inhibitor Tunicamycin in the Therapeutic Implication of Cancers" Cells 13, no. 5: 395. https://doi.org/10.3390/cells13050395