

The Contribution of Innate Immunity in Large-Vessel Vasculitis: Detangling New Pathomechanisms beyond the Onset of Vascular Inflammation
 ,
,  and
and
Abstract
:1. Introduction
2. Dendritic Cells Drive the Dysregulated Immune Response within the Arterial Wall
3. Macrophages Are Critically Involved across All Phases of Vascular Inflammation
4. Polymorphonuclear Leukocytes: An Heterogenous Group of Cells Deeply Involved in LVV Pathogenesis
5. Mast Cells Display Pleiotropic Functions in LVV with a Possible Dichotomic Behavior in Boosting Inflammation
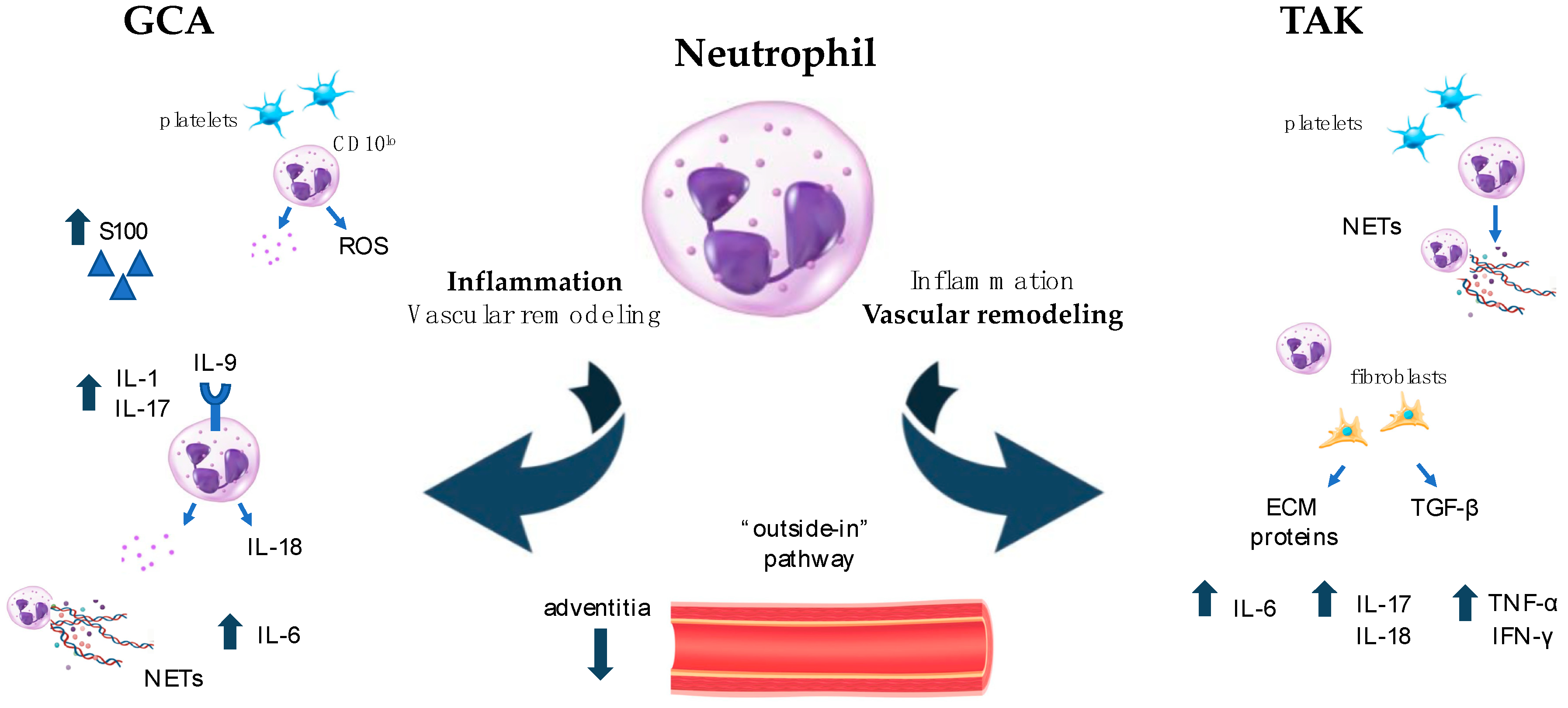
6. Neutrophils: Old Cells with Newly Described Functions and Plasticity in Driving Both Inflammation and Fibrosis in LVV
7. Other Immune Pathways Involved in Innate Immunity: Focus on Complement System and Inflammasome Activation in LVV
7.1. Complement System
7.2. Inflammasome
8. Conclusions and Future Therapeutic Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Pugh, D.; Karabayas, M.; Basu, N.; Cid, M.C.; Goel, R.; Goodyear, C.S.; Grayson, P.C.; McAdoo, S.P.; Mason, J.C.; Owen, C.; et al. Large-Vessel Vasculitis. Nat. Rev. Dis. Primers 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef]
- Svensson, L.G.; Arafat, A.; Roselli, E.E.; Idrees, J.; Clifford, A.; Tan, C.; Hoffman, G.; Eng, C.; Langford, C.; Rodriguez, E.R.; et al. Inflammatory Disease of the Aorta: Patterns and Classification of Giant Cell Aortitis, Takayasu Arteritis, and Nonsyndromic Aortitis. J. Thorac. Cardiovasc. Surg. 2015, 149, S170–S175. [Google Scholar] [CrossRef]
- Salvarani, C.; Macchioni, P.; Rossi, F.; Castri, C.; Capozzoli, N.; Baricchi, R.; Boiardi, L.; Chiaravalloti, F.; Portioli, I.; Zizzi, F.; et al. Epidemiologic and Immunogenetic Aspects of Polymyalgia Rheumatica and Giant Cell Arteritis in Northern Italy. Arthritis Rheum. 1991, 34, 351–356. [Google Scholar] [CrossRef]
- Zaldivar Villon, M.L.F.; De La Rocha, J.A.L.; Espinoza, L.R. Takayasu Arteritis: Recent Developments. Curr. Rheumatol. Rep. 2019, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.V.; Liao, Y.J.; Kim, J.W.; Goronzy, J.J.; Weyand, C.M. Giant Cell Arteritis: Immune and Vascular Aging as Disease Risk Factors. Arthritis Res. Ther. 2011, 13, 231. [Google Scholar] [CrossRef]
- Gruver, A.; Hudson, L.; Sempowski, G. Immunosenescence of Ageing. J. Pathol. 2007, 211, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Brandes, J.C.; Schmidt, D.; Fulbright, J.W.; Goronzy, J.J. Functional Properties of CD4+CD28− T Cells in the Aging Immune System. Mech. Ageing Dev. 1998, 102, 131–147. [Google Scholar] [CrossRef]
- Adrover, J.M.; Nicolás-Ávila, J.A.; Hidalgo, A. Aging: A Temporal Dimension for Neutrophils. Trends Immunol. 2016, 37, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory Cytokines, Aging, and Age-Related Diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Agrawal, S.; Gupta, S. Role of Dendritic Cells in Inflammation and Loss of Tolerance in the Elderly. Front. Immunol. 2017, 8, 896. [Google Scholar] [CrossRef]
- Akiyama, M.; Ohtsuki, S.; Berry, G.J.; Liang, D.H.; Goronzy, J.J.; Weyand, C.M. Innate and Adaptive Immunity in Giant Cell Arteritis. Front. Immunol. 2021, 11, 621098. [Google Scholar] [CrossRef]
- Galli, E.; Muratore, F.; Boiardi, L.; Restuccia, G.; Cavazza, A.; Catanoso, M.; Macchioni, P.; Spaggiari, L.; Casali, M.; Pipitone, N.; et al. Significance of Inflammation Restricted to Adventitial/Periadventitial Tissue on Temporal Artery Biopsy. Semin. Arthritis Rheum. 2020, 50, 1064–1072. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Goronzy, J.J. Immune Mechanisms in Medium and Large-Vessel Vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Kermani, T.A. Takayasu Arteritis and Giant Cell Arteritis: Are They a Spectrum of the Same Disease? Int. J. Rheum. Dis. 2019, 22, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Younge, B.R.; Olshen, R.A.; Goronzy, J.J.; Weyand, C.M. Th17 and Th1 T-Cell Responses in Giant Cell Arteritis. Circulation 2010, 121, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, D.; Garrido, M.; Comarmond, C.; Desbois, A.C.; Domont, F.; Savey, L.; Terrier, B.; Geri, G.; Rosenzwajg, M.; Klatzmann, D.; et al. Th1 and Th17 Cytokines Drive Inflammation in Takayasu Arteritis. Arthritis Rheumatol. 2015, 67, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Watanabe, R.; Zhang, H.; Akiyama, M.; Berry, G.J.; Goronzy, J.J. Cytokines, Growth Factors and Proteases in Medium and Large Vessel Vasculitis. Clin. Immunol. 2019, 206, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Watanabe, R.; Berry, G.J.; Vaglio, A.; Liao, Y.J.; Warrington, K.J.; Goronzy, J.J.; Weyand, C.M. Immunoinhibitory Checkpoint Deficiency in Medium and Large Vessel Vasculitis. Proc. Natl. Acad. Sci. USA 2017, 114, E970–E979. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, C.; La Barbera, L.; Miceli, G.; Tuttolomondo, A.; Guggino, G. The Innate Face of Giant Cell Arteritis: Insight into Cellular and Molecular Innate Immunity Pathways to Unravel New Possible Biomarkers of Disease. Front. Mol. Med. 2022, 2, 933161. [Google Scholar] [CrossRef]
- Ma-Krupa, W.; Jeon, M.-S.; Spoerl, S.; Tedder, T.F.; Goronzy, J.J.; Weyand, C.M. Activation of Arterial Wall Dendritic Cells and Breakdown of Self-Tolerance in Giant Cell Arteritis. J. Exp. Med. 2004, 199, 173–183. [Google Scholar] [CrossRef]
- Watanabe, R.; Berry, G.J.; Liang, D.H.; Goronzy, J.J.; Weyand, C.M. Cellular Signaling Pathways in Medium and Large Vessel Vasculitis. Front. Immunol. 2020, 11, 587089. [Google Scholar] [CrossRef]
- Hoffman, G.S.; Getz, T.M.; Padmanabhan, R.; Villa-Forte, A.; Clifford, A.H.; Funchain, P.; Sankunny, M.; Perry, J.D.; Blandford, A.; Kosmorsky, G.; et al. The Microbiome of Temporal Arteries. Pathog. Immun. 2019, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Piggott, K.; Biousse, V.; Newman, N.J.; Goronzy, J.J.; Weyand, C.M. Vascular Damage in Giant Cell Arteritis. Autoimmunity 2009, 42, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Ma-Krupa, W.; Gewirtz, A.T.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Toll-Like Receptors 4 and 5 Induce Distinct Types of Vasculitis. Circ. Res. 2009, 104, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Rhee, R.L.; Grayson, P.C.; Merkel, P.A.; Tomasson, G. Infections and the Risk of Incident Giant Cell Arteritis: A Population-Based, Case-Control Study. Ann. Rheum. Dis. 2017, 76, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- La Barbera, L.; Macaluso, F.; Fasano, S.; Grasso, G.; Ciccia, F.; Guggino, G. Microbiome Changes in Connective Tissue Diseases and Vasculitis: Focus on Metabolism and Inflammation. Int. J. Mol. Sci. 2022, 23, 6532. [Google Scholar] [CrossRef] [PubMed]
- Weyand, C.M.; Ma-Krupa, W.; Pryshchep, O.; Gröschel, S.; Bernardino, R.; Goronzy, J.J. Vascular Dendritic Cells in Giant Cell Arteritis. Ann. N. Y. Acad. Sci. 2005, 1062, 195–208. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Brahmer, J.R.; Callahan, M.K.; Flores-Chávez, A.; Keegan, N.; Khamashta, M.A.; Lambotte, O.; Mariette, X.; Prat, A.; Suárez-Almazor, M.E. Immune-Related Adverse Events of Checkpoint Inhibitors. Nat. Rev. Dis. Primers 2020, 6, 38. [Google Scholar] [CrossRef]
- Micaily, I.; Chernoff, M. An Unknown Reaction to Pembrolizumab: Giant Cell Arteritis. Ann. Oncol. 2017, 28, 2621–2622. [Google Scholar] [CrossRef] [PubMed]
- Narala, R.; Reddy, S.A.; Mruthyunjaya, P. Giant Cell Arteritis Manifesting as Retinal Arterial Occlusion and Paracentral Acute Middle Maculopathy in a Patient on Pembrolizumab for Metastatic Uveal Melanoma. Am. J. Ophthalmol. Case Rep. 2020, 20, 100891. [Google Scholar] [CrossRef]
- Betrains, A.E.; Blockmans, D.E. Immune Checkpoint Inhibitor-Associated Polymyalgia Rheumatica/Giant Cell Arteritis Occurring in a Patient after Treatment with Nivolumab. J. Clin. Rheumatol. 2021, 27, S555–S556. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Zhang, H.; Berry, G.; Goronzy, J.J.; Weyand, C.M. Immune Checkpoint Dysfunction in Large and Medium Vessel Vasculitis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1052–H1059. [Google Scholar] [CrossRef] [PubMed]
- Mirault, T.; Guillet, H.; Messas, E. Immune response in Takayasu arteritis. Presse Médicale 2017, 46, e189–e196. [Google Scholar] [CrossRef] [PubMed]
- Inder, S.J.; Bobryshev, Y.V.; Cherian, S.M.; Albert Lord, R.S.; Masuda, K.; Yutani, C. Accumulation of Lymphocytes, Dendritic Cells, and Granulocytes in the Aortic Wall Affected by Takayasu’s Disease. Angiology 2000, 51, 565–579. [Google Scholar] [CrossRef]
- Kurata, A.; Saito, A.; Hashimoto, H.; Fujita, K.; Ohno, S.; Kamma, H.; Nagao, T.; Kobayashi, S.; Yamashina, A.; Kuroda, M. Difference in Immunohistochemical Characteristics between Takayasu Arteritis and Giant Cell Arteritis: It May Be Better to Distinguish Them in the Same Age. Mod. Rheumatol. 2019, 29, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Cavazza, A.; Muratore, F.; Boiardi, L.; Restuccia, G.; Pipitone, N.; Pazzola, G.; Tagliavini, E.; Ragazzi, M.; Rossi, G.; Salvarani, C. Inflamed Temporal Artery: Histologic Findings in 354 Biopsies, with Clinical Correlations. Am. J. Surg. Pathol. 2014, 38, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Esen, I.; Jiemy, W.F.; Van Sleen, Y.; Van Der Geest, K.S.M.; Sandovici, M.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Functionally Heterogenous Macrophage Subsets in the Pathogenesis of Giant Cell Arteritis: Novel Targets for Disease Monitoring and Treatment. J. Clin. Med. 2021, 10, 4958. [Google Scholar] [CrossRef]
- Van Sleen, Y.; Wang, Q.; Van Der Geest, K.S.M.; Westra, J.; Abdulahad, W.H.; Heeringa, P.; Boots, A.M.H.; Brouwer, E. Involvement of Monocyte Subsets in the Immunopathology of Giant Cell Arteritis. Sci. Rep. 2017, 7, 6553. [Google Scholar] [CrossRef]
- Santos, J.P.; Artigiani Neto, R.; Mangueira, C.L.P.; Filippi, R.Z.; Gutierrez, P.S.; Westra, J.; Brouwer, E.; De Souza, A.W.S. Associations between Clinical Features and Therapy with Macrophage Subpopulations and T Cells in Inflammatory Lesions in the Aorta from Patients with Takayasu Arteritis. Clin. Exp. Immunol. 2020, 202, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Seo, N.; Torii, M.; Ma, N.; Muraoka, D.; Tawara, I.; Masuya, M.; Tanaka, K.; Takei, Y.; Shiku, H.; et al. Interleukin-17 Induces an Atypical M2-Like Macrophage Subpopulation That Regulates Intestinal Inflammation. PLoS ONE 2014, 9, e108494. [Google Scholar] [CrossRef] [PubMed]
- Van Sleen, Y.; Jiemy, W.F.; Pringle, S.; Van Der Geest, K.S.M.; Abdulahad, W.H.; Sandovici, M.; Brouwer, E.; Heeringa, P.; Boots, A.M.H. A Distinct Macrophage Subset Mediating Tissue Destruction and Neovascularization in Giant Cell Arteritis: Implication of the YKL-40/Interleukin-13 Receptor A2 Axis. Arthritis Rheumatol. 2021, 73, 2327–2337. [Google Scholar] [CrossRef] [PubMed]
- Jiemy, W.F.; Van Sleen, Y.; Van Der Geest, K.S.; Ten Berge, H.A.; Abdulahad, W.H.; Sandovici, M.; Boots, A.M.; Heeringa, P.; Brouwer, E. Distinct Macrophage Phenotypes Skewed by Local Granulocyte Macrophage Colony-stimulating Factor (GM-CSF) and Macrophage Colony-stimulating Factor (M-CSF) Are Associated with Tissue Destruction and Intimal Hyperplasia in Giant Cell Arteritis. Clin. Transl. Immunol. 2020, 9, e1164. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pla, A.; Bosch-Gil, J.A.; Rosselló-Urgell, J.; Huguet-Redecilla, P.; Stone, J.H.; Vilardell-Tarres, M. Metalloproteinase-2 and -9 in Giant Cell Arteritis: Involvement in Vascular Remodeling. Circulation 2005, 112, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Maeda, T.; Zhang, H.; Berry, G.J.; Zeisbrich, M.; Brockett, R.; Greenstein, A.E.; Tian, L.; Goronzy, J.J.; Weyand, C.M. MMP (Matrix Metalloprotease)-9–Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis. Circ. Res. 2018, 123, 700–715. [Google Scholar] [CrossRef]
- Kaiser, M.; Younge, B.; Björnsson, J.; Goronzy, J.J.; Weyand, C.M. Formation of New Vasa Vasorum in Vasculitis. Am. J. Pathol. 1999, 155, 765–774. [Google Scholar] [CrossRef]
- Watanabe, R.; Hilhorst, M.; Zhang, H.; Zeisbrich, M.; Berry, G.J.; Wallis, B.B.; Harrison, D.G.; Giacomini, J.C.; Goronzy, J.J.; Weyand, C.M. Glucose Metabolism Controls Disease-Specific Signatures of Macrophage Effector Functions. JCI Insight 2018, 3, e123047. [Google Scholar] [CrossRef]
- Ohtsuki, S.; Wang, C.; Watanabe, R.; Zhang, H.; Akiyama, M.; Bois, M.C.; Maleszewski, J.J.; Warrington, K.J.; Berry, G.J.; Goronzy, J.J.; et al. Deficiency of the CD155-CD96 Immune Checkpoint Controls IL-9 Production in Giant Cell Arteritis. Cell Rep. Med. 2023, 4, 101012. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Rizzo, A.; Guggino, G.; Cavazza, A.; Alessandro, R.; Maugeri, R.; Cannizzaro, A.; Boiardi, L.; Iacopino, D.G.; Salvarani, C.; et al. Difference in the Expression of IL-9 and IL-17 Correlates with Different Histological Pattern of Vascular Wall Injury in Giant Cell Arteritis. Rheumatology 2015, 54, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Khoury, P.; Grayson, P.C.; Klion, A.D. Eosinophils in Vasculitis: Characteristics and Roles in Pathogenesis. Nat. Rev. Rheumatol. 2014, 10, 474–483. [Google Scholar] [CrossRef]
- Schnabel, A.; Csernok, E.; Braun, J.; Gross, W.L. Activation of Neutrophils, Eosinophils, and Lymphocytes in the Lower Respiratory Tract in Wegener’s Granulomatosis. Am. J. Respir. Crit. Care Med. 2000, 161, 399–405. [Google Scholar] [CrossRef]
- Terai, M.; Yasukawa, K.; Honda, T.; Jibiki, T.; Hirano, K.; Sato, J.; Ishiwada, N.; Seguchi, M.; Ueda, S.; Kohno, Y. Peripheral Blood Eosinophilia and Eosinophil Accumulation in Coronary Microvessels in Acute Kawasaki Disease. Pediatr. Infect. Dis. J. 2002, 21, 777–780. [Google Scholar] [CrossRef]
- Bahrami, S.; Malone, J.C.; Webb, K.G.; Callen, J.P. Tissue Eosinophilia as an Indicator of Drug-Induced Cutaneous Small-Vessel Vasculitis. Arch. Dermatol. 2006, 142, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Fujiya, M.; Kashima, S.; Sugiyama, Y.; Iwama, T.; Ijiri, M.; Tanaka, K.; Takahashi, K.; Ando, K.; Nomura, Y.; Ueno, N.; et al. Takayasu’s Arteritis Associated with Eosinophilic Gastroenteritis, Possibly via the Overactivation of Th17. Gut Pathog. 2018, 10, 22. [Google Scholar] [CrossRef] [PubMed]
- Benavides, R.; Ramírez-Peralta, A.F.; Muñoz-Urbano, M.; Mejía, L.; Cardona-Cardona, A.F.; Muñoz-Vahos, C.H. Temporal Arteritis Caused by Eosinophilic Vasculitis Associated with a Lymphocytic Variant of the Hypereosinophilic Syndrome: A Case Report. Rev. Colomb. Reumatol. 2023; in press. [Google Scholar] [CrossRef]
- Beaven, M.A. Our Perception of the Mast Cell from Paul Ehrlich to Now. Eur. J. Immunol. 2009, 39, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.F. The Role of Mast Cells in Wound Healing. Int. Wound J. 2010, 7, 55–61. [Google Scholar] [CrossRef] [PubMed]
- De Souza Junior, D.; Mazucato, V.; Santana, A.; Oliver, C.; Jamur, M. Mast Cells Interact with Endothelial Cells to Accelerate In Vitro Angiogenesis. Int. J. Mol. Sci. 2017, 18, 2674. [Google Scholar] [CrossRef] [PubMed]
- Mäyränpää, M.I.; Trosien, J.A.; Fontaine, V.; Folkesson, M.; Kazi, M.; Eriksson, P.; Swedenborg, J.; Hedin, U. Mast Cells Associate with Neovessels in the Media and Adventitia of Abdominal Aortic Aneurysms. J. Vasc. Surg. 2009, 50, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Sibilano, R.; Frossi, B.; Pucillo, C.E. Mast Cell Activation: A Complex Interplay of Positive and Negative Signaling Pathways. Eur. J. Immunol. 2014, 44, 2558–2566. [Google Scholar] [CrossRef]
- Levick, S.P.; Melendez, G.C.; Plante, E.; McLarty, J.L.; Brower, G.L.; Janicki, J.S. Cardiac Mast Cells: The Centrepiece in Adverse Myocardial Remodelling. Cardiovasc. Res. 2011, 89, 12–19. [Google Scholar] [CrossRef]
- Bot, I.; Biessen, E. Mast Cells in Atherosclerosis. Thromb. Haemost. 2011, 106, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Alessandro, R.; Rizzo, A.; Raimondo, S.; Giardina, A.; Raiata, F.; Boiardi, L.; Cavazza, A.; Guggino, G.; De Leo, G.; et al. IL-33 Is Overexpressed in the Inflamed Arteries of Patients with Giant Cell Arteritis. Ann. Rheum. Dis. 2013, 72, 258–264. [Google Scholar] [CrossRef]
- Nakae, S.; Suto, H.; Iikura, M.; Kakurai, M.; Sedgwick, J.D.; Tsai, M.; Galli, S.J. Mast Cells Enhance T Cell Activation: Importance of Mast Cell Costimulatory Molecules and Secreted TNF. J. Immunol. 2006, 176, 2238–2248. [Google Scholar] [CrossRef] [PubMed]
- Mäyränpää, M.I.; Trosien, J.A.; Nikkari, S.T.; Kovanen, P.T. Mast Cells Associate with T-Cells and Neointimal Microvessels in Giant Cell Arteritis. Clin. Exp. Rheumatol. 2008, 26, S63–S66. [Google Scholar] [PubMed]
- Misra, D.P.; Singh, K.; Sharma, A.; Agarwal, V. Arterial Wall Fibrosis in Takayasu Arteritis and Its Potential for Therapeutic Modulation. Front. Immunol. 2023, 14, 1174249. [Google Scholar] [CrossRef]
- Le Joncour, A.; Desbois, A.-C.; Leroyer, A.S.; Tellier, E.; Régnier, P.; Maciejewski-Duval, A.; Comarmond, C.; Barete, S.; Arock, M.; Bruneval, P.; et al. Mast Cells Drive Pathologic Vascular Lesions in Takayasu Arteritis. J. Allergy Clin. Immunol. 2022, 149, 292–301.e3. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.-Y.; Smrž, D.; Desai, A.; Bandara, G.; Ito, T.; Iwaki, S.; Kang, J.-H.; Andrade, M.V.; Hilderbrand, S.C.; Brown, J.M.; et al. IL-33 Induces a Hyporesponsive Phenotype in Human and Mouse Mast Cells. J. Immunol. 2013, 190, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-F.; Lind, E.F.; Gondek, D.C.; Bennett, K.A.; Gleeson, M.W.; Pino-Lagos, K.; Scott, Z.A.; Coyle, A.J.; Reed, J.L.; Van Snick, J.; et al. Mast Cells Are Essential Intermediaries in Regulatory T-Cell Tolerance. Nature 2006, 442, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, D.; Mustelin, T.; Lood, C. Role of Neutrophils in Systemic Vasculitides. Front. Immunol. 2020, 11, 619705. [Google Scholar] [CrossRef]
- Aymonnier, K.; Amsler, J.; Lamprecht, P.; Salama, A.; Witko-Sarsat, V. The Neutrophil: A Key Resourceful Agent in Immune-mediated Vasculitis. Immunol. Rev. 2023, 314, 326–356. [Google Scholar] [CrossRef]
- Nakazawa, D.; Masuda, S.; Tomaru, U.; Ishizu, A. Pathogenesis and Therapeutic Interventions for ANCA-Associated Vasculitis. Nat. Rev. Rheumatol. 2019, 15, 91–101. [Google Scholar] [CrossRef]
- Michailidou, D.; Duvvuri, B.; Kuley, R.; Cuthbertson, D.; Grayson, P.C.; Khalidi, N.A.; Koening, C.L.; Langford, C.A.; McAlear, C.A.; Moreland, L.W.; et al. Neutrophil Activation in Patients with Anti-Neutrophil Cytoplasmic Autoantibody-Associated Vasculitis and Large-Vessel Vasculitis. Arthritis Res. Ther. 2022, 24, 160. [Google Scholar] [CrossRef]
- Foell, D.; Hernández-Rodríguez, J.; Sánchez, M.; Vogl, T.; Cid, M.C.; Roth, J. Early Recruitment of Phagocytes Contributes to the Vascular Inflammation of Giant Cell Arteritis. J. Pathol. 2004, 204, 311–316. [Google Scholar] [CrossRef]
- Springer, J.M.; Monach, P.; Cuthbertson, D.; Carette, S.; Khalidi, N.A.; McAlear, C.A.; Pagnoux, C.; Seo, P.; Warrington, K.J.; Ytterberg, S.R.; et al. Serum S100 Proteins as a Marker of Disease Activity in Large Vessel Vasculitis. J. Clin. Rheumatol. 2018, 24, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Ly, K.-H.; Stirnemann, J.; Liozon, E.; Michel, M.; Fain, O.; Fauchais, A.-L. Interleukin-1 Blockade in Refractory Giant Cell Arteritis. Jt. Bone Spine 2014, 81, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Lindén, A. Interleukin-17 Family Members and Inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Haroche, J.; Mathian, A.; Gorochov, G.; Amoura, Z. Pathogenesis of Takayasu’s Arteritis: A 2011 Update. Autoimmun. Rev. 2011, 11, 61–67. [Google Scholar] [CrossRef]
- Michailidou, D.; Kuley, R.; Wang, T.; Hermanson, P.; Grayson, P.C.; Cuthbertson, D.; Khalidi, N.A.; Koening, C.L.; Langford, C.A.; McAlear, C.A.; et al. Neutrophil Extracellular Trap Formation in Anti-Neutrophil Cytoplasmic Antibody-Associated and Large-Vessel Vasculitis. Clin. Immunol. 2023, 249, 109274. [Google Scholar] [CrossRef] [PubMed]
- Palamidas, D.A.; Argyropoulou, O.D.; Georgantzoglou, N.; Karatza, E.; Xingi, E.; Kapsogeorgou, E.K.; Anagnostopoulos, C.D.; Lazaris, A.C.; Ritis, K.; Goules, A.V.; et al. Neutrophil Extracellular Traps in Giant Cell Arteritis Biopsies: Presentation, Localization and Co-Expression with Inflammatory Cytokines. Rheumatology 2022, 61, 1639–1644. [Google Scholar] [CrossRef]
- Matsumoto, K.; Yasuoka, H.; Yoshimoto, K.; Suzuki, K.; Takeuchi, T. Platelet CXCL4 Mediates Neutrophil Extracellular Traps Formation in ANCA-Associated Vasculitis. Sci. Rep. 2021, 11, 222. [Google Scholar] [CrossRef]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant Neutrophil Extracellular Traps in Thrombus of Patient with Microscopic Polyangiitis. Front. Immun. 2012, 3, 333. [Google Scholar] [CrossRef] [PubMed]
- Pillay, J.; Kamp, V.M.; Van Hoffen, E.; Visser, T.; Tak, T.; Lammers, J.-W.; Ulfman, L.H.; Leenen, L.P.; Pickkers, P.; Koenderman, L. A Subset of Neutrophils in Human Systemic Inflammation Inhibits T Cell Responses through Mac-1. J. Clin. Investig. 2012, 122, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, S.; Dalli, J.; Hollywood, J.; Mason, J.C.; Dasgupta, B.; Perretti, M. Investigational Analysis Reveals a Potential Role for Neutrophils in Giant-Cell Arteritis Disease Progression. Circ. Res. 2014, 114, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ai, Z.; Khoyratty, T.; Zec, K.; Eames, H.L.; Van Grinsven, E.; Hudak, A.; Morris, S.; Ahern, D.; Monaco, C.; et al. ROS-Producing Immature Neutrophils in Giant Cell Arteritis Are Linked to Vascular Pathologies. JCI Insight 2020, 5, e139163. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Wannemacher, J.; Christ, S.; Koopmans, T.; Kadri, S.; Zhao, J.; Gouda, M.; Ye, H.; Mück-Häusl, M.; Krenn, P.W.; et al. Neutrophils Direct Preexisting Matrix to Initiate Repair in Damaged Tissues. Nat. Immunol. 2022, 23, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Curaj, A.; Schumacher, D.; Rusu, M.; Staudt, M.; Li, X.; Simsekyilmaz, S.; Jankowski, V.; Jankowski, J.; Dumitraşcu, A.R.; Hausenloy, D.J.; et al. Neutrophils Modulate Fibroblast Function and Promote Healing and Scar Formation after Murine Myocardial Infarction. Int. J. Mol. Sci. 2020, 21, 3685. [Google Scholar] [CrossRef] [PubMed]
- Pulli, R.; Dorigo, W.; Pratesi, G.; Fargion, A.; Pratesi, C. Single-Center Experience on Endovascular Repair of Noninfected Extracranial Internal Carotid Artery Pseudoaneurysms. Ann. Vasc. Surg. 2013, 27, e13–e672. [Google Scholar] [CrossRef] [PubMed]
- Vignesh, P.; Rawat, A.; Sharma, M.; Singh, S. Complement in Autoimmune Diseases. Clin. Chim. Acta 2017, 465, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Trivioli, G.; Vaglio, A. The Rise of Complement in ANCA-Associated Vasculitis: From Marginal Player to Target of Modern Therapy. Clin. Exp. Immunol. 2020, 202, 403–406. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Hou, R.; Xu, K.; Han, Y.; Hu, J.; Zhang, Y.; Su, Y.; Gao, J.; Zhang, G.; Zhang, L. Pentraxin 3 Is More Accurate than C-Reactive Protein for Takayasu Arteritis Activity Assessment: A Systematic Review and Meta-Analysis. PLoS ONE 2021, 16, e0245612. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, V.S.; Brossart, P.; Warrington, K.J.; Kurts, C.; Sendtner, G.W.; Aden, C.A. The Role of Autoimmunity and Autoinflammation in Giant Cell Arteritis: A Systematic Literature Review. Autoimmun. Rev. 2023, 22, 103328. [Google Scholar] [CrossRef] [PubMed]
- Jayakanthan, K.; Gupta, A.N.; Mathew, J.; Ravindran, R.; Mahasampth, G.; Danda, D. Clinical Utility of Anti-C1q Antibody in Primary and Secondary Vasculitic Conditions. Int. J. Health Sci. 2017, 11, 3–6. [Google Scholar]
- Potlukova, E.; Kralikova, P. Complement Component C1q and Anti-C1q Antibodies in Theory and in Clinical Practice. Scand. J. Immunol. 2008, 67, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Rongyi, C.; Xiaojuan, D.; Jinghua, W.; Lingying, M.; Xiaomin, D.; Lili, M.; Huiyong, C.; Lindi, J.; Ying, S. High Level of Serum Complement 3 Is a Risk Factor for Vascular Stenosis Progression in TA Patients Receiving Tocilizumab: A Prospective Observational Study. Arthritis Res. Ther. 2023, 25, 137. [Google Scholar] [CrossRef]
- Chen, R.; Ma, L.; Lv, P.; Lin, J.; Li, C.; Yan, Y.; Jin, X.; Dai, X.; Ji, Z.; Chen, H.; et al. Serum Complement 3 Is a Potential Biomarker for Assessing Disease Activity in Takayasu Arteritis. Arthritis Res. Ther. 2021, 23, 63. [Google Scholar] [CrossRef]
- Ma, J.; Luo, X.; Wu, Q.; Chen, Z.; Kou, L.; Wang, H. Circulation Levels of Acute Phase Proteins in Patients with Takayasu Arteritis. J. Vasc. Surg. 2010, 51, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Takahashi, M. NLRP3 Inflammasome as a Key Driver of Vascular Disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef]
- Wortmann, M.; Peters, A.S.; Erhart, P.; Körfer, D.; Böckler, D.; Dihlmann, S. Inflammasomes in the Pathophysiology of Aortic Disease. Cells 2021, 10, 2433. [Google Scholar] [CrossRef] [PubMed]
- Sollberger, G.; Strittmatter, G.E.; Garstkiewicz, M.; Sand, J.; Beer, H.-D. Caspase-1: The Inflammasome and Beyond. Innate Immun. 2014, 20, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Jiang, W.; Liu, L.; Wang, X.; Ding, C.; Tian, Z.; Zhou, R. Dopamine Controls Systemic Inflammation through Inhibition of NLRP3 Inflammasome. Cell 2015, 160, 62–73. [Google Scholar] [CrossRef]
- Kimura, T.; Jain, A.; Choi, S.W.; Mandell, M.A.; Schroder, K.; Johansen, T.; Deretic, V. TRIM-Mediated Precision Autophagy Targets Cytoplasmic Regulators of Innate Immunity. J. Cell Biol. 2015, 210, 973–989. [Google Scholar] [CrossRef] [PubMed]
- Tamura, N.; Maejima, Y.; Matsumura, T.; Vega, R.B.; Amiya, E.; Ito, Y.; Shiheido-Watanabe, Y.; Ashikaga, T.; Komuro, I.; Kelly, D.P.; et al. Single-Nucleotide Polymorphism of the MLX Gene Is Associated With Takayasu Arteritis. Circ. Genom. Precis. Med. 2018, 11, e002296. [Google Scholar] [CrossRef] [PubMed]
- Agostini, L.; Martinon, F.; Burns, K.; McDermott, M.F.; Hawkins, P.N.; Tschopp, J. NALP3 Forms an IL-1β-Processing Inflammasome with Increased Activity in Muckle-Wells Autoinflammatory Disorder. Immunity 2004, 20, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Carmona, F.D.; Castañeda, S.; Solans, R.; Hernández-Rodríguez, J.; Cid, M.C.; Prieto-González, S.; Miranda-Filloy, J.A.; Rodríguez-Rodríguez, L.; Morado, I.C.; et al. Evidence of Association of the NLRP1 Gene with Giant Cell Arteritis. Ann. Rheum. Dis. 2013, 72, 628–630. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, Y.; Isobe, M.; Takei, S.; Tanaka, Y.; Ishii, T.; Yokota, S.; Nomura, A.; Yoshida, S.; Nishimoto, N. Efficacy and Safety of Tocilizumab in Patients with Refractory Takayasu Arteritis: Results from a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial in Japan (the TAKT Study). Ann. Rheum. Dis. 2018, 77, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; Brouwer, E.; Cid, M.C.; Dasgupta, B.; Rech, J.; et al. Trial of Tocilizumab in Giant-Cell Arteritis. N. Engl. J. Med. 2017, 377, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Cid, M.C.; Unizony, S.H.; Blockmans, D.; Brouwer, E.; Dagna, L.; Dasgupta, B.; Hellmich, B.; Molloy, E.; Salvarani, C.; Trapnell, B.C.; et al. Efficacy and Safety of Mavrilimumab in Giant Cell Arteritis: A Phase 2, Randomised, Double-Blind, Placebo-Controlled Trial. Ann. Rheum. Dis. 2022, 81, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and Regulation of Endothelial VEGF Receptor Signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Sano, M. Complexity of Inflammation in the Trajectory of Vascular Disease: Interleukin 6 and Beyond. Ann. Vasc. Dis. 2023, 16, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Terkeltaub, R.A. Colchicine Update: 2008. Semin. Arthritis Rheum. 2009, 38, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Abbate, A.; Toldo, S.; Marchetti, C.; Kron, J.; Van Tassell, B.W.; Dinarello, C.A. Interleukin-1 and the Inflammasome as Therapeutic Targets in Cardiovascular Disease. Circ. Res. 2020, 126, 1260–1280. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, F.; Marvisi, C.; Castrignanò, P.; Pipitone, N.; Salvarani, C. Comparing Treatment Options for Large Vessel Vasculitis. Expert. Rev. Clin. Immunol. 2022, 18, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Matsumoto, H.; Temmoku, J.; Fujita, Y.; Matsuoka, N.; Furuya, M.; Gunji, N.; Fujiwara, T.; Asano, T.; Onizawa, M.; et al. A Case of Takayasu Arteritis Complicated by Refractory Ulcerative Colitis Successfully Treated with Tofacitinib. Rheumatology 2020, 59, 1773–1775. [Google Scholar] [CrossRef]
- Kuwabara, S.; Tanimura, S.; Matsumoto, S.; Nakamura, H.; Horita, T. Successful Remission with Tofacitinib in a Patient with Refractory Takayasu Arteritis Complicated by Ulcerative Colitis. Ann. Rheum. Dis. 2020, 79, 1125–1126. [Google Scholar] [CrossRef]
- Watanabe, R.; Hashimoto, M. Perspectives of JAK Inhibitors for Large Vessel Vasculitis. Front. Immunol. 2022, 13, 881705. [Google Scholar] [CrossRef]


{kind=link}
| GCA | TAK | |
|---|---|---|
| Mast cells | ||
 VEGF-ST2 receptor VEGF-ST2 receptorPresence in the neointima | PDGF and TGF-β Wall fibrosis | |
| Neutrophils | ||
| Vasa vasorum and small vessels in the temporal arteries S100A12, IL-1β, IL-17, IL-9, IL-8, IL-6 | Aorta walls; IL-17, IL-8, IFN-γ, TNF-α Anti-histone antibodies Wall fibrosis | |
| NET activation Partnership with platelet activation | ||
| Macrophages | ||
| M1 in adventitia and media M2 in the media-intima border CD206+/YKL-40+/MMP-9+ in media FRβ+/CD206- in adventitia and intima  PD-L1 expression CD155-CD96 checkpoint PD-L1 expression CD155-CD96 checkpoint | All layers of the vessel M1 is more frequent in the aorta | |
| Dendritic cells (vasDCs) | ||
| Expression of CCR7, CCL19 and CCL21 TLR4 and TLR5 ligands promote vascular damage IL-6, IL-18, IL-23, IL-32, and IL-33 PD-L1 expression | T cells co-localize with DC in the adventitia of the aortic wall The involvement of DC remains marginal | |
| Complement | ||
| C3, C4b, and C4BP correlate with disease activity and vascular stenosis progression | ||
| Inflammasome | ||
| Genetic association of NLRP1 with GCA was found genotypinge a single-nucleotide polymorphism (rs8182352) | MLX-Q139R mutation promotes NLRP3 inflammasome formation, leading to increased IL-1β production | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Barbera, L.; Rizzo, C.; Camarda, F.; Miceli, G.; Tuttolomondo, A.; Guggino, G. The Contribution of Innate Immunity in Large-Vessel Vasculitis: Detangling New Pathomechanisms beyond the Onset of Vascular Inflammation. Cells 2024, 13, 271. https://doi.org/10.3390/cells13030271
La Barbera L, Rizzo C, Camarda F, Miceli G, Tuttolomondo A, Guggino G. The Contribution of Innate Immunity in Large-Vessel Vasculitis: Detangling New Pathomechanisms beyond the Onset of Vascular Inflammation. Cells. 2024; 13(3):271. https://doi.org/10.3390/cells13030271
Chicago/Turabian StyleLa Barbera, Lidia, Chiara Rizzo, Federica Camarda, Giuseppe Miceli, Antonino Tuttolomondo, and Giuliana Guggino. 2024. "The Contribution of Innate Immunity in Large-Vessel Vasculitis: Detangling New Pathomechanisms beyond the Onset of Vascular Inflammation" Cells 13, no. 3: 271. https://doi.org/10.3390/cells13030271