
The Activation of the Fibrodysplasia Ossificans Progressiva-Inducing ALK2-R206H Mutant Depends on the Distinct Homo-Oligomerization Patterns of ACVR2B and ACVR2A

 , , , and
, , , and 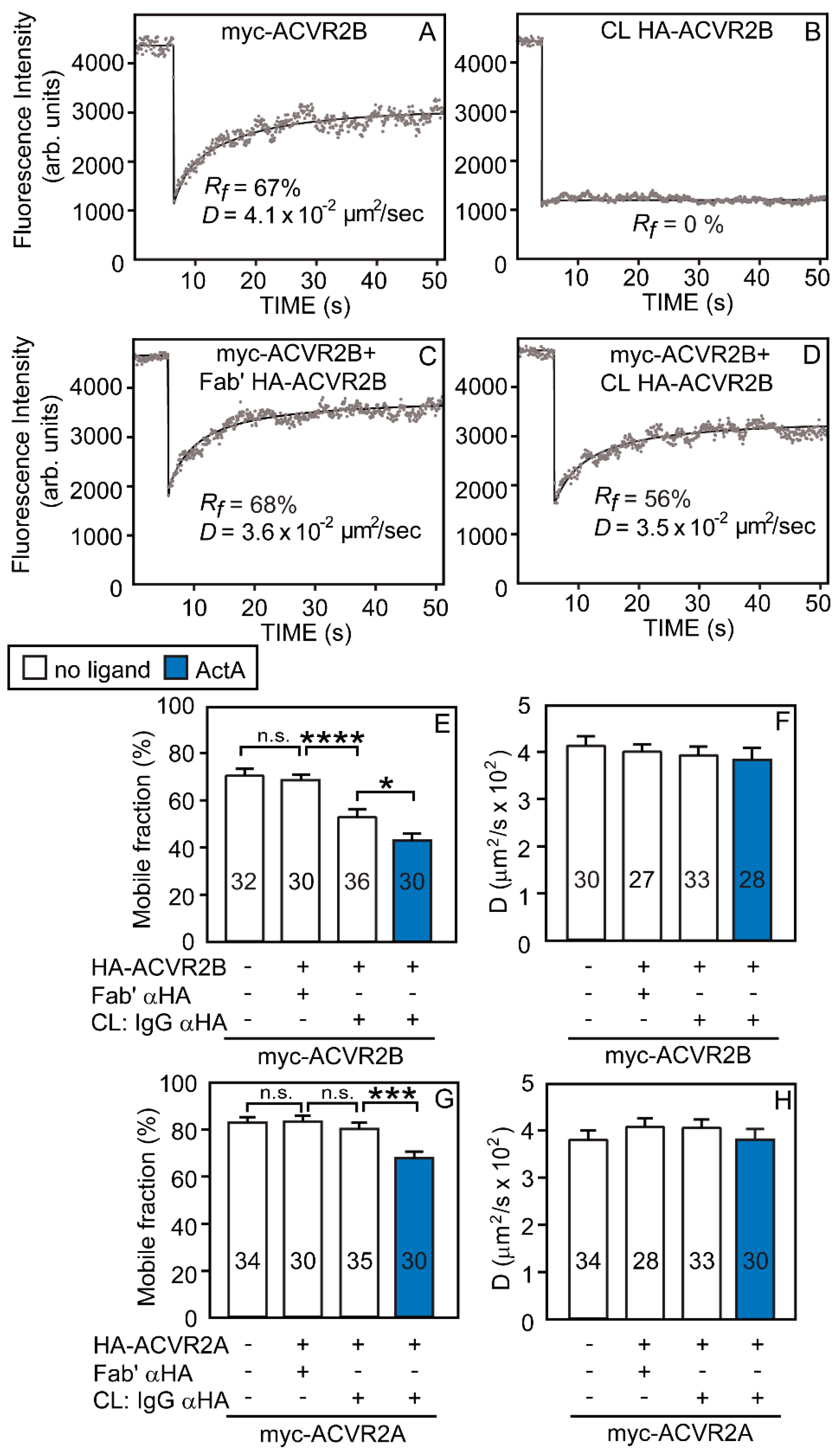{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Antibodies
2.3. Plasmids
2.4. Cell Culture
2.5. Transfection
2.6. Fluorescent Labeling of Cells and IgG-Induced Crosslinking for FRAP and Patch/FRAP
2.7. FRAP and Patch/FRAP
2.8. SMAD1/5/8 Phosphorylation Measurements by Western Blot Analysis
2.9. Transcriptional Activation Assays
2.10. Statistical Analysis
2.11. Illustration
3. Results
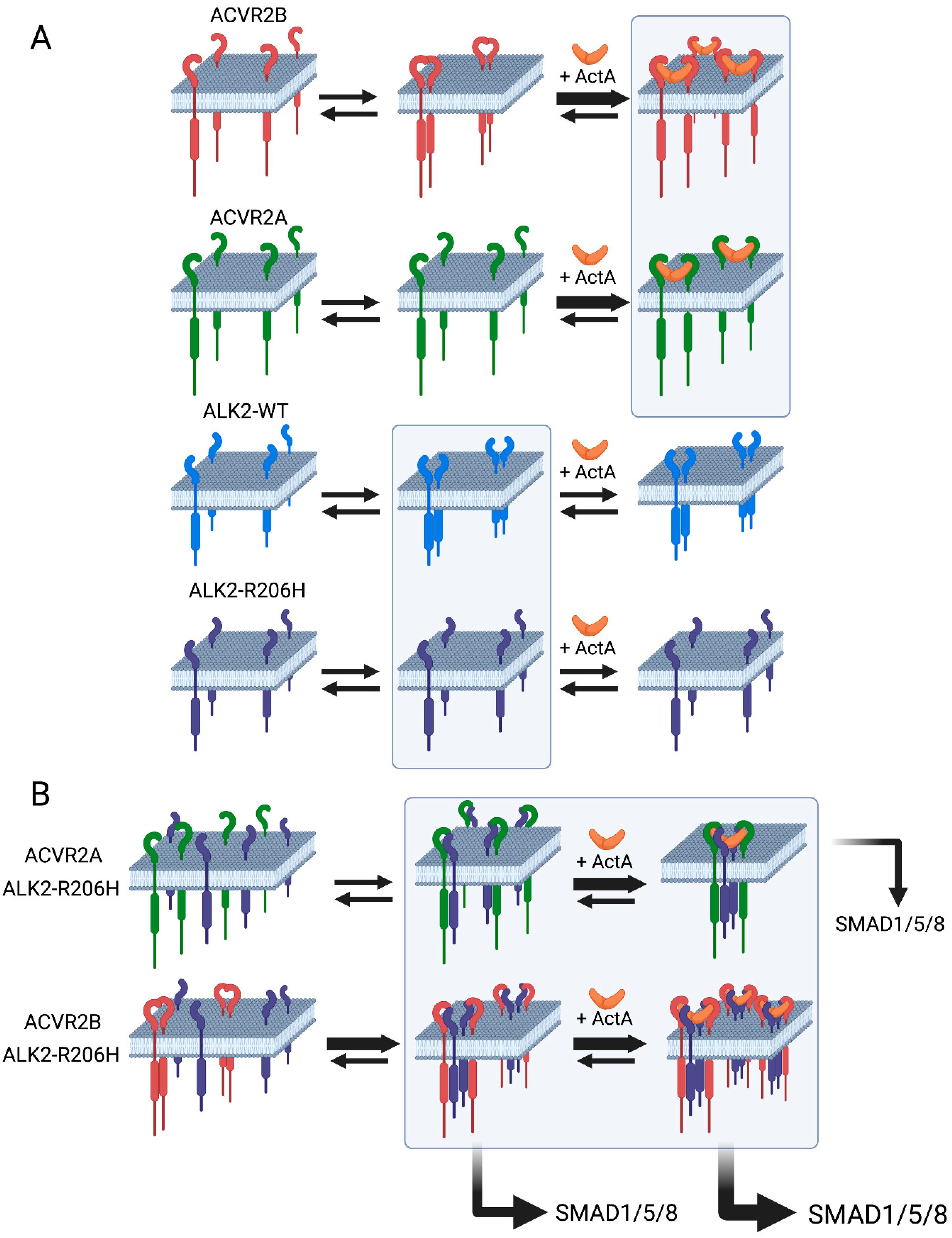
3.1. ACVR2B vs. ACVR2A and ALK2-WT vs. ALK2-R206H Display Different Tendencies to Form Homomeric Complexes
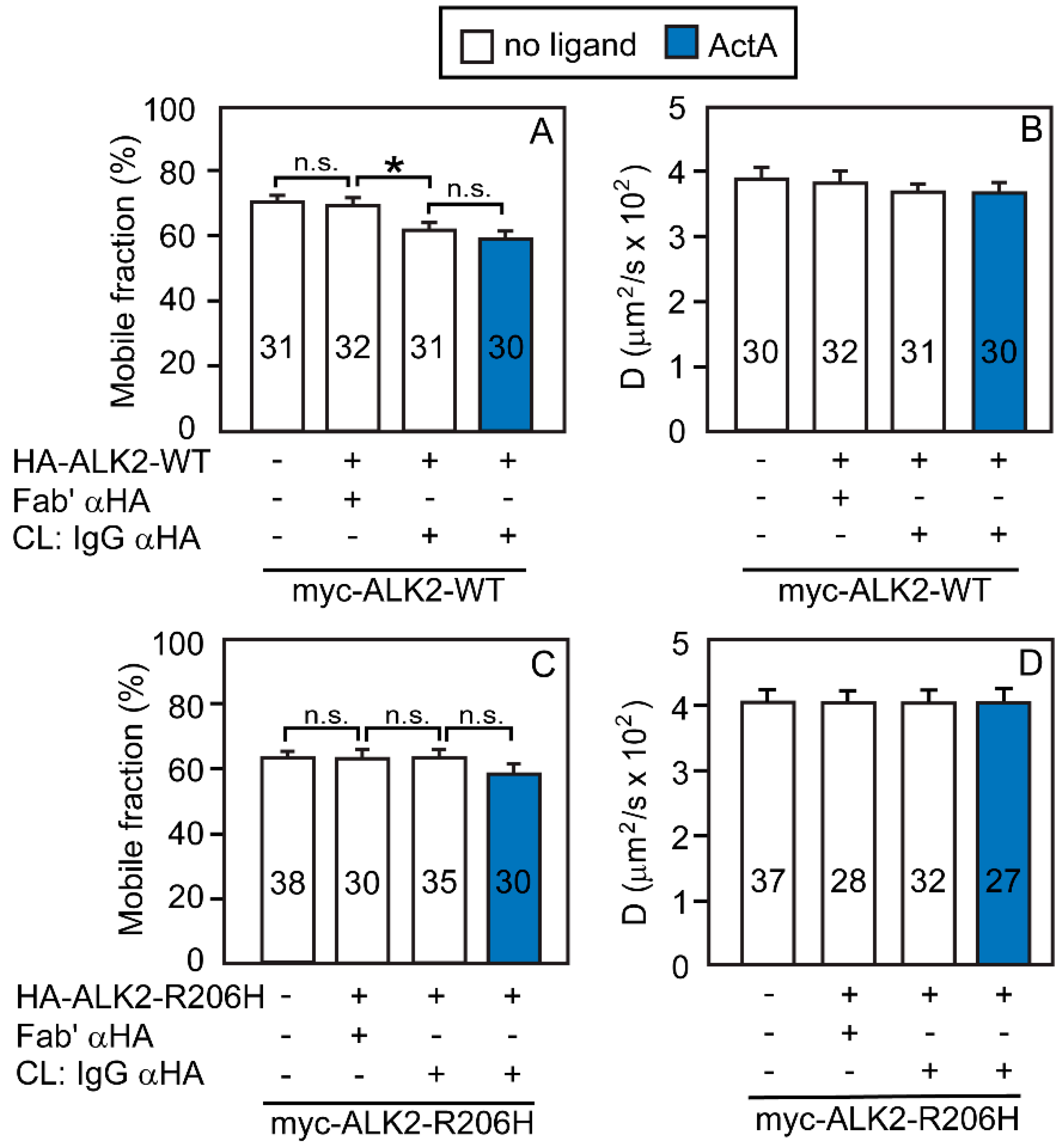
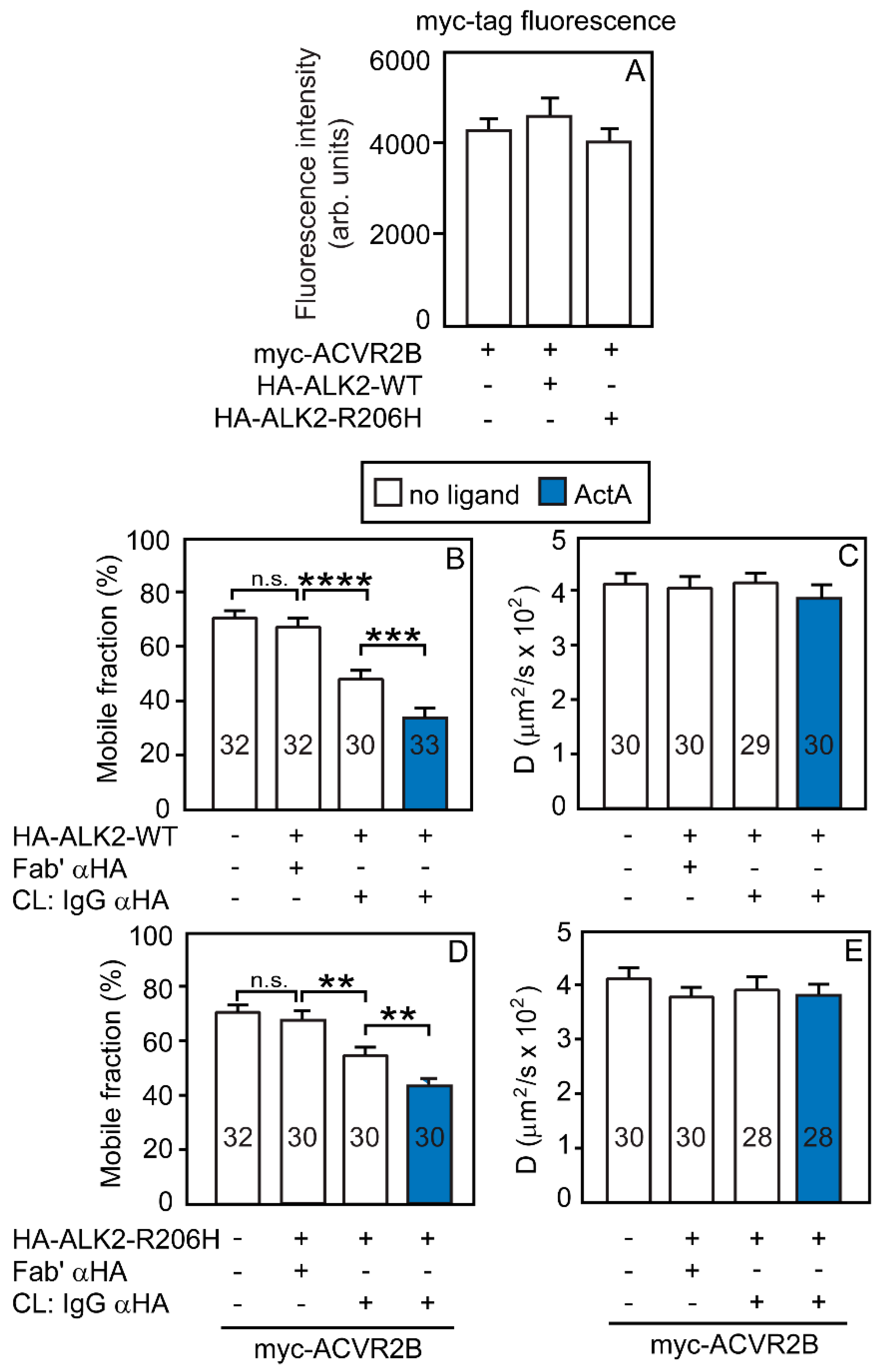
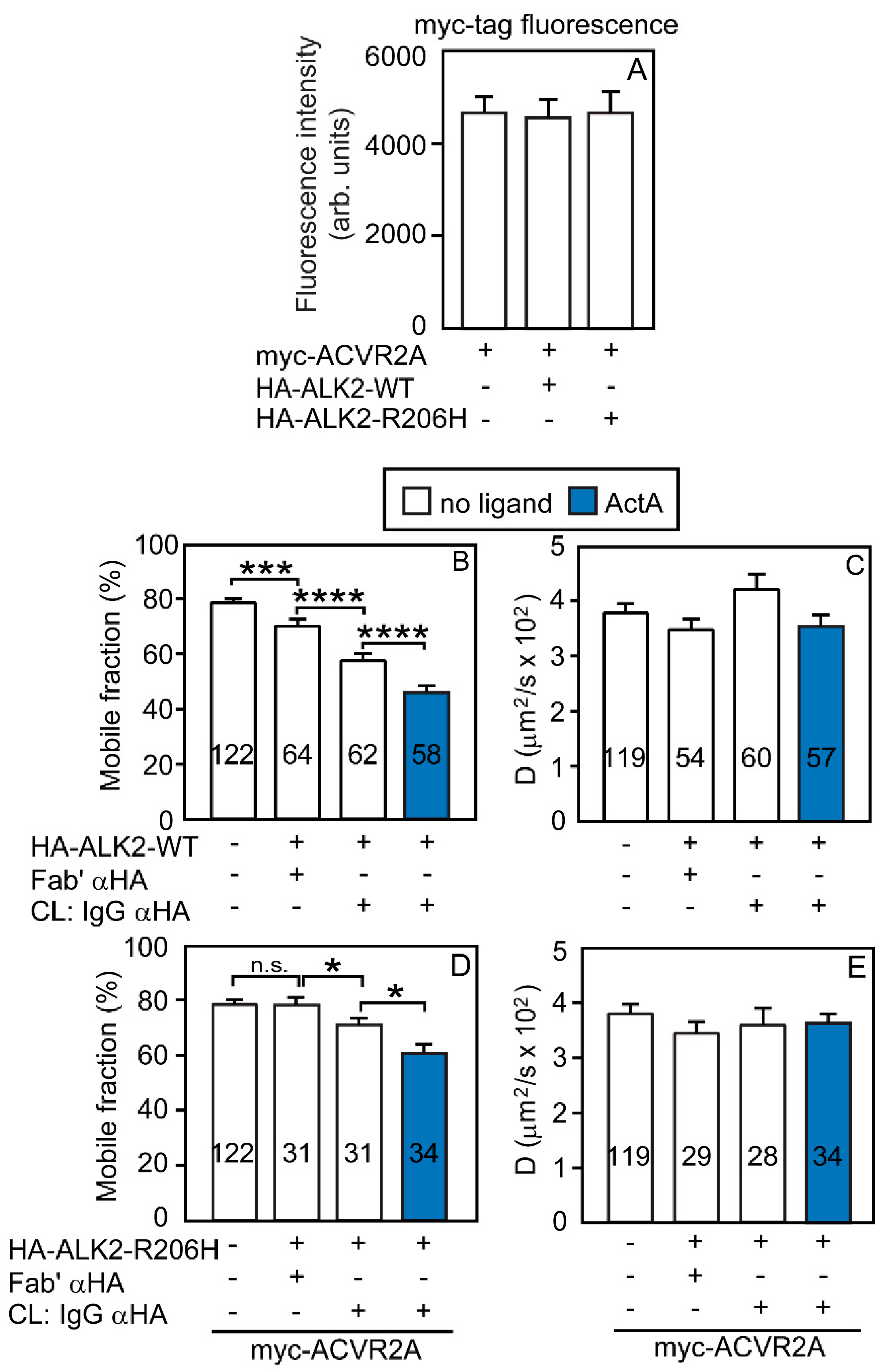
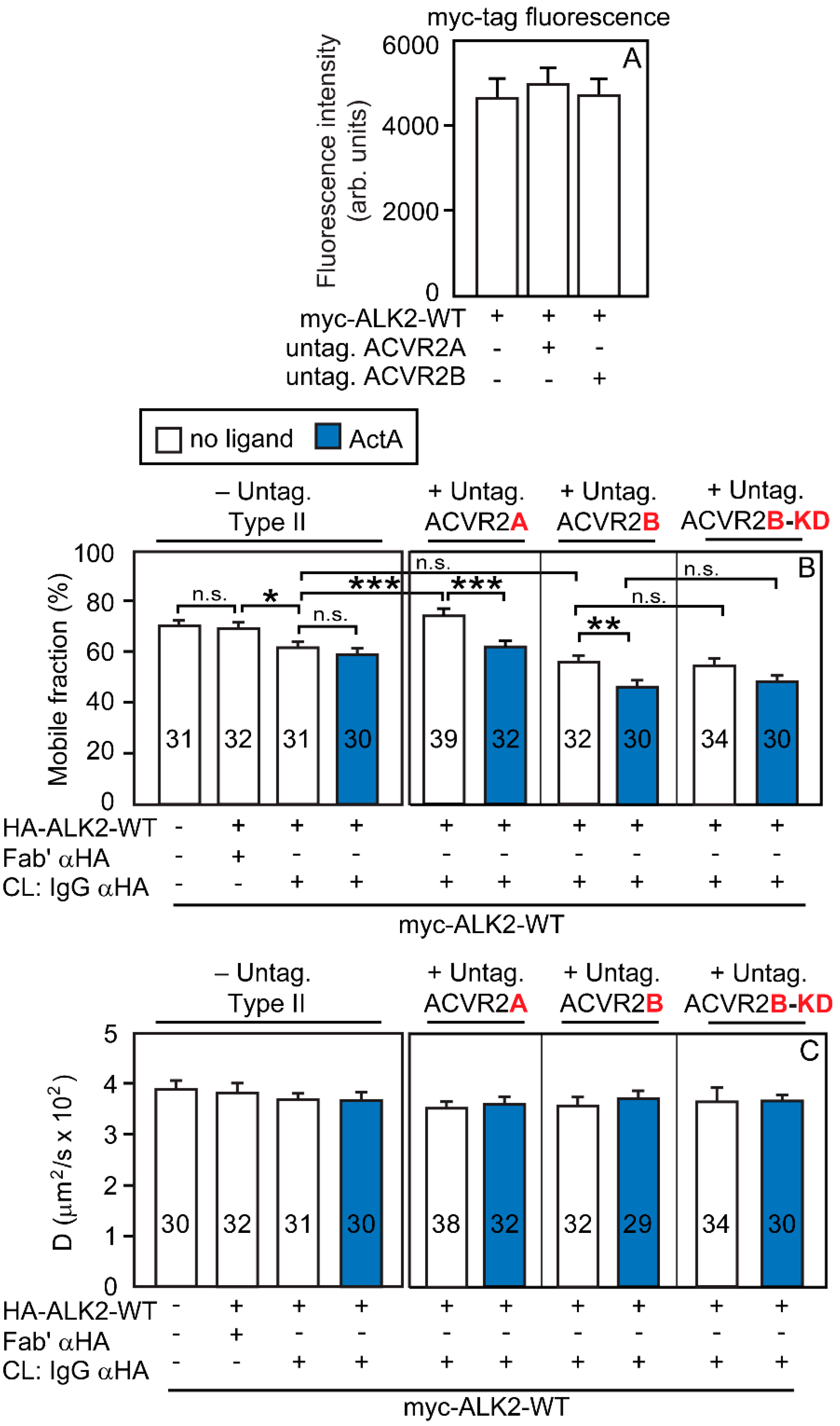
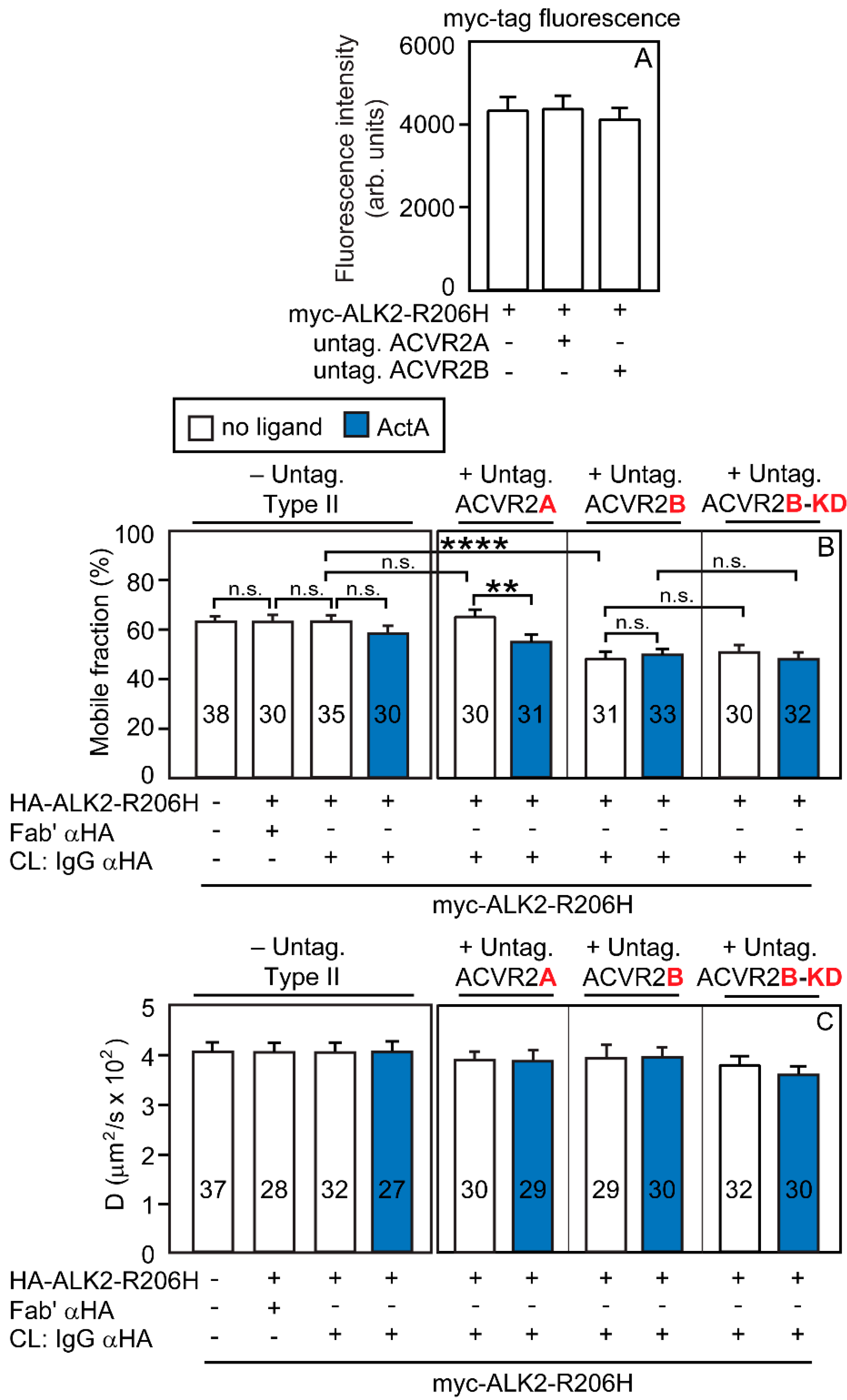
3.2. Heterocomplex Formation of ACVR2A/B with ALK2 (WT or R206H) and the Effects on ALK2 Homomeric Interactions
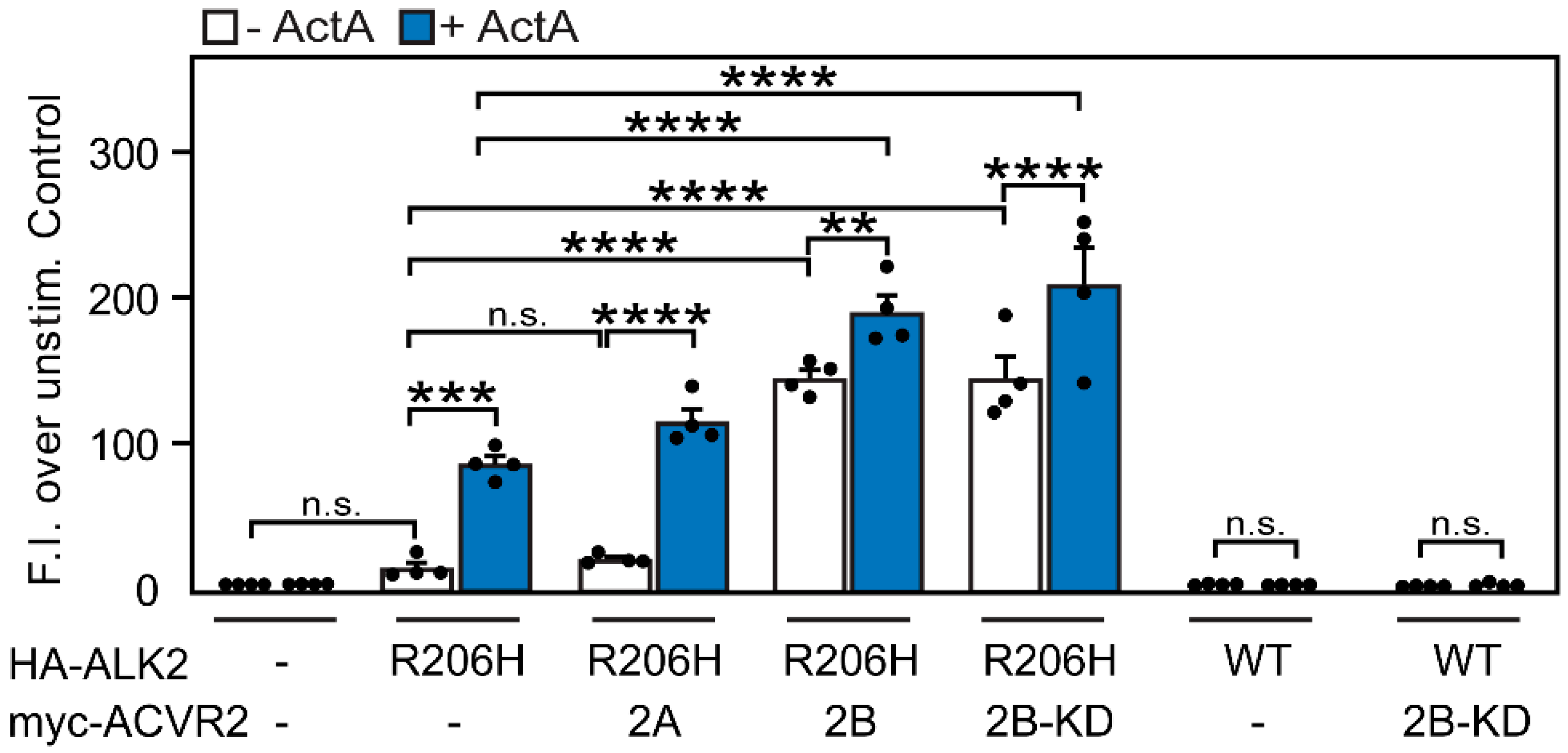
3.3. ACVR2B Is More Effective than ACVR2A in Inducing Constitutive and ActA-Mediated Activation of ALK2-R206H Signaling to SMAD1/5/8
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Massague, J.; Sheppard, D. TGF-b signaling in health and disease. Cell 2023, 186, 4007–4037. [Google Scholar] [CrossRef]
- Davis, H.; Raja, E.; Miyazono, K.; Tsubakihara, Y.; Moustakas, A. Mechanisms of action of bone morphogenetic proteins in cancer. Cytokine Growth Factor Rev. 2016, 27, 81–92. [Google Scholar] [CrossRef]
- Reddi, A.H. Role of morphogenetic proteins in skeletal tissue engineering and regeneration. Nat. Biotechnol. 1998, 16, 247–252. [Google Scholar] [CrossRef]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Parvani, J.G.; Taylor, M.A.; Schiemann, W.P. Noncanonical TGF-b signaling during mammary tumorigenesis. J. Mammary Gland Biol. Neoplasia 2011, 16, 127–146. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-b and the TGF-b Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef]
- Allen, R.S.; Tajer, B.; Shore, E.M.; Mullins, M.C. Fibrodysplasia ossificans progressiva mutant ACVR1 signals by multiple modalities in the developing zebrafish. eLife 2020, 9, e53761. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-b signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Drabsch, Y.; Ten Dijke, P. TGF-b signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Yu, P.B. In Search of the Second Hit in Pulmonary Arterial Hypertension. Circ. Res. 2019, 124, 6–8. [Google Scholar] [CrossRef] [PubMed]
- Yung, L.M.; Nikolic, I.; Paskin-Flerlage, S.D.; Pearsall, R.S.; Kumar, R.; Yu, P.B. A Selective TGFb Ligand Trap Attenuates Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2016, 194, 1140–1151. [Google Scholar] [CrossRef]
- Kaplan, F.S.; Smith, R.M. Fibrodysplasia ossificans progressiva (FOP). J. Bone Miner. Res. 1997, 12, 855. [Google Scholar] [CrossRef] [PubMed]
- Groppe, J.C.; Shore, E.M.; Kaplan, F.S. Functional modeling of the ACVR1 (R206H) mutation in FOP. Clin. Orthop. Relat. Res. 2007, 462, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Shore, E.M.; Xu, M.; Feldman, G.J.; Fenstermacher, D.A.; Cho, T.J.; Choi, I.H.; Connor, J.M.; Delai, P.; Glaser, D.L.; LeMerrer, M.; et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat. Genet. 2006, 38, 525–527. [Google Scholar] [CrossRef] [PubMed]
- Bagarova, J.; Vonner, A.J.; Armstrong, K.A.; Borgermann, J.; Lai, C.S.; Deng, D.Y.; Beppu, H.; Alfano, I.; Filippakopoulos, P.; Morrell, N.W.; et al. Constitutively active ALK2 receptor mutants require type II receptor cooperation. Mol. Cell. Biol. 2013, 33, 2413–2424. [Google Scholar] [CrossRef] [PubMed]
- Hatsell, S.J.; Idone, V.; Wolken, D.M.; Huang, L.; Kim, H.J.; Wang, L.; Wen, X.; Nannuru, K.C.; Jimenez, J.; Xie, L.; et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci. Transl. Med. 2015, 7, 303ra137. [Google Scholar] [CrossRef] [PubMed]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-b superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell 2008, 29, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Radaev, S.; Zou, Z.; Huang, T.; Lafer, E.M.; Hinck, A.P.; Sun, P.D. Ternary complex of transforming growth factor-b1 reveals isoform-specific ligand recognition and receptor recruitment in the superfamily. J. Biol. Chem. 2010, 285, 14806–14814. [Google Scholar] [CrossRef]
- Townson, S.A.; Martinez-Hackert, E.; Greppi, C.; Lowden, P.; Sako, D.; Liu, J.; Ucran, J.A.; Liharska, K.; Underwood, K.W.; Seehra, J.; et al. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J. Biol. Chem. 2012, 287, 27313–27325. [Google Scholar] [CrossRef]
- Allendorph, G.P.; Vale, W.W.; Choe, S. Structure of the ternary signaling complex of a TGF-b superfamily member. Proc. Natl. Acad. Sci. USA 2006, 103, 7643–7648. [Google Scholar] [CrossRef]
- Weber, D.; Kotzsch, A.; Nickel, J.; Harth, S.; Seher, A.; Mueller, U.; Sebald, W.; Mueller, T.D. A silent H-bond can be mutationally activated for high-affinity interaction of BMP-2 and activin type IIB receptor. BMC Struct. Biol. 2007, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Goebel, E.J.; Corpina, R.A.; Hinck, C.S.; Czepnik, M.; Castonguay, R.; Grenha, R.; Boisvert, A.; Miklossy, G.; Fullerton, P.T.; Matzuk, M.M.; et al. Structural characterization of an activin class ternary receptor complex reveals a third paradigm for receptor specificity. Proc. Natl. Acad. Sci. USA 2019, 116, 15505–15513. [Google Scholar] [CrossRef] [PubMed]
- Rechtman, M.M.; Nakaryakov, A.; Shapira, K.E.; Ehrlich, M.; Henis, Y.I. Different domains regulate homomeric and heteromeric complex formation among type I and type II transforming growth factor-b receptors. J. Biol. Chem. 2009, 284, 7843–7852. [Google Scholar] [CrossRef] [PubMed]
- Marom, B.; Heining, E.; Knaus, P.; Henis, Y.I. Formation of stable homomeric and transient heteromeric bone morphogenetic protein (BMP) receptor complexes regulates Smad protein signaling. J. Biol. Chem. 2011, 286, 19287–19296. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gutman, O.; Knaus, P.; Henis, Y.I. Oligomeric interactions of TGF-b and BMP receptors. FEBS Lett. 2012, 586, 1885–1896. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-b signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-b. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Ozden, O.; Akagi, N.; Carroll, T.; Principe, D.R.; Staudacher, J.J.; Spehlmann, M.E.; Eckmann, L.; Grippo, P.J.; Jung, B. Activin and TGFb use diverging mitogenic signaling in advanced colon cancer. Mol. Cancer 2015, 14, 182. [Google Scholar] [CrossRef]
- Miyazono, K.; Kamiya, Y.; Morikawa, M. Bone morphogenetic protein receptors and signal transduction. J. Biochem. 2010, 147, 35–51. [Google Scholar] [CrossRef]
- Massague, J. TGFb signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFb signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming growth factor-b receptors and Smads: Regulatory complexity and functional versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Shi, Y.; Massague, J. Mechanisms of TGF-b signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Lebrin, F.; Deckers, M.; Bertolino, P.; Ten Dijke, P. TGF-b receptor function in the endothelium. Cardiovasc. Res. 2005, 65, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. The regulation of TGFb signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Ikeya, M.; Horigome, K.; Matsumoto, Y.; Ebise, H.; Nishio, M.; Sekiguchi, K.; Shibata, M.; Nagata, S.; Matsuda, S.; et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc. Natl. Acad. Sci. USA 2015, 112, 15438–15443. [Google Scholar] [CrossRef] [PubMed]
- Olsen, O.E.; Sankar, M.; Elsaadi, S.; Hella, H.; Buene, G.; Darvekar, S.R.; Misund, K.; Katagiri, T.; Knaus, P.; Holien, T. BMPR2 inhibits activin and BMP signaling via wild-type ALK2. J. Cell Sci. 2018, 131, jcs213512. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Mehic, M.; Wasim, L.; Malinova, D.; Gori, I.; Blaszczyk, B.K.; Carvalho, D.M.; Shore, E.M.; Jones, C.; Hyvonen, M.; et al. Pathogenic ACVR1(R206H) activation by Activin A-induced receptor clustering and autophosphorylation. EMBO J. 2021, 40, e106317. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, A.; Vizan, P.; Das, D.; Chakravarty, P.; Vogt, J.; Rogers, K.W.; Muller, P.; Hinck, A.P.; Sapkota, G.P.; Hill, C.S. TGF-b uses a novel mode of receptor activation to phosphorylate SMAD1/5 and induce epithelial-to-mesenchymal transition. eLife 2018, 7, e31756. [Google Scholar] [CrossRef] [PubMed]
- Holtzhausen, A.; Golzio, C.; How, T.; Lee, Y.H.; Schiemann, W.P.; Katsanis, N.; Blobe, G.C. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J. 2014, 28, 1248–1267. [Google Scholar] [CrossRef]
- Star, G.P.; Giovinazzo, M.; Langleben, D. Bone morphogenic protein-9 stimulates endothelin-1 release from human pulmonary microvascular endothelial cells: A potential mechanism for elevated ET-1 levels in pulmonary arterial hypertension. Microvasc. Res. 2010, 80, 349–354. [Google Scholar] [CrossRef]
- Upton, P.D.; Davies, R.J.; Trembath, R.C.; Morrell, N.W. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J. Biol. Chem. 2009, 284, 15794–15804. [Google Scholar] [CrossRef]
- Olsen, O.E.; Wader, K.F.; Hella, H.; Mylin, A.K.; Turesson, I.; Nesthus, I.; Waage, A.; Sundan, A.; Holien, T. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell Commun. Signal. 2015, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Corpina, R.A.; Goebel, E.J.; Cunanan, C.J.; Dimitriou, A.; Kim, H.J.; Zhang, Q.; Rafique, A.; Leidich, R.; Wang, X.; et al. Activin A forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife 2020, 9, e54582. [Google Scholar] [CrossRef] [PubMed]
- Haupt, J.; Xu, M.; Shore, E.M. Variable signaling activity by FOP ACVR1 mutations. Bone 2018, 109, 232–240. [Google Scholar] [CrossRef] [PubMed]
- van Dinther, M.; Visser, N.; de Gorter, D.J.; Doorn, J.; Goumans, M.J.; de Boer, J.; ten Dijke, P. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J. Bone Miner. Res. 2010, 25, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, T.; Kohda, M.; Kanomata, K.; Nojima, J.; Nakamura, A.; Kamizono, J.; Noguchi, Y.; Iwakiri, K.; Kondo, T.; Kurose, J.; et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J. Biol. Chem. 2009, 284, 7149–7156. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Little, S.C.; Xu, M.; Haupt, J.; Ast, C.; Katagiri, T.; Mundlos, S.; Seemann, P.; Kaplan, F.S.; Mullins, M.C.; et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J. Clin. Investig. 2009, 119, 3462–3472. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, T.; Tsukamoto, S.; Kuratani, M.; Tsuji, S.; Nakamura, K.; Ohte, S.; Kawaguchi, Y.; Takaishi, K. A blocking monoclonal antibody reveals dimerization of intracellular domains of ALK2 associated with genetic disorders. Nat. Commun. 2023, 14, 2960. [Google Scholar] [CrossRef]
- Szilágyi, S.S.; Amsalem-Zafran, A.R.; Shapira, K.E.; Ehrlich, M.; Henis, Y.I. Competition between type I activin and BMP receptors for binding to ACVR2A regulates signaling to distinct Smad pathways. BMC Biol. 2022, 20, 50. [Google Scholar] [CrossRef]
- Evan, G.I.; Lewis, G.K.; Ramsay, G.; Bishop, J.M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 1985, 5, 3610–3616. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Ehrlich, M.; Zhang, M.; Blobe, G.C.; Henis, Y.I. NRP1 interacts with endoglin and VEGFR2 to modulate VEGF signaling and endothelial cell sprouting. Res. Sq. 2023, 7, 112. [Google Scholar] [CrossRef]
- Korchynskyi, O.; ten Dijke, P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J. Biol. Chem. 2002, 277, 4883–4891. [Google Scholar] [CrossRef]
- Petersen, N.O.; Felder, S.; Elson, E.L. Measurement of lateral diffusion by fluorescence photobleaching recovery. In Handbook of Experimental Immunology; Weir, D.M., Herzenberg, L.A., Blackwell, C.C., Herzenberg, L.A., Eds.; Blackwell Scientific Publications: Edinburgh, UK, 1986; pp. 24.21–24.23. [Google Scholar]
- Chaikuad, A.; Alfano, I.; Kerr, G.; Sanvitale, C.E.; Boergermann, J.H.; Triffitt, J.T.; von Delft, F.; Knapp, S.; Knaus, P.; Bullock, A.N. Structure of the bone morphogenetic protein receptor ALK2 and implications for fibrodysplasia ossificans progressiva. J. Biol. Chem. 2012, 287, 36990–36998. [Google Scholar] [CrossRef]
- Groppe, J.C.; Lu, G.; Tandang-Silvas, M.R.; Pathi, A.; Konda, S.; Wu, J.; Le, V.Q.; Culbert, A.L.; Shore, E.M.; Wharton, K.A.; et al. Polypeptide substrate accessibility hypothesis: Gain-of-function R206H mutation allosterically affects activin receptor-like protein kinase activity. Biomolecules 2023, 13, 1129. [Google Scholar] [CrossRef]
- Gomez-Puerto, M.C.; Iyengar, P.V.; Garcia de Vinuesa, A.; Ten Dijke, P.; Sanchez-Duffhues, G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019, 247, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-b family action. Cytokine Growth Factor Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Jatzlau, J.; Burdzinski, W.; Trumpp, M.; Obendorf, L.; Rossmann, K.; Ravn, K.; Hyvonen, M.; Bottanelli, F.; Broichhagen, J.; Knaus, P. A versatile Halo- and SNAP-tagged BMP/TGFb receptor library for quantification of cell surface ligand binding. Commun. Biol. 2023, 6, 34. [Google Scholar] [CrossRef]
- Pignolo, R.J.; Shore, E.M.; Kaplan, F.S. Fibrodysplasia ossificans progressiva: Clinical and genetic aspects. Orphanet J. Rare Dis. 2011, 6, 80. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szilágyi, S.S.; Burdzinski, W.; Jatzlau, J.; Ehrlich, M.; Knaus, P.; Henis, Y.I. The Activation of the Fibrodysplasia Ossificans Progressiva-Inducing ALK2-R206H Mutant Depends on the Distinct Homo-Oligomerization Patterns of ACVR2B and ACVR2A. Cells 2024, 13, 221. https://doi.org/10.3390/cells13030221
Szilágyi SS, Burdzinski W, Jatzlau J, Ehrlich M, Knaus P, Henis YI. The Activation of the Fibrodysplasia Ossificans Progressiva-Inducing ALK2-R206H Mutant Depends on the Distinct Homo-Oligomerization Patterns of ACVR2B and ACVR2A. Cells. 2024; 13(3):221. https://doi.org/10.3390/cells13030221
Chicago/Turabian StyleSzilágyi, Szabina Szófia, Wiktor Burdzinski, Jerome Jatzlau, Marcelo Ehrlich, Petra Knaus, and Yoav I. Henis. 2024. "The Activation of the Fibrodysplasia Ossificans Progressiva-Inducing ALK2-R206H Mutant Depends on the Distinct Homo-Oligomerization Patterns of ACVR2B and ACVR2A" Cells 13, no. 3: 221. https://doi.org/10.3390/cells13030221