
Disruption of Mitophagy Flux through the PARL-PINK1 Pathway by CHCHD10 Mutations or CHCHD10 Depletion


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Ethics Approval
2.2. Human Brain Samples
2.3. Mice
2.4. Antibodies and Reagents
2.5. Cell Culture
2.6. DNA Constructs, Transfections, and rAAV9 Transduction
2.7. Mitochondria Isolation, Cell/Tissue Lysis, and SDS-PAGE
2.8. Immunocytochemistry, Proximity Ligation Assay, and Fluorescent Microscopy
2.9. Statistical Analysis
3. Results
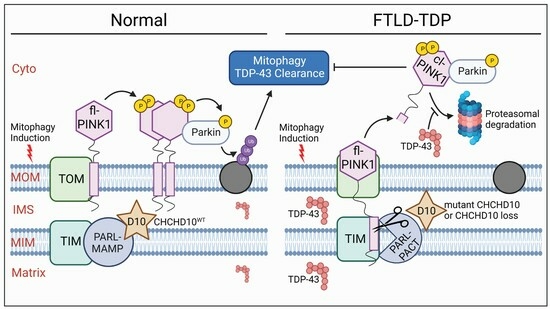
3.1. CHCHD10WT Promotes and ALS/FTD-Linked CHCHD10 Mutations Suppress Mitophagy Flux and Mitochondrial Parkin Recruitment In Vivo
3.2. ALS/FTD-Linked CHCHD10 Mutations (R15L and S59L) Increase PARL Activity and Reduce Activated Full-Length PINK1 In Vivo
3.3. CHCHD10 Interactions with PARL and PINK1 Are Suppressed by CHCHD10R15L and CHCHD10S59L Mutations
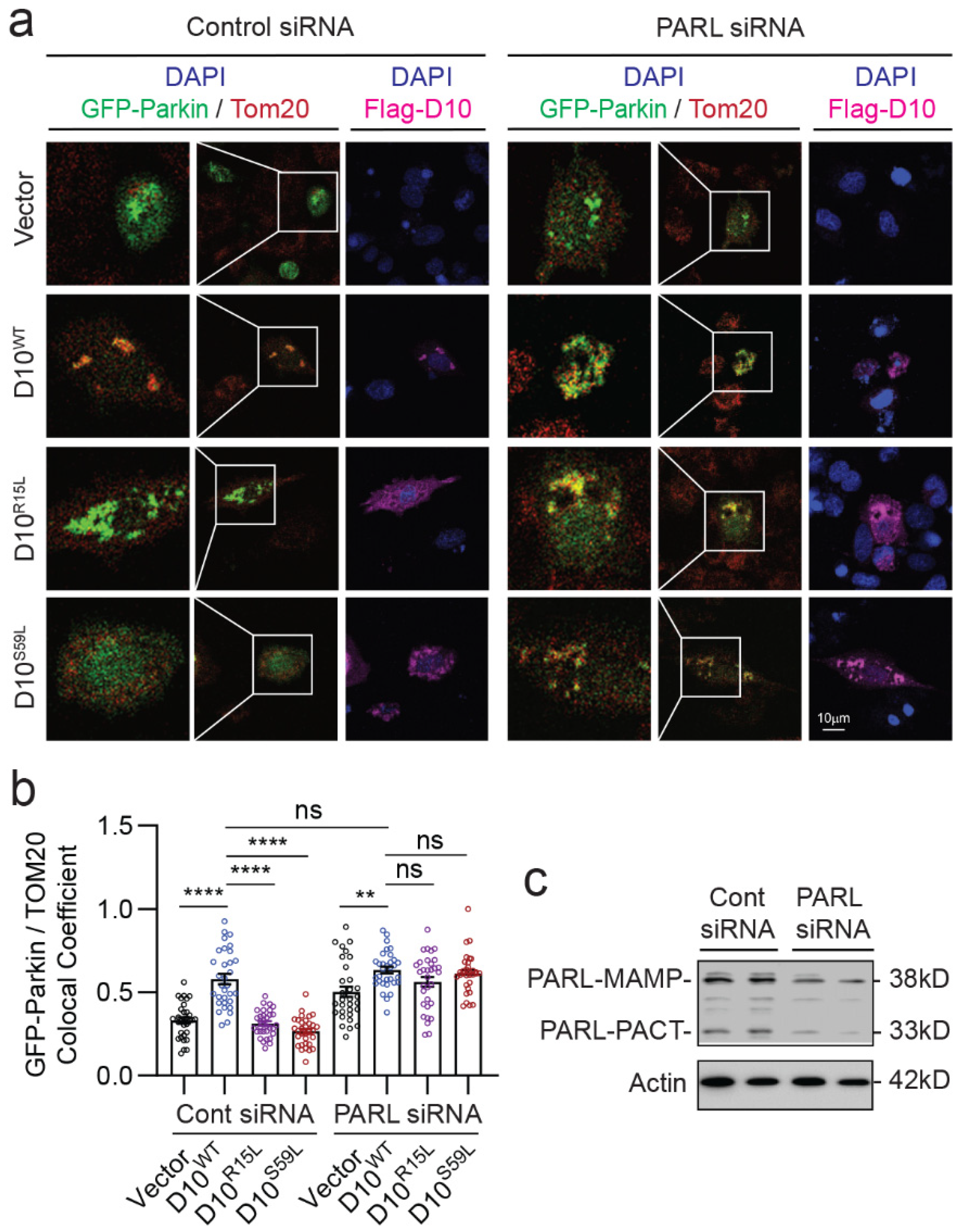
3.4. CHCHD10 Variants Depend on PARL to Regulate Mitochondrial Parkin Recruitment
3.5. CHCHD10 Depletion Enhances PARL Cleavage and Reduces PINK1, Suppressing Parkin Recruitment and Mitophagy Flux
3.6. Disruption of the PARL–PINK1 Pathway in FTLD-TDP Patient Brains
3.7. Depletion of PINK1, CHCHD10, or MIC60 Promotes TDP-43 Aggregation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Redmann, M.; Darley-Usmar, V.; Zhang, J. The Role of Autophagy, Mitophagy and Lysosomal Functions in Modulating Bioenergetics and Survival in the Context of Redox and Proteotoxic Damage: Implications for Neurodegenerative Diseases. Aging Dis. 2016, 7, 150–162. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Palomo, V.; Tosat-Bitrian, C.; Nozal, V.; Nagaraj, S.; Martin-Requero, A.; Martinez, A. TDP-43: A Key Therapeutic Target beyond Amyotrophic Lateral Sclerosis. ACS Chem. Neurosci. 2019, 10, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Hergesheimer, R.C.; Chami, A.A.; de Assis, D.R.; Vourc’h, P.; Andres, C.R.; Corcia, P.; Lanznaster, D.; Blasco, H. The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: A resolution in sight? Brain 2019, 142, 1176–1194. [Google Scholar] [CrossRef]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the balance of TDP-43, mitochondria, and autophagy: A promising therapeutic strategy for neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 40. [Google Scholar] [CrossRef]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e618. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Glynn, S.M.; Gibbs, J.R.; Nalls, M.A.; Sabatelli, M.; Restagno, G.; Drory, V.E.; Chio, A.; Rogaeva, E.; Traynor, B.J. Mutations in the CHCHD10 gene are a common cause of familial amyotrophic lateral sclerosis. Brain 2014, 137, e311. [Google Scholar] [CrossRef]
- Muller, K.; Andersen, P.M.; Hubers, A.; Marroquin, N.; Volk, A.E.; Danzer, K.M.; Meitinger, T.; Ludolph, A.C.; Strom, T.M.; Weishaupt, J.H. Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 2014, 137, e309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xi, Z.; Zinman, L.; Bruni, A.C.; Maletta, R.G.; Curcio, S.A.; Rainero, I.; Rubino, E.; Pinessi, L.; Nacmias, B.; et al. Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 2015, 138, e380. [Google Scholar] [CrossRef] [PubMed]
- Chaussenot, A.; Le Ber, I.; Ait-El-Mkadem, S.; Camuzat, A.; de Septenville, A.; Bannwarth, S.; Genin, E.C.; Serre, V.; Augé, G.; Brice, A.; et al. Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 2014, 35, 2884.e1–2884.e4. [Google Scholar] [CrossRef] [PubMed]
- Dols-Icardo, O.; Nebot, I.; Gorostidi, A.; Ortega-Cubero, S.; Hernandez, I.; Rojas-Garcia, R.; Garcia-Redondo, A.; Povedano, M.; Llado, A.; Alvarez, V.; et al. Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 2015, 138, e400. [Google Scholar] [CrossRef]
- Jiao, B.; Xiao, T.; Hou, L.; Gu, X.; Zhou, Y.; Zhou, L.; Tang, B.; Xu, J.; Shen, L. High prevalence of CHCHD10 mutation in patients with frontotemporal dementia from China. Brain 2016, 139, e21. [Google Scholar] [CrossRef]
- Genin, E.C.; Plutino, M.; Bannwarth, S.; Villa, E.; Cisneros-Barroso, E.; Roy, M.; Ortega-Vila, B.; Fragaki, K.; Lespinasse, F.; Pinero-Martos, E.; et al. CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 2016, 8, 58–72. [Google Scholar] [CrossRef]
- Ruan, Y.; Hu, J.; Che, Y.; Liu, Y.; Luo, Z.; Cheng, J.; Han, Q.; He, H.; Zhou, Q. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022, 13, 156. [Google Scholar] [CrossRef]
- Liu, T.; Woo, J.A.; Bukhari, M.Z.; LePochat, P.; Chacko, A.; Selenica, M.B.; Yan, Y.; Kotsiviras, P.; Buosi, S.C.; Zhao, X.; et al. CHCHD10-regulated OPA1-mitofilin complex mediates TDP-43-induced mitochondrial phenotypes associated with frontotemporal dementia. FASEB J. 2020, 34, 8493–8509. [Google Scholar] [CrossRef]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghanel, M.; Exner, N.; Riemenschneider, H.; van der Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; LePochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; Choe, Y.J.; Bannwarth, S.; Kim, J.; Maitra, S.; Dorn, G.W., 2nd; Taylor, J.P.; Paquis-Flucklinger, V.; Kim, N.C. TDP-43 and PINK1 mediate CHCHD10(S59L) mutation-induced defects in Drosophila and in vitro. Nat. Commun. 2021, 12, 1924. [Google Scholar] [CrossRef]
- Liu, T.; Woo, J.A.; Bukhari, M.Z.; Wang, X.; Yan, Y.; Buosi, S.C.; Ermekbaeva, A.; Sista, A.; Kotsiviras, P.; LePochat, P.; et al. Modulation of synaptic plasticity, motor unit physiology, and TDP-43 pathology by CHCHD10. Acta Neuropathol. Commun. 2022, 10, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Wang, J.; Levichkin, I.V.; Stasinopoulos, S.; Ryan, M.T.; Hoogenraad, N.J. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002, 21, 4411–4419. [Google Scholar] [CrossRef] [PubMed]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-mediated mitophagy at a glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef]
- Aerts, L.; Craessaerts, K.; De Strooper, B.; Morais, V.A. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J. Biol. Chem. 2015, 290, 2798–2811. [Google Scholar] [CrossRef]
- Akabane, S.; Uno, M.; Tani, N.; Shimazaki, S.; Ebara, N.; Kato, H.; Kosako, H.; Oka, T. PKA Regulates PINK1 Stability and Parkin Recruitment to Damaged Mitochondria through Phosphorylation of MIC60. Mol. Cell 2016, 62, 371–384. [Google Scholar] [CrossRef]
- Cazzaro, S.; Woo, J.A.; Wang, X.; Liu, T.; Rego, S.; Kee, T.R.; Koh, Y.; Vazquez-Rosa, E.; Pieper, A.A.; Kang, D.E. Slingshot homolog-1-mediated Nrf2 sequestration tips the balance from neuroprotection to neurodegeneration in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2023, 120, e2217128120. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, X.; Chaput, D.; Shin, M.K.; Koh, Y.; Gan, L.; Pieper, A.A.; Woo, J.A.; Kang, D.E. X-linked ubiquitin-specific peptidase 11 increases tauopathy vulnerability in women. Cell 2022, 185, 3913–3930.e3919. [Google Scholar] [CrossRef]
- Fang, C.; Woo, J.A.; Liu, T.; Zhao, X.; Cazzaro, S.; Yan, Y.; Matlack, J.; Kee, T.; LePochat, P.; Kang, D.E. SSH1 impedes SQSTM1/p62 flux and MAPT/Tau clearance independent of CFL (cofilin) activation. Autophagy 2021, 17, 2144–2165. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, W.; Wang, C.; Zhang, B.; Li, R.; Zhang, L.; Zhao, K.; Li, Y.; Tian, L.; Li, B.; et al. IRGM promotes the PINK1-mediated mitophagy through the degradation of Mitofilin in SH-SY5Y cells. FASEB J. 2020, 34, 14768–14779. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Cazzaro, S.; Zhao, X.; Zhao, V.K.; Kim, Y.K.; Woo, J.A. Slingshot homolog-1 amplifies mitochondrial abnormalities by distinctly impairing health and clearance of mitochondria. Hum. Mol. Genet. 2023, 32, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Sik, A.; Passer, B.J.; Koonin, E.V.; Pellegrini, L. Self-regulated cleavage of the mitochondrial intramembrane-cleaving protease PARL yields Pbeta, a nuclear-targeted peptide. J. Biol. Chem. 2004, 279, 15323–15329. [Google Scholar] [CrossRef] [PubMed]
- Lysyk, L.; Brassard, R.; Arutyunova, E.; Siebert, V.; Jiang, Z.; Takyi, E.; Morrison, M.; Young, H.S.; Lemberg, M.K.; O’Donoghue, A.J.; et al. Insights into the catalytic properties of the mitochondrial rhomboid protease PARL. J. Biol. Chem. 2021, 296, 100383. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraju, D.V.; Xu, L.; Letellier, M.C.; Bandaru, S.; Zunino, R.; Berg, E.A.; McBride, H.M.; Pellegrini, L. Phosphorylation and cleavage of presenilin-associated rhomboid-like protein (PARL) promotes changes in mitochondrial morphology. Proc. Natl. Acad. Sci. USA 2006, 103, 18562–18567. [Google Scholar] [CrossRef] [PubMed]
- Fedorowicz, M.A.; de Vries-Schneider, R.L.; Rub, C.; Becker, D.; Huang, Y.; Zhou, C.; Alessi Wolken, D.M.; Voos, W.; Liu, Y.; Przedborski, S. Cytosolic cleaved PINK1 represses Parkin translocation to mitochondria and mitophagy. EMBO Rep. 2014, 15, 86–93. [Google Scholar] [CrossRef] [PubMed]
- McCann, E.P.; Fifita, J.A.; Grima, N.; Galper, J.; Mehta, P.; Freckleton, S.E.; Zhang, K.Y.; Henden, L.; Hogan, A.L.; Chan Moi Fat, S.; et al. Genetic and immunopathological analysis of CHCHD10 in Australian amyotrophic lateral sclerosis and frontotemporal dementia and transgenic TDP-43 mice. J. Neurol. Neurosurg. Psychiatry 2020, 91, 162–171. [Google Scholar] [CrossRef]
- Sun, N.; Yun, J.; Liu, J.; Malide, D.; Liu, C.; Rovira, I.I.; Holmström, K.M.; Fergusson, M.M.; Yoo, Y.H.; Combs, C.A.; et al. Measuring In Vivo Mitophagy. Mol. Cell 2015, 60, 685–696. [Google Scholar] [CrossRef]
- Liu, Y.T.; Sliter, D.A.; Shammas, M.K.; Huang, X.; Wang, C.; Calvelli, H.; Maric, D.S.; Narendra, D.P. Mt-Keima detects PINK1-PRKN mitophagy in vivo with greater sensitivity than mito-QC. Autophagy 2021, 17, 3753–3762. [Google Scholar] [CrossRef]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed]
- Gaweda-Walerych, K.; Sitek, E.J.; Narozanska, E.; Buratti, E. Parkin beyond Parkinson’s Disease-A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies. Cells 2021, 10, 3389. [Google Scholar] [CrossRef]
- Deas, E.; Plun-Favreau, H.; Wood, N.W. PINK1 function in health and disease. EMBO Mol. Med. 2009, 1, 152–165. [Google Scholar] [CrossRef]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Wetzel, L.; Zhu, Z.; Kumaraguru, P.; Gorthi, V.; Yan, Y.; Bukhari, M.Z.; Ermekbaeva, A.; Jeon, H.; Kee, T.R.; et al. Disruption of Mitophagy Flux through the PARL-PINK1 Pathway by CHCHD10 Mutations or CHCHD10 Depletion. Cells 2023, 12, 2781. https://doi.org/10.3390/cells12242781
Liu T, Wetzel L, Zhu Z, Kumaraguru P, Gorthi V, Yan Y, Bukhari MZ, Ermekbaeva A, Jeon H, Kee TR, et al. Disruption of Mitophagy Flux through the PARL-PINK1 Pathway by CHCHD10 Mutations or CHCHD10 Depletion. Cells. 2023; 12(24):2781. https://doi.org/10.3390/cells12242781
Chicago/Turabian StyleLiu, Tian, Liam Wetzel, Zexi Zhu, Pavan Kumaraguru, Viraj Gorthi, Yan Yan, Mohammed Zaheen Bukhari, Aizara Ermekbaeva, Hanna Jeon, Teresa R. Kee, and et al. 2023. "Disruption of Mitophagy Flux through the PARL-PINK1 Pathway by CHCHD10 Mutations or CHCHD10 Depletion" Cells 12, no. 24: 2781. https://doi.org/10.3390/cells12242781