
Development of a Polymeric Pharmacological Nanocarrier System as a Potential Therapy for Spinocerebellar Ataxia Type 7

 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of NPs
2.3. Physicochemical Characterization
2.3.1. Scanning Electron Microscope (SEM)
2.3.2. Particle Size, PDI and Z-Potential Measurement
2.3.3. Fourier Transform-Infrared Spectroscopy (FTIR)
2.3.4. Differential Scanning Calorimetry (DSC)
2.3.5. Thermogravimetric Analysis (TGA)
2.3.6. Entrapment Efficiency and Drug Loading
2.3.7. Determination of Rapamycin Release Kinetics from NPs
2.3.8. Stability of NPs in Dispersion
2.4. Biological Characterization
2.4.1. Cell Culture
2.4.2. Nanoparticle Uptake
2.4.3. Cell Viability Assays
2.4.4. Analysis of Cell Morphology by Confocal Microscopy
2.4.5. Annexin V Assays
2.4.6. Analysis of Caspases Activation
2.5. Therapeutic Effect
2.5.1. NPs Treatment
2.5.2. Analysis of PolyQ Ataxin-7 Subcellular Localization and Autophagy Activation by Confocal Microscopy
2.6. Statistical Analysis
3. Results and Discussion
3.1. Design, Physicochemical, and Structural Characterization of Nanocarriers for SCA7 Therapy
3.1.1. Nanocarriers Morphology
3.1.2. Physical Characterization of NPs
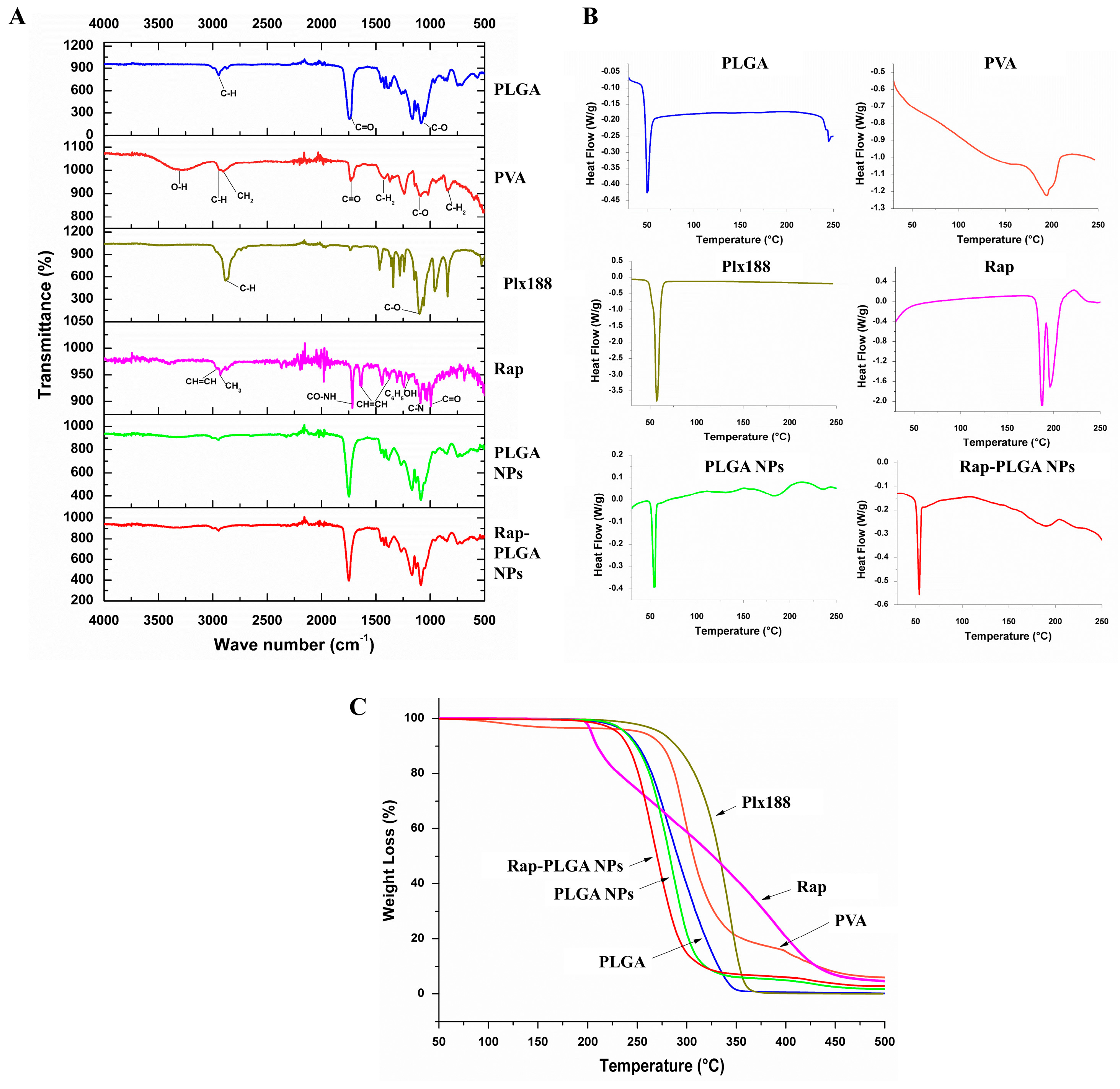
3.1.3. Chemical Characterization of NPs
3.1.4. Entrapment Efficiency and Drug Loading
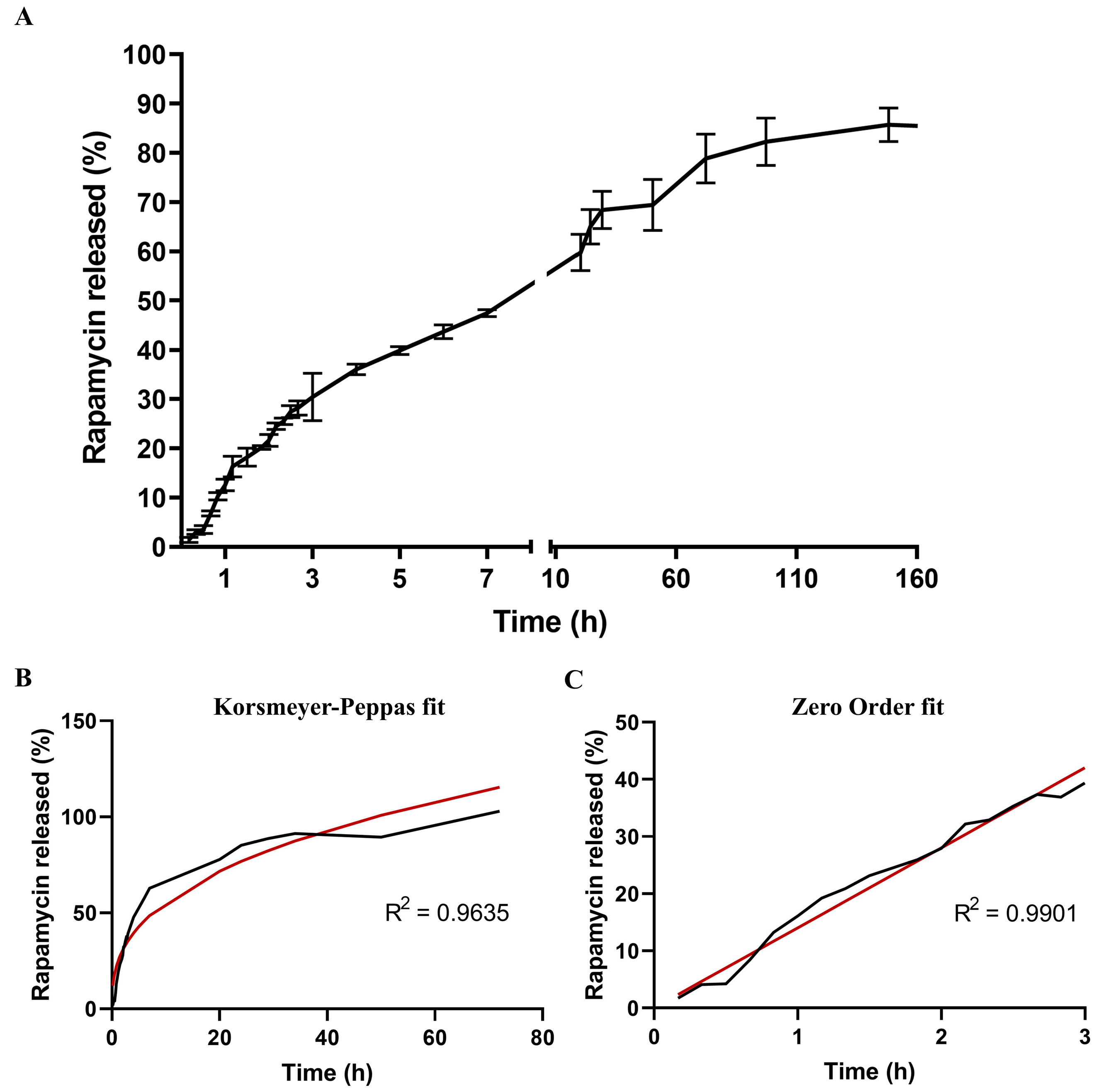
3.1.5. Kinetic Analysis of Rapamycin Release
3.2. Biological Characterization of NPs
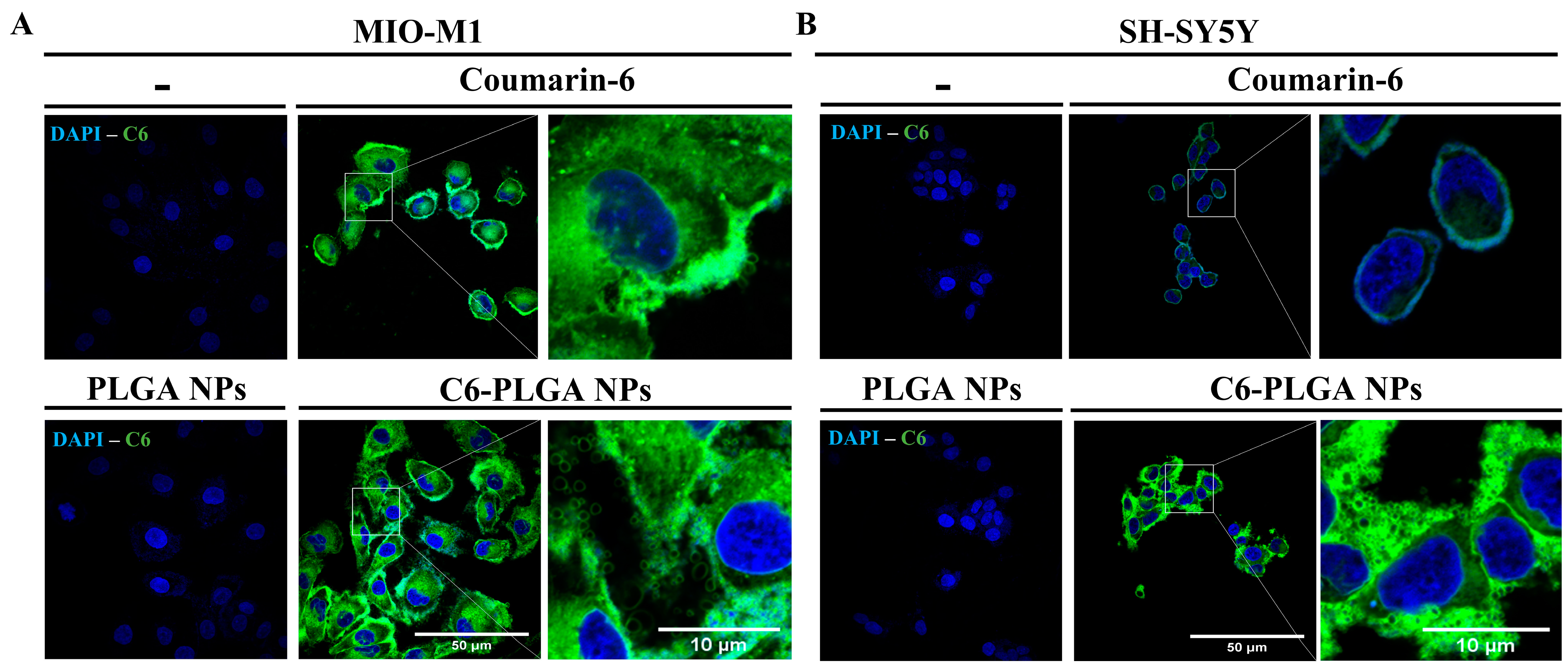
3.2.1. Uptake of NPs by Neuronal and Glial Cells
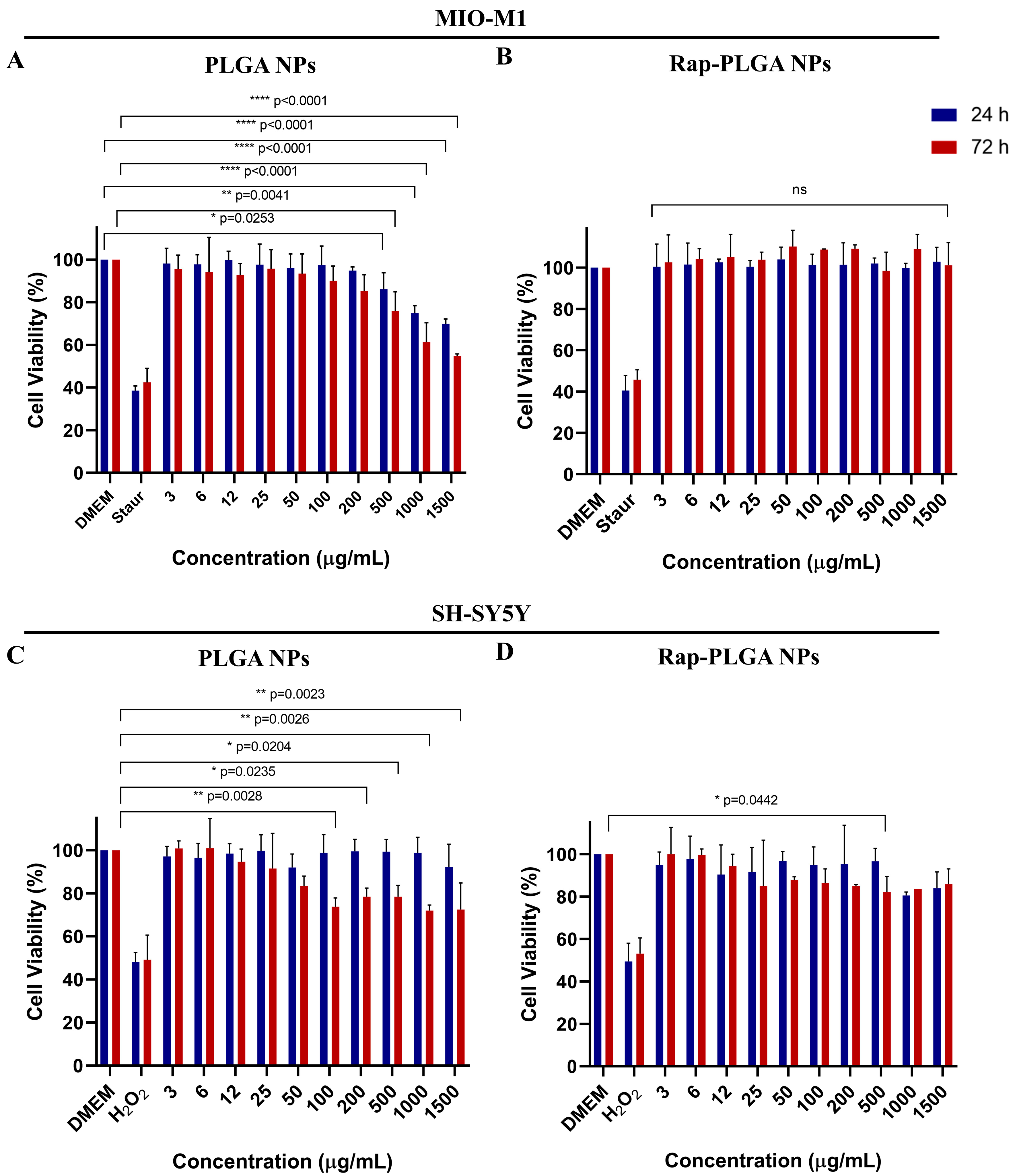
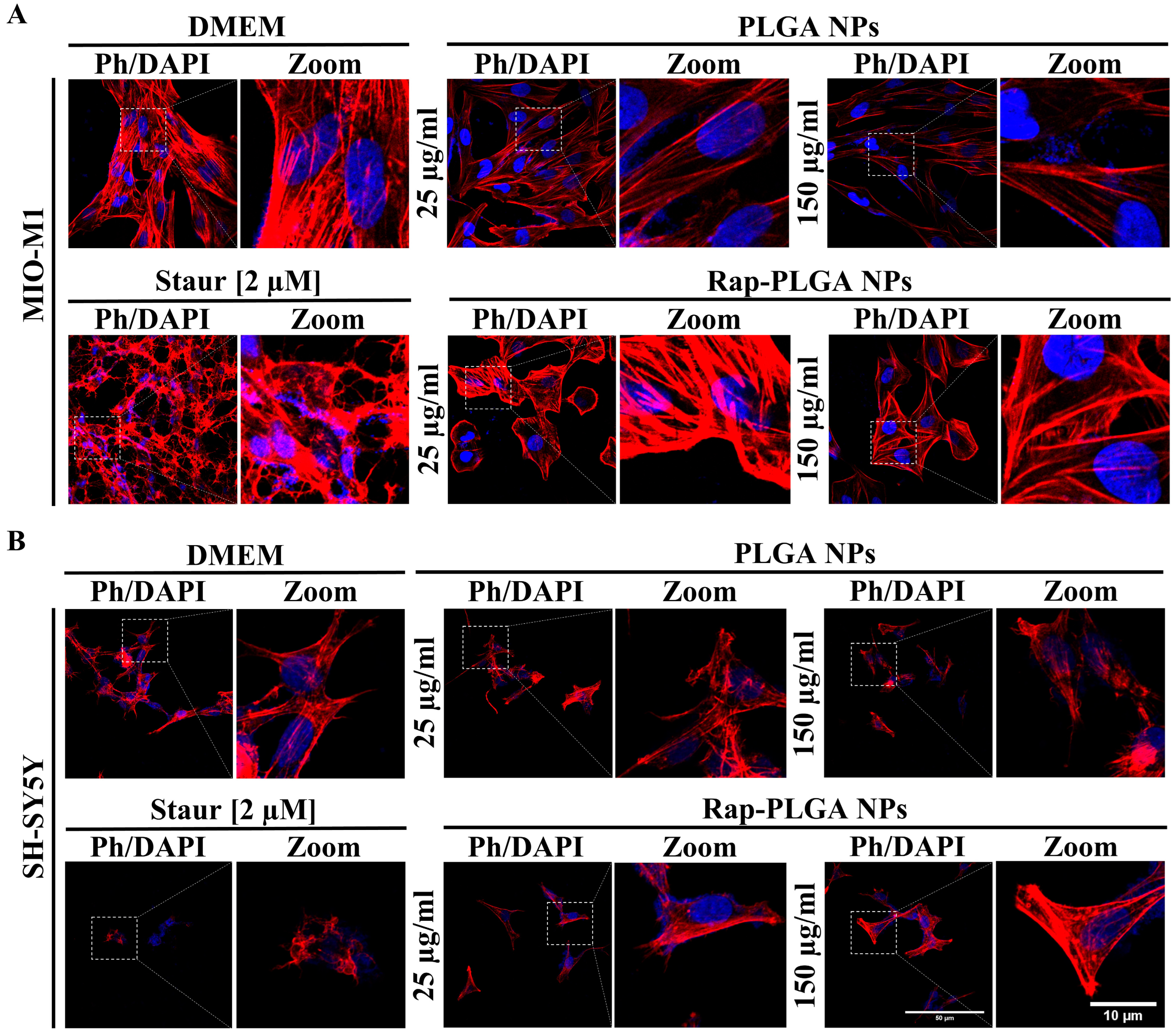
3.2.2. Effect of NPs on Cell Viability and Morphology
3.2.3. Analysis of NPs Cytotoxicity in Glial Cells
3.3. Application of Rapamycin-Loaded NPs to Alleviate the SCA7 Phenotype in a Glial Cell Model
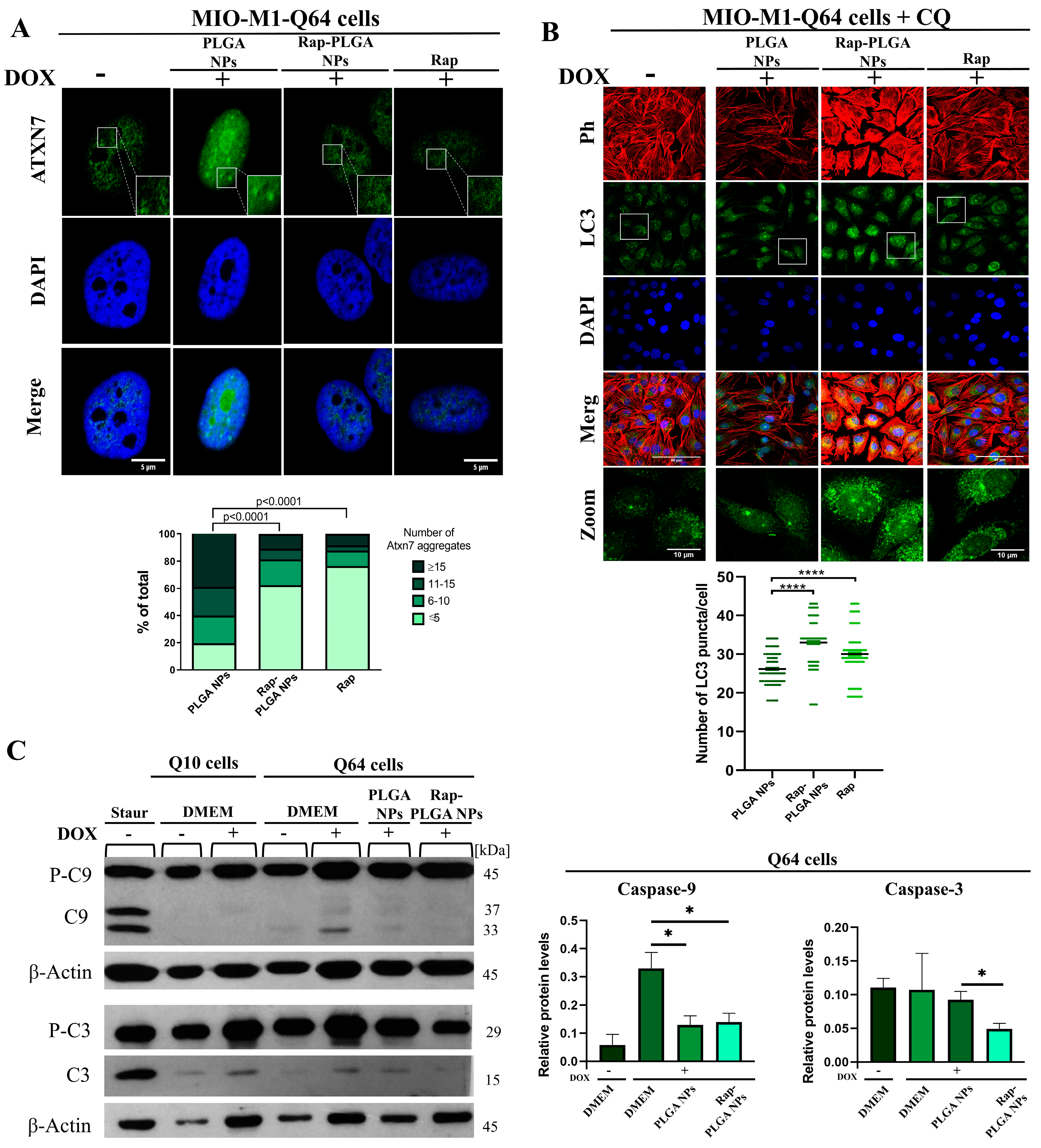
3.3.1. Treatment with Rap-PLGA NPs Promotes the Clearance of Mutant Ataxin-7 Aggregates by Inducing Autophagy in a SCA7 Glial Cell Model
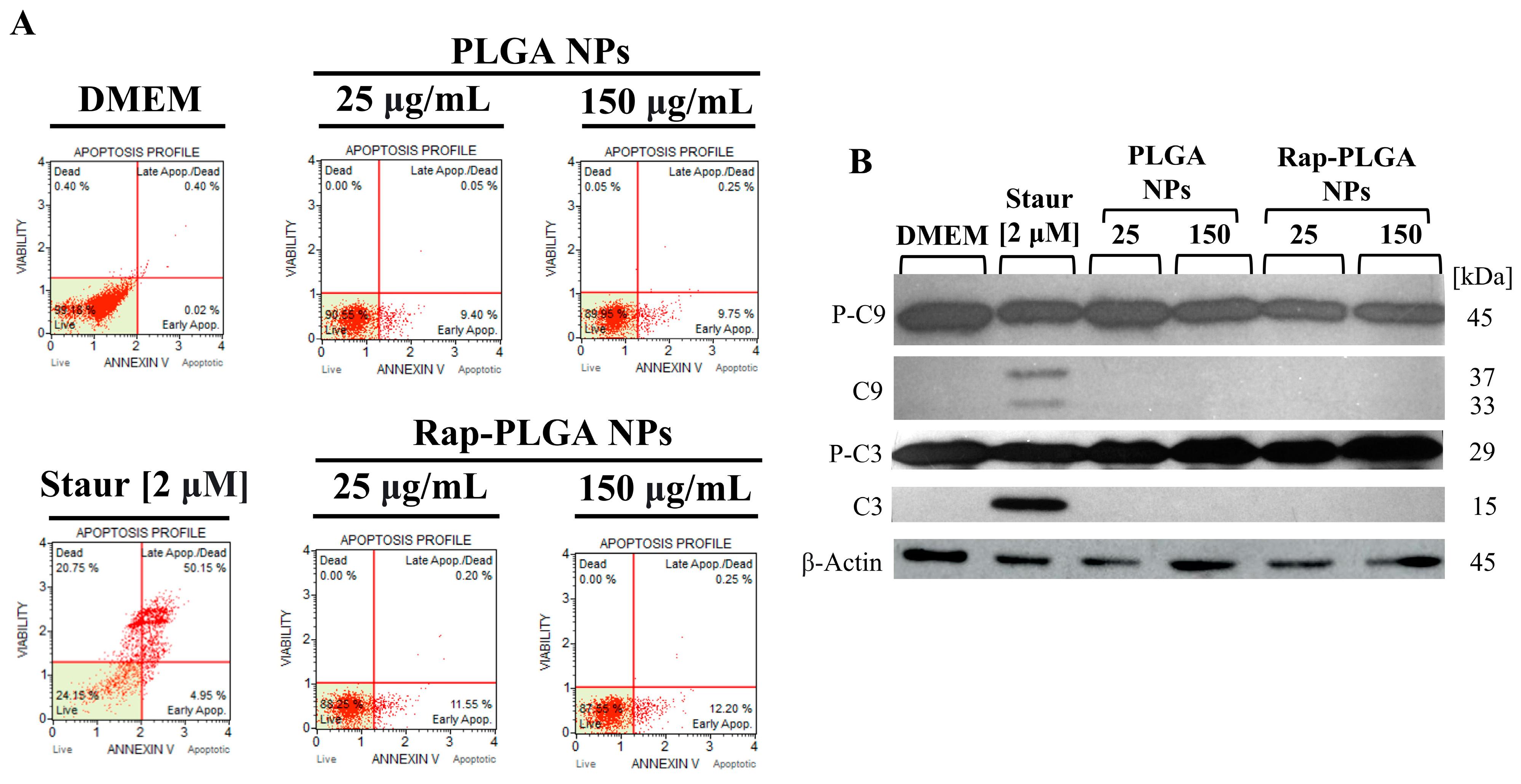
3.3.2. Treatment with Rap-PLGA NPs Prevented Apoptosis in a SCA7 Glial Cell Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carvalho Donis, K.; Preusser Mattos, E.; Anjos Silva, A.; Vasata Furtado, G.; Luiza Saraiva-Pereira, M.; Bannach Jardim, L.; Alex Saute, J. Infantile Spinocerebellar Ataxia Type 7: Case Report and a Review of the Literature. J. Neurol. Sci. 2015, 354, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Durr, A. Autosomal Dominant Cerebellar Ataxias: Polyglutamine Expansions and Beyond. Lancet Neurol. 2010, 9, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Rüb, U.; Schöls, L.; Paulson, H.; Auburger, G.; Kermer, P.; Jen, J.C.; Seidel, K.; Korf, H.W.; Deller, T. Clinical Features, Neurogenetics and Neuropathology of the Polyglutamine Spinocerebellar Ataxias Type 1, 2, 3, 6 and 7. Prog. Neurobiol. 2013, 104, 38–66. [Google Scholar] [CrossRef]
- Rodríguez-Labrada, R.; Martins, A.C.; Magaña, J.J.; Vazquez-Mojena, Y.; Medrano-Montero, J.; Fernandez-Ruíz, J.; Cisneros, B.; Teive, H.; McFarland, K.N.; Saraiva-Pereira, M.L.; et al. Founder Effects of Spinocerebellar Ataxias in the American Continents and the Caribbean. Cerebellum 2020, 19, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Jonasson, J.; Juvonen, V.; Sistonen, P.; Ignatius, J.; Johansson, D.; Björck, E.J.; Wahlström, J.; Melberg, A.; Holmgren, G.; Forsgren, L.; et al. Evidence for a Common Spinocerebellar Ataxia Type 7 (SCA7) Founder Mutation in Scandinavia. Eur. J. Hum. Genet. 2000, 8, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C.; Atadzhanov, M.; Mwaba, M.; Greenberg, J. Evidence for a Common Founder Effect amongst South African and Zambian Individuals with SCA7. J. Neurol. Sci. 2015, 354, 75–78. [Google Scholar] [CrossRef]
- Magaña, J.; Tapia-Guerrero, Y.; Velázquez-Pérez, L.; Cerecedo-Zapata, C.; Maldonado-Rodríguez, M.; Jano-Ito, J.; Leyva-García, N.; González-Piña, R.; Martínez-Cruz, E.; Hernández-Hernández, O.; et al. Analysis of CAG Repeats in Five SCA Loci in Mexican Population: Epidemiological Evidence of a SCA7 Founder Effect. Clin. Genet. 2014, 85, 159–165. [Google Scholar] [CrossRef]
- Horton, L.C.; Frosch, M.P.; Vangel, M.G.; Weigel-Difranco, C.; Berson, E.L.; Schmahmann, J.D. Spinocerebellar Ataxia Type 7: Clinical Course, Phenotype-Genotype Correlations, and Neuropathology. Cerebellum 2013, 12, 176–193. [Google Scholar] [CrossRef]
- Latouche, M.; Fragner, P.; Martin, E.; El Hachimi, K.H.; Zander, C.; Sittler, A.; Ruberg, M.; Brice, A.; Stevanin, G. Polyglutamine and polyalanine expansions in ataxin7 result in different types of aggregation and levels of toxicity. Mol. Cell. Neurosci. 2006, 31, 438–445. [Google Scholar] [CrossRef]
- Ström, A.L.; Forsgren, L.; Holmberg, M. A Role for Both Wild-Type and Expanded Ataxin-7 in Transcriptional Regulation. Neurobiol. Dis. 2005, 20, 646–655. [Google Scholar] [CrossRef]
- Ajayi, A.; Yu, X.; Wahlo-Svedin, C.; Tsirigotaki, G.; Karlström, V.; Ström, A.-L. Altered P53 and NOX1 Activity Cause Bioenergetic Defects in a SCA7 Polyglutamine Disease Model. Biochim. Biophys. Acta BBA-Bioenergy 2015, 1847, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Furrer, S.A.; Mohanachandran, M.S.; Waldherr, S.M.; Chang, C.; Damian, V.A.; Sopher, B.L.; Garden, G.A.; la Spada, A.R. SCA7 Cerebellar Disease Requires the Coordinated Action of Mutant Ataxin-7 in Neurons and Glia, and Displays Non-Cell Autonomous Bergmann Glia Degeneration. J. Neurosci. 2011, 3, 16269–16278. [Google Scholar] [CrossRef] [PubMed]
- Garden, G.A.; la Spada, A.R. Molecular Pathogenesis and Cellular Pathology of Spinocerebellar Ataxia Type 7 Neurodegeneration. Cerebellum 2008, 7, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Matos, C.A.; Almeida, L.P.; Nobrega, C. Proteolytic Cleavage of Polyglutamine Disease-Causing Proteins: Revisiting the Toxic Fragment Hypothesis. Curr. Pharm. Des. 2017, 23, 753–775. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; Stoyas, C.A.; Switonski, P.M.; Ichou, F.; Fan, W.; Collins, B.; Wall, C.E.; Adanyeguh, I.; Niu, C.; Sopher, B.L.; et al. Metabolic and Organelle Morphology Defects in Mice and Human Patients Define Spinocerebellar Ataxia Type 7 as a Mitochondrial Disease. Cell Rep. 2019, 26, 1189–1202. [Google Scholar] [CrossRef]
- Torres-Ramos, Y.; Montoya-Estrada, A.; Cisneros, B.; Tercero-Pérez, K.; León-Reyes, G.; Leyva-García, N.; Hernández-Hernández, O.; Magaña, J.J. Oxidative Stress in Spinocerebellar Ataxia Type 7 Is Associated with Disease Severity. Cerebellum 2018, 17, 601–609. [Google Scholar] [CrossRef]
- Weber, J.J.; Sowa, A.S.; Binder, T.; Hübener, J. From Pathways to Targets: Understanding the Mechanisms behind Polyglutamine Disease. Biomed. Res. Int. 2014, 2014, 701758. [Google Scholar] [CrossRef]
- Xu, Z.; Joel Tito, A.; Rui, Y.-N.; Zhang, S. Studying Polyglutamine Diseases in Drosophila. Exp. Neurol. 2015, 274, 25–41. [Google Scholar] [CrossRef]
- Zander, C.; Takahashi, J.; el Hachimi, K.H.; Fujigasaki, H.; Albanese, V.; Lebre, A.S.; Stevanin, G.; Duyckaerts, C.; Brice, A. Similarities between Spinocerebellar Ataxia Type 7 (SCA7) Cell Models and Human Brain: Proteins Recruited in Inclusions and Activation of Caspase-3. Hum. Mol. Genet. 2001, 10, 2569–2579. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Trottier, Y. Molecular Targets and Therapeutic Strategies in Spinocerebellar Ataxia Type 7. Neurotherapeutics 2019, 16, 1074–1096. [Google Scholar] [CrossRef]
- Cancel, G.; Duyckaerts, C.; Holmberg, M.; Zander, C.; Yvert, G.; Lebre, A.S.; Ruberg, M.; Faucheux, B.; Agid, Y.; Hirsch, E.; et al. Distribution of Ataxin-7 in Normal Human Brain and Retina. Brain 2000, 123, 2519–2530. [Google Scholar] [CrossRef] [PubMed]
- Guyenet, S.J.; Mookerjee, S.S.; Lin, A.; Custer, S.K.; Chen, S.F.; Sopher, B.L.; la Spada, A.R.; Ellerby, L.M. Proteolytic Cleavage of Ataxin-7 Promotes SCA7 Retinal Degeneration and Neurological Dysfunction. Hum. Mol. Genet. 2015, 24, 3908–3917. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; la Spada, A.R. Autophagy in Polyglutamine Disease: Imposing Order on Disorder or Contributing to the Chaos? Mol. Cell. Neurosci. 2015, 66, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Sambataro, F.; Pennuto, M. Cell-Autonomous and Non-Cell-Autonomous Toxicity in Polyglutamine Diseases. Prog. Neurobiol. 2012, 97, 152–172. [Google Scholar] [CrossRef] [PubMed]
- Borbolla-Jiménez, F.V.; Del Prado-Audelo, M.L.; Cisneros, B.; Caballero-Florán, I.H.; Leyva-Gómez, G.; Magaña, J.J. New Perspectives of Gene Therapy on Polyglutamine Spinocerebellar Ataxias: From Molecular Targets to Novel Nanovectors. Pharmaceutics 2021, 13, 1018. [Google Scholar] [CrossRef]
- Chort, A.; Alves, S.; Marinello, M.; Dufresnois, B.; Dornbierer, J.G.; Tesson, C.; Latouche, M.; Baker, D.P.; Barkats, M.; el Hachimi, K.H.; et al. Interferon Beta Induces Clearance of Mutant Ataxin 7 and Improves Locomotion in SCA7 Knock-in Mice. Brain 2013, 136, 1732–1745. [Google Scholar] [CrossRef]
- Patel, M.M.; Patel, B.M. Crossing the Blood–Brain Barrier: Recent Advances in Drug Delivery to the Brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef]
- Escalona-rayo, O.; Fuentes-vázquez, P.; Leyva-gómez, G.; Cisneros, B.; Villalobos, R.; Magaña, J.J.; Quintanar-, D. Nanoparticulate Strategies for the Treatment of Polyglutamine Diseases by Halting the Protein Aggregation Process. Drug Dev. Ind. Pharm. 2017, 43, 871–888. [Google Scholar] [CrossRef]
- Grabrucker, A.M.; Ruozi, B.; Belletti, D.; Pederzoli, F.; Forni, F.; Vandelli, M.A.; Tosi, G. Nanoparticle Transport across the Blood Brain Barrier. Tissue Barriers 2016, 4, e1153568. [Google Scholar] [CrossRef]
- del Prado-Audelo, M.L.; Magaña, J.J.; Mejía-Contreras, B.A.; Borbolla-Jiménez, F.V.; Giraldo-Gomez, D.M.; Piña-Barba, M.C.; Quintanar-Guerrero, D.; Leyva-Gómez, G. In Vitro Cell Uptake Evaluation of Curcumin-Loaded PCL/F68 Nanoparticles for Potential Application in Neuronal Diseases. J. Drug Deliv. Sci. Technol. 2019, 52, 905–914. [Google Scholar] [CrossRef]
- Nikalje, A.P. Nanotechnology and Its Applications in Medicine. Med. Chem. 2015, 5, 2. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Nair, S.; Bhatnagar, P.; Karmakar, M.; Kumari, M.; Chauhan, L.K.S.; Patel, D.K.; et al. Curcumin-Loaded Nanoparticles Potently Induce Adult Neurogenesis and Reverse Cognitive Deficits in Alzheimer’s Disease Model via Canonical Wnt/β-Catenin Pathway. ACS Nano 2014, 8, 76–103. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Guerrero, Y.S.; Del Prado-Audelo, M.L.; Borbolla-Jiménez, F.V.; Giraldo Gomez, D.M.; García-Aguirre, I.; Colín-Castro, C.A.; Morales-González, J.A.; Leyva-Gómez, G.; Magaña, J.J. Effect of UV and Gamma Irradiation Sterilization Processes in the Properties of Different Polymeric Nanoparticles for Biomedical Applications. Materials 2020, 13, 1090. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Sánchez, R.; Ávila-Avilés, R.D.; Hernández-Hernández, J.M.; Sánchez-Celis, D.; Azotla-Vilchis, C.N.; Gómez-Macías, E.R.; Leyva-García, N.; Ortega, A.; Magaña, J.J.; Cisneros, B.; et al. RNA Foci Formation in a Retinal Glial Model for Spinocerebellar Ataxia Type 7. Life 2022, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, U.; Sharma, R.; Gupta, M.; Vyas, S.P. Is Nanotechnology a Boon for Oral Drug Delivery? Drug Discov. Today 2014, 19, 1530–1546. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Kumar, S.; Lather, V. Hybrid Poly(Lactic-Co-Glycolic Acid) Nanoparticles: Design and Delivery Prospectives. Drug Discov. Today 2015, 20, 95–104. [Google Scholar] [CrossRef]
- Huang, R.; Ke, W.; Liu, Y.; Jiang, C.; Pei, Y. The Use of Lactoferrin as a Ligand for Targeting the Polyamidoamine-Based Gene Delivery System to the Brain. Biomaterials 2008, 29, 238–246. [Google Scholar] [CrossRef]
- Kreuter, J. Influence of the Surface Properties on Nanoparticle-Mediated Transport of Drugs to the Brain. Proc. J. Nanosci. Nanotechnol. 2004, 4, 484–488. [Google Scholar] [CrossRef]
- Domínguez, A.; Suárez-Merino, B.; Goñi-De-Cerio, F. Nanoparticles and Blood-Brain Barrier: The Key to Central Nervous System Diseases. J. Nanosci. Nanotechnol. 2014, 14, 766–779. [Google Scholar] [CrossRef]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A Rational Mechanism for Combination Treatment of Huntington’s Disease Using Lithium and Rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of MTOR Induces Autophagy and Reduces Toxicity of Polyglutamine Expansions in Fly and Mouse Models of Huntington Disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Mansoor, S.; Rafi, Z.; Kumari, B.; Shoaib, A.; Saeed, M.; Alshehri, S.; Ghoneim, M.M.; Rahamathulla, M.; Hani, U.; et al. A review on nanotechnology: Properties, applications, and mechanistic insights of cellular uptake mechanisms. J. Mol. Liq. 2022, 348, 118008. [Google Scholar] [CrossRef]
- Petros, R.A.; Desimone, J.M. Strategies in the Design of Nanoparticles for Therapeutic Applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Lv, X.; Le, Y. Chitosan-Modified PLGA Nanoparticles for Control-Released Drug Delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Li, J.; Shen, Y.; Lu, W.; Gao, X.; Zhang, Q.; Jiang, X. Lactoferrin-Conjugated PEG-PLA Nanoparticles with Improved Brain Delivery: In Vitro and in Vivo Evaluations. J. Control. Release 2009, 134, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Crucho, C.I.C.; Barros, M.T. Polymeric Nanoparticles: A Study on the Preparation Variables and Characterization Methods. Mater. Sci. Eng. C 2017, 80, 771–784. [Google Scholar] [CrossRef]
- Tsai, Y.M.; Chien, C.F.; Lin, L.C.; Tsai, T.H. Curcumin and Its Nano-Formulation: The Kinetics of Tissue Distribution and Blood-Brain Barrier Penetration. Int. J. Pharm. 2011, 416, 331–338. [Google Scholar] [CrossRef]
- Nasiri, F.; Faghfouri, L.; Hamidi, M. Preparation, Optimization, and in-Vitro Characterization of α-Tocopherol-Loaded Solid Lipid Nanoparticles (SLNs). Drug Dev. Ind. Pharm. 2020, 46, 159–171. [Google Scholar] [CrossRef]
- Caro, N.; Bruna, T.; Guerreiro, A.; Alvarez-Tejos, P.; Garretón, V.; Piletsky, S.; González-Casanova, J.; Rojas-Gómez, D.; Ehrenfeld, N. Florfenicol Binding to Molecularly Imprinted Polymer Nanoparticles in Model and Real Samples. Nanomaterials 2020, 10, 306. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Hunter, A.C. Poloxamers and Poloxamines in Nanoparticle Engineering and Experimental Medicine. Trends Biotechnol. 2000, 18, 412–420. [Google Scholar] [CrossRef]
- Pitto-Barry, A.; Barry, N.P.E. Pluronic® Block-Copolymers in Medicine: From Chemical and Biological Versatility to Rationalisation and Clinical Advances. Polym. Chem. 2014, 5, 3291–3297. [Google Scholar] [CrossRef]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The Mechanisms of Drug Release in Poly(Lactic-Co-Glycolic Acid)-Based Drug Delivery Systems—A Review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Pascolo, L.; Bortot, B.; Benseny-Cases, N.; Gianoncelli, A.; Tosi, G.; Ruozi, B.; Rizzardi, C.; de Martino, E.; Vandelli, M.A.; Severini, G.M. Detection of PLGA-Based Nanoparticles at a Single-Cell Level by Synchrotron Radiation FTIR Spectromicroscopy and Correlation with X-Ray Fluorescence Microscopy. Int. J. Nanomed. 2014, 9, 2791–2801. [Google Scholar] [CrossRef]
- D’Avila Carvalho Erbetta, C. Synthesis and Characterization of Poly(D,L-Lactide-Co-Glycolide) Copolymer. J. Biomater. Nanobiotechnol. 2012, 03, 208–225. [Google Scholar] [CrossRef]
- Raposo, C.D.; Costa, R.; Petrova, K.T.; Brito, C.; Scotti, M.T.; Cardoso, M.M. Development of Novel Galactosylated PLGA Nanoparticles for Hepatocyte Targeting Using Molecular Modelling. Polymers 2020, 12, 94. [Google Scholar] [CrossRef]
- Mandegari, M.; Ghasemi-Mobarakeh, L.; Zamani, M. Manipulating the Degradation Rate of PVA Nanoparticles by a Novel Chemical-Free Method. Polym. Adv. Technol. 2019, 30, 2381–2391. [Google Scholar] [CrossRef]
- Pereira, V.A.; de Arruda, I.N.Q.; Stefani, R. Active Chitosan/PVA Films with Anthocyanins from Brassica Oleraceae (Red Cabbage) as Time-Temperature Indicators for Application in Intelligent Food Packaging. Food Hydrocoll. 2015, 43, 180–188. [Google Scholar] [CrossRef]
- Patel, A.R.; Vavia, P.R. Evaluation of Synthesized Cross Linked Polyvinyl Alcohol as Potential Disintegrant. J. Pharm. Pharm. Sci. 2010, 13, 114–127. [Google Scholar] [CrossRef]
- Girotra, P.; Singh, S.K.; Kumar, G. Development of Zolmitriptan Loaded PLGA/Poloxamer Nanoparticles for Migraine Using Quality by Design Approach. Int. J. Biol. Macromol. 2016, 85, 92–101. [Google Scholar] [CrossRef]
- Zhu, C.; Yang, H.; Shen, L.; Zheng, Z.; Zhao, S.; Li, Q.; Yu, F.; Cen, L. Microfluidic Preparation of PLGA Microspheres as Cell Carriers with Sustainable Rapa Release. J. Biomater. Sci. Polym. Ed. 2019, 30, 737–755. [Google Scholar] [CrossRef]
- Patel, R.R.; Khan, G.; Chaurasia, S.; Kumar, N.; Mishra, B. Rationally Developed Core-Shell Polymeric-Lipid Hybrid Nanoparticles as a Delivery Vehicle for Cromolyn Sodium: Implications of Lipid Envelop on in Vitro and in Vivo Behaviour of Nanoparticles upon Oral Administration. RSC Adv. 2015, 5, 76491–76506. [Google Scholar] [CrossRef]
- Keles, H.; Naylor, A.; Clegg, F.; Sammon, C. Investigation of Factors Influencing the Hydrolytic Degradation of Single PLGA Microparticles. Polym. Degrad. Stab. 2015, 119, 228–241. [Google Scholar] [CrossRef]
- Emami, S.; Valizadeh, H.; Islambulchilar, Z.; Zakeri-Milani, P. Development and Physicochemical Characterization of Sirolimus Solid Dispersions Prepared by Solvent Evaporation Method. Adv. Pharm. Bull. 2014, 4, 369–374. [Google Scholar] [CrossRef]
- Kim, M.S.; Kim, J.S.; Park, H.J.; Cho, W.K.; Cha, K.H.; Hwang, S.J. Enhanced Bioavailability of Sirolimus via Preparation of Solid Dispersion Nanoparticles Using a Supercritical Antisolvent Process. Int. J. Nanomed. 2011, 6, 2997–3009. [Google Scholar] [CrossRef]
- Bhadra, D.; Bhadra, S.; Jain, P.; Jain, N.K. Pegnology: A Review of PEG-Ylated Systems. Pharmazie 2002, 57, 5–29. [Google Scholar] [PubMed]
- Abdelwahed, W.; Degobert, G.; Stainmesse, S.; Fessi, H. Freeze-Drying of Nanoparticles: Formulation, Process and Storage Considerations. Adv. Drug Deliv. Rev. 2006, 58, 1688–1713. [Google Scholar] [CrossRef]
- Li, Q.; Cai, T.; Huang, Y.; Xia, X.; Cole, S.P.C.; Cai, Y. A Review of the Structure, Preparation, and Application of NLCs, PNPs, and PLNs. Nanomaterials 2017, 7, 122. [Google Scholar] [CrossRef]
- Bruschi, M.L. Mathematical Models of Drug Release. Strateg. Modify Drug Release Pharm. Syst. 2015, 63, 63–86. [Google Scholar] [CrossRef]
- Pourtalebi Jahromi, L.; Ghazali, M.; Ashrafi, H.; Azadi, A. A Comparison of Models for the Analysis of the Kinetics of Drug Release from PLGA-Based Nanoparticles. Heliyon 2020, 6, e03451. [Google Scholar] [CrossRef]
- Chourasiya, V.; Bohrey, S.; Pandey, A. Formulation, Optimization, Characterization and in-Vitro Drug Release Kinetics of Atenolol Loaded PLGA Nanoparticles Using 33 Factorial Design for Oral Delivery. Mater. Discov. 2016, 5, 1–13. [Google Scholar] [CrossRef]
- Rüb, U.; Brunt, E.R.; Gierga, K.; Seidel, K.; Schultz, C.; Schöls, L.; Auburger, G.; Heinsen, H.; Ippel, P.F.; Glimmerveen, W.F.; et al. Spinocerebellar Ataxia Type 7 (SCA7): First Report of a Systematic Neuropathological Study of the Brain of a Patient with a Very Short Expanded CAG-Repeat. Brain Pathol. 2005, 15, 287–295. [Google Scholar] [CrossRef]
- Rüb, U.; Brunt, E.R.; Seidel, K.; Gierga, K.; Mooy, C.M.; Kettner, M.; Van Broeckhoven, C.; Bechmann, I.; La Spada, A.R.; Schöls, L.; et al. Spinocerebellar ataxia type 7 (SCA7): Widespread brain damage in an adult-onset patient with progressive visual impairments in comparison with an adult-onset patient without visual impairments. Neuropathol. Appl. Neurobiol. 2008, 34, 155–168. [Google Scholar] [CrossRef]
- Martin, J.; van Regemorter, N.; Krols, L.; Brucher, J.; de Barsy, T.; Szliwowski, H.; Evrard, P.; Ceuterick, C.; Tassignon, M.; Smet-Dieleman, H.; et al. On an Autosomal Dominant Form of Retinal-Cerebellar Degeneration: An Autopsy Study of Five Patients in One Family. Acta Neuropathol. 1994, 88, 277–286. [Google Scholar] [CrossRef]
- Michalik, A.; Martin, J.J.; van Broeckhoven, C. Spinocerebellar Ataxia Type 7 Associated with Pigmentary Retinal Dystrophy. Eur. J. Hum. Genet. 2004, 12, 2–15. [Google Scholar] [CrossRef]
- Hernandez-Castillo, C.R.; Galvez, V.; Diaz, R.; Fernandez-Ruiz, J. Specific Cerebellar and Cortical Degeneration Correlates with Ataxia Severity in Spinocerebellar Ataxia Type 7. Brain Imaging Behav. 2016, 10, 252–257. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Pennesi, M.E.; Weeber, E.J.; Xu, B.; Atkinson, R.; Chen, S.; Armstrong, D.L.; Wu, S.M.; Sweatt, J.D.; Zoghbi, H.Y. SCA7 Knockin Mice Model Human SCA7 and Reveal Gradual Accumulation of Mutant Ataxin-7 in Neurons and Abnormalities in Short-Term Plasticity. Neuron 2003, 37, 383–401. [Google Scholar] [CrossRef]
- Furrer, S.A.; Waldherr, S.M.; Mohanachandran, M.S.; Baughn, T.D.; Nguyen, K.-T.; Sopher, B.L.; Damian, V.A.; Garden, G.A.; Spada, A.R. La Reduction of Mutant Ataxin-7 Expression Restores Motor Function and Prevents Cerebellar Synaptic Reorganization in a Conditional Mouse Model of SCA7. Hum. Mol. Genet. 2013, 22, 890–903. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.-B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B.; et al. SCA7 Mouse Cerebellar Pathology Reveals Preferential Downregulation of Key Purkinje Cell-Identity Genes and Shared Disease Signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef]
- Helmlinger, D.; Hardy, S.; Abou-Sleymane, G.; Eberlin, A.; Bowman, A.B.; Gansmüller, A.; Picaud, S.; Zoghbi, H.Y.; Trottier, Y.; Tora, L.; et al. Glutamine-Expanded Ataxin-7 Alters TFTC/STAGA Recruitment and Chromatin Structure Leading to Photoreceptor Dysfunction. PLoS Biol. 2006, 4, 0432–0445. [Google Scholar] [CrossRef]
- Liu, P.; Sun, Y.; Wang, Q.; Sun, Y.; Li, H.; Duan, Y. Intracellular Trafficking and Cellular Uptake Mechanism of MPEG-PLGA-PLL and MPEG-PLGA-PLL-Gal Nanoparticles for Targeted Delivery to Hepatomas. Biomaterials 2014, 35, 760–770. [Google Scholar] [CrossRef]
- Malinovskaya, Y.; Melnikov, P.; Baklaushev, V.; Gabashvili, A.; Osipova, N.; Mantrov, S.; Ermolenko, Y.; Maksimenko, O.; Gorshkova, M.; Balabanyan, V.; et al. Delivery of Doxorubicin-Loaded PLGA Nanoparticles into U87 Human Glioblastoma Cells. Int. J. Pharm. 2017, 524, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Santos, B.; Gremião, M.P.D.; Chorilli, M. Nanotechnology-Based Drug Delivery Systems for the Treatment of Alzheimer’s Disease. Int. J. Nanomed. 2015, 10, 4981–5003. [Google Scholar] [CrossRef] [PubMed]
- Batrakova, E.V.; Kabanov, A.V. Pluronic Block Copolymers: Evolution of Drug Delivery Concept from Inert Nanocarriers to Biological Response Modifiers. J. Control. Release 2008, 130, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Petri, B.; Bootz, A.; Khalansky, A.; Hekmatara, T.; Müller, R.; Uhl, R.; Kreuter, J.; Gelperina, S. Chemotherapy of Brain Tumour Using Doxorubicin Bound to Surfactant-Coated Poly(Butyl Cyanoacrylate) Nanoparticles: Revisiting the Role of Surfactants. J. Control. Release 2007, 117, 51–58. [Google Scholar] [CrossRef]
- Gelperina, S.; Maksimenko, O.; Khalansky, A.; Vanchugova, L.; Shipulo, E.; Abbasova, K.; Kreuter, J. Drug Delivery to the Brain Using Surfactant-Coated Poly(Lactide-Co-Glycolide) Nanoparticles: Influence of the Formulation Parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163. [Google Scholar] [CrossRef]
- Huerta García, E.; Pérez-Arizti, J.A.; Márquez-Ramírez, S.G.; Delgado-Buenrostro, N.L.; Chirino, Y.I.; Iglesias, G.G.; López-Marure, R. Titanium Dioxide Nanoparticles Induce Strong Oxidative Stress and Mitochondrial Damage in Glial Cells. Free Radic. Biol. Med. 2014, 73, 84–94. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, J.; Zuo, Y.; Gu, Z. Rapamycin Protects the Mitochondria against Oxidative Stress and Apoptosis in a Rat Model of Parkinson’s Disease. Int. J. Mol. Med. 2013, 31, 825–832. [Google Scholar] [CrossRef]
- Chauhan, A.; Sharma, U.; Jagannathan, N.R.; Reeta, K.H.; Gupta, Y.K. Rapamycin Protects against Middle Cerebral Artery Occlusion Induced Focal Cerebral Ischemia in Rats. Behav. Brain Res. 2011, 225, 603–609. [Google Scholar] [CrossRef]
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Astrocyte-Neuron Metabolic Relationships: For Better and for Worse. Trends Neurosci. 2011, 34, 76–87. [Google Scholar] [CrossRef]
- Brand, A.; Richter-Landsberg, C.; Leibfritz, D. Multinuclear NMR Studies on the Energy Metabolism of Glial and Neuronal Cells. Dev. Neurosci. 1993, 15, 289–298. [Google Scholar] [CrossRef]
- Poeck, B.; Fischer, S.; Gunning, D.; Zipursky, S.L.; Salecker, I. Glial Cells Mediate Target Layer Selection of Retinal Axons in the Developing Visual System of Drosophila. Neuron 2001, 29, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Custer, S.K.; Garden, G.A.; Gill, N.; Rueb, U.; Libby, R.T.; Schultz, C.; Guyenet, S.J.; Deller, T.; Westrum, L.E.; Sopher, B.L.; et al. Bergmann Glia Expression of Polyglutamine-Expanded Ataxin-7 Produces Neurodegeneration by Impairing Glutamate Transport. Nat. Neurosci. 2006, 9, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Ravikumar, B.; Menzies, F.M.; Oroz, L.G.; Underwood, B.R.; Pangalos, M.N.; Schmitt, I.; Wullner, U.; Evert, B.O.; O’Kane, C.J.; et al. Rapamycin Alleviates Toxicity of Different Aggregate-Prone Proteins. Hum. Mol. Genet. 2006, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ajayi, A.; Boga, N.R.; Ström, A.L. Differential Degradation of Full-Length and Cleaved Ataxin-7 Fragments in a Novel Stable Inducible SCA7 Model. J. Mol. Neurosci. 2012, 47, 219–233. [Google Scholar] [CrossRef]
- Masserini, M. Nanoparticles for Brain Drug Delivery. ISRN Biochem. 2013, 2013, 238428. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in Healthy Aging and Disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- LaSpada, A.R.; Fu, Y.; Chen, S. Polyglutamine-Expanded Ataxin-7 Antagonizes CRX Function and Induces Cone-Rod Dystrophy in a Mouse Model of SCA7. Neuron 2001, 31, 913–927. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Messaddeq, N.; Karam, A.; Jacquard, C.; Weber, C.; Jonet, L.; Wolfrum, U.; Jeanny, J.C.; Trottier, Y. Polyglutamine Toxicity Induces Rod Photoreceptor Division, Morphological Transformation or Death in Spinocerebellar Ataxia 7 Mouse Retina. Neurobiol. Dis. 2010, 40, 311–324. [Google Scholar] [CrossRef]
- Wang, H.L.; Yeh, T.H.; Chou, A.H.; Kuo, Y.L.; Luo, L.J.; He, C.Y.; Huang, P.C.; Li, A.H. Polyglutamine-Expanded Ataxin-7 Activates Mitochondrial Apoptotic Pathway of Cerebellar Neurons by Upregulating Bax and Downregulating Bcl-XL. Cell Signal. 2006, 18, 541–552. [Google Scholar] [CrossRef]
- Dong, H.; Qin, Y.; Huang, Y.; Ji, D.; Wu, F. Poloxamer 188 Rescues MPTP-Induced Lysosomal Membrane Integrity Impairment in Cellular and Mouse Models of Parkinson’s Disease. Neurochem. Int. 2019, 126, 178–186. [Google Scholar] [CrossRef]
- Dubaj, T.; Kozics, K.; Sramkova, M.; Manova, A.; Bastús, N.G.; Moriones, O.H.; Kohl, Y.; Dusinska, M.; Runden-Pran, E.; Puntes, V.; et al. Pharmacokinetics of PEGylated Gold Nanoparticles: In Vitro—In Vivo Correlation. Nanomaterials 2022, 12, 511. [Google Scholar] [CrossRef] [PubMed]
- Di Gioacchino, M.; Petrarca, C.; Lazzarin, F.; Di Giampaolo, L.; Sabbioni, E.; Boscolo, P.; Mariani-Costantini, R.; Bernardini, G. Immunotoxicity of Nanoparticles. Int. J. Immunopathol. Pharmacol. 2011, 24, 65S–71S. [Google Scholar] [PubMed]
- Mohanty, C.; Sahoo, S.K. The in Vitro Stability and in Vivo Pharmacokinetics of Curcumin Prepared as an Aqueous Nanoparticulate Formulation. Biomaterials 2010, 31, 6597–6611. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Size (nm) ± SD | PDI ± SD | Z-Potential (mV) ± SD |
|---|---|---|---|
| PLGA NPs (lyophilized) | 212.73 ± 4.87 | 0.053 ± 0.02 | −17.66 ± 0.40 |
| Rap-PLGA NPs (lyophilized) | 211.86 ± 4.037 | 0.045 ± 0.017 | −18.53 ± 0.288 |
| Stability Assay (PLGA NPs dispersed) | |||
| Time of storage | Size (nm) ± SD | PDI ± SD | Z-Potential (mV) ± SD |
| 1 month | 226.5 ± 5.074 | 0.031 ± 0.03 | −17.9 ± 0.436 |
| 2 months | 221 ± 7.016 | 0.048 ± 0.031 | −20.6 ± 0.503 |
| 3 months | 217.2 ± 4.359 | 0.079 ± 0.037 | −19.3 ± 0.755 |
| 4 months | 208.7 ± 2.868 | 0.041 ± 0.007 | −13.6 ± 0.557 |
| 6 months | 205.7 ± 2.723 | 0.037 ± 0.019 | −14 ± 0.379 |
| Rapamycin release profile (entire time range) | ||||
| Mathematical model | Equation | R2 | K | n |
| Zero order | Qt = Q0 + K0t | 0.83 | 2.13 | 0 |
| First Order | ln Qt = lnQ0 + K1t | 0.52 | 0.066 | 0 |
| Higuchi | Qt = KHt1/2 | 0.94 | 15.60 | 0 |
| Korsmeyer–Peppas | Qt/Q∞ = Kktn | 0.96 | 23.57 | 0.37 |
| Rapamycin release profile (burst effect) | ||||
| Mathematical model | Equation | R2 | K | n |
| Zero order | Qt = Q0 + K0t | 0.99 | 14.01 | 0 |
| First Order | ln Qt = lnQ0 + K1t | 0.85 | 1.32 | 0 |
| Higuchi | Qt = KHt1/2 | 0.99 | 19.89 | 0 |
| Korsmeyer–Peppas | Qt/Q∞ = Kktn | 0.99 | 15.05 | 0.91 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borbolla-Jiménez, F.V.; García-Aguirre, I.A.; Del Prado-Audelo, M.L.; Hernández-Hernández, O.; Cisneros, B.; Leyva-Gómez, G.; Magaña, J.J. Development of a Polymeric Pharmacological Nanocarrier System as a Potential Therapy for Spinocerebellar Ataxia Type 7. Cells 2023, 12, 2735. https://doi.org/10.3390/cells12232735
Borbolla-Jiménez FV, García-Aguirre IA, Del Prado-Audelo ML, Hernández-Hernández O, Cisneros B, Leyva-Gómez G, Magaña JJ. Development of a Polymeric Pharmacological Nanocarrier System as a Potential Therapy for Spinocerebellar Ataxia Type 7. Cells. 2023; 12(23):2735. https://doi.org/10.3390/cells12232735
Chicago/Turabian StyleBorbolla-Jiménez, Fabiola V., Ian A. García-Aguirre, María Luisa Del Prado-Audelo, Oscar Hernández-Hernández, Bulmaro Cisneros, Gerardo Leyva-Gómez, and Jonathan J. Magaña. 2023. "Development of a Polymeric Pharmacological Nanocarrier System as a Potential Therapy for Spinocerebellar Ataxia Type 7" Cells 12, no. 23: 2735. https://doi.org/10.3390/cells12232735