1. Introduction
In recent years, the demand for the production of biopharmaceuticals, such as therapeutic antibodies for cancer and rheumatoid arthritis, has rapidly expanded due to their high therapeutic efficacy [
1]. More than 70% of therapeutic antibodies are currently produced in cell culture using Chinese hamster ovary (CHO) cells as host production cells [
2]. CHO cells can produce bioactive, non-immunogenic recombinant antibodies with glycosylation patterns similar to those of human antibodies [
3]. However, the slow growth of cells compared with that of microorganisms and the consumption of large amounts of medium lead to increased production costs [
4]. To reduce manufacturing costs, cell line development [
5], medium development [
6], and culture environment and process optimization [
7], among others, have been performed to improve production titers by increasing specific productivity or viable cell density. Several studies have achieved high-titer antibody production at concentrations on the order of grams per liter by improving culture processes such as high-cell-density fed-batch culture and continuous culture [
8,
9]. Cell line development, which is the most upstream process of antibody manufacturing, is very important because the cell line determines the performance of the entire manufacturing process. Improving the system for expressing the target gene is effective to further elevate the antibody production level.
Various studies have been conducted on the constituent elements of gene expression cassettes to achieve high expression of the target gene, including insulators that are less susceptible to gene silencing [
10,
11], improved transcription termination at gene insertion sites in the genome [
12], stabilization of target transcripts [
13], and codon optimization [
14]. Because gene expression is triggered by transcription from promoters, the ability to induce strong gene expression has been developed from both natural and synthetic promoters [
15]. The tetracycline-dependent transcription activation system is a synthetic promoter that works in animal cells [
16]. This system consists of a fusion protein of the tetracycline repressor (TetR), as a DNA-binding domain, the transcriptional activation domain (TAD), as an artificial transcription factor (aTF), and an artificial promoter with a TetR-responsive element (TRE) sequence. The gene expression system can strongly induce the expression of a target gene. We have constructed gene expression control systems that work in animal cells using a synthetic biology approach. In these systems, the overexpression of aTF is induced by transcriptional amplification using a positive feedback loop of the aTF expression in the Tet-transcription activation system, inducing high-level expression of target genes under the control of an artificial promoter that responds to aTF. This system not only regulates target gene expression by adding inducer drugs, which is the original induction method of the Tet-transcription activation system, but also allows for autonomous induction according to external environmental factors by selecting a trigger promoter that induces aTF expression. In fact, we have succeeded in generating cell lines that can express target genes in response to various external environmental factors, such as hypoxia [
17] and heat treatment caused by direct heating or heat generation of magnetic nanoparticles upon the application of a magnetic field [
18,
19].
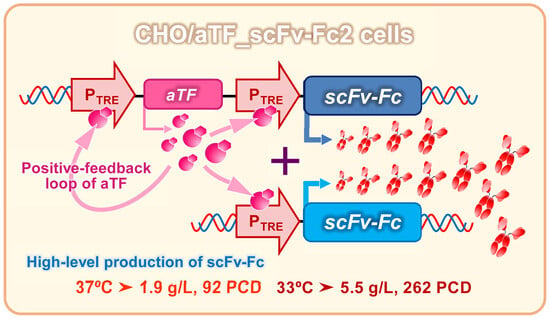
In this study, we applied an artificial gene expression system with the amplification of aTF expression via a positive feedback loop to CHO cells, and aimed to develop a method for the high production of recombinant antibodies. An anti-prion single-chain antibody fragment fused with a human IgG1-derived Fc region (scFv-Fc) was used as a model antibody. To increase the number of transgenes, a PiggyBac transposon vector system was used to integrate an aTF expression unit with a positive feedback loop and a target gene expression unit into the CHO cell genome. Recombinant protein production via CHO cell culture is improved by culturing at lower temperatures [
20,
21]. Therefore, the scFv-Fc antibody productivity of our artificial gene expression system in combination with low-temperature culture was evaluated. After the constructed recombinant cells were adapted to serum-free suspension culture, the efficacy of the gene expression system in a serum-free medium, the effect of enhancing production in low-temperature culture, and the structures of glycans modified to scFv-Fc were evaluated. Furthermore, antibody productivity, glucose usage, and the transcriptome in semi-continuous culture with high cell density were analyzed under normal- and low-temperature conditions.
2. Materials and Methods
2.1. Cells and Media
CHO-K1 (RIKEN Cell Bank, Tsukuba, Japan) and recombinant CHO cells were cultured using F12 medium (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal calf serum (FCS; BioWest, Nuaillé, France) and antibiotics (Penicillin-Streptomycin, #15140122; Invitrogen, Waltham, MA, USA). The cells were cultured in a 5% CO2 incubator at 37 °C or 33 °C. Serum-free adaptation was performed by gradually reducing the serum concentration using a mixture of 10% FCS-containing F12 medium and serum-free medium. The serum-free medium was prepared as follows: CD CHO AGT Medium (#12490025; Invitrogen), CHO custom medium (#ISJGp014; Fujifilm Wako Pure Chemical, Osaka, Japan), and EX-CELL Advanced CHO Fed-batch Medium (#14366C-1000ML; Sigma-Aldrich) were mixed at a 3:1:1 ratio. The medium was supplemented with 0.2% anti-clumping reagent (#0010057AE; Invitrogen), 4 mM L-alanyl-L-glutamine (#G8541; Sigma-Aldrich), and antibiotics (Penicillin-Streptomycin). When culturing cells in serum-containing medium, 100 mm cell culture dishes (BioLite #130182; Thermo Fisher Scientific, Waltham, MA, USA) or 24-well tissue culture plates (BioLite #130186; Thermo Fisher Scientific) were used. For serum-free suspension culture, the cells were cultured with 10 mL of culture medium in 50 mL bioreactor tubes equipped with sterile gas exchange caps (#87050, TPP; Techno Plastic Products AG, Trasadingen, Switzerland). The tubes were placed at a 45° angle and rotated at a speed of 180 rpm in a shaking incubator (Model #0081704-000; Taitec, Koshigaya, Japan). All cell cultures were conducted within a 5% CO2 incubator at 37 °C or 33 °C.
2.2. Plasmid Construction
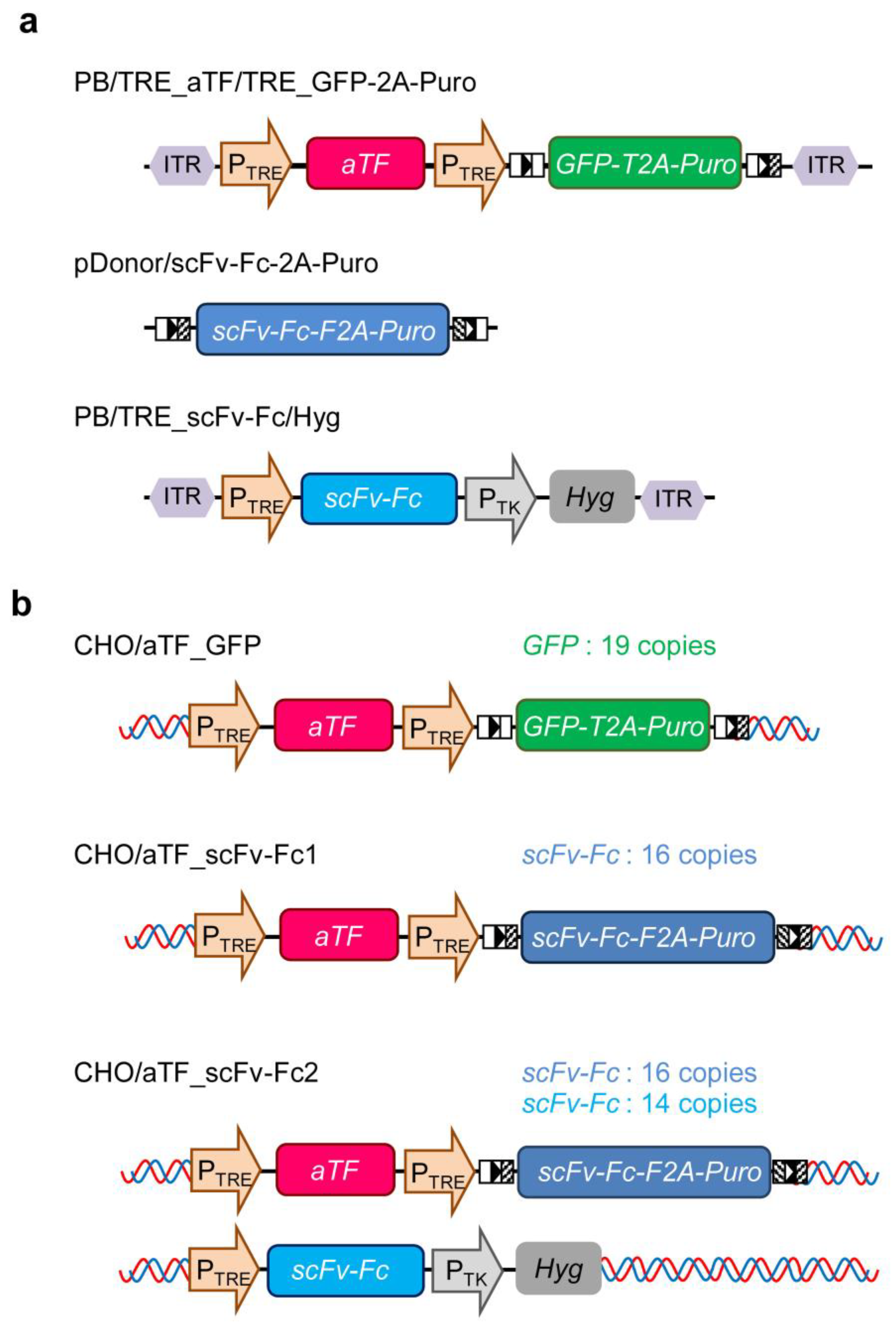
DNA fragments for the TRE promoter consisting of TRE and cytomegalovirus (CMV) minimal promoter, aTF, and the reporter (GFP-2A-Puro) genes were chemically synthesized (GeneArt Gene Synthesis; Thermo Fisher Scientific). For GFP-2A-Puro, two loxP sites (5′-ATA ACT TCG TAT AGC ATA CAT TAT ACG AAG TTA T-3′ and 5′-ATA ACT TCG TAT AGG ATA CTT TAT ACG AAC GGT A-3′) were added before and after the gene. The DNA fragments encoding expression units for aTF and GFP-2A-Puro under the control of TRE promoter were prepared via digestion with SpeI (Nippon Gene, Tokyo, Japan) and XhoI (Nippon gene), and XhoI and BstXI (Takara Bio, Kusatsu, Japan) from the custom recombinant plasmids containing chemically synthesized genes, respectively. The resultant fragments were arranged in tandem and inserted into the SpeI-BstXI-digested PiggyBac transposon vector plasmid (#PB513B-1; System Biosciences, Palo Alto, CA, USA), to generate PB/TRE_aTF/TRE_GFP-2A-Puro (
Figure 1a). A DNA fragment for the scFv-Fc-2A-Puro gene optimized using human codons was chemically synthesized (GeneArt Gene Synthesis; Thermo Fisher Scientific), flanked by compatible loxP sites (5′-ATA ACT TCG TAT AGC ATA CAT TAT ACG AAG TTA T-3′ and 5′-TAC CGT TCG TAT AGG ATA CTT TAT ACG AAG TTA T-3′), and ligated into pMK-RQ (Thermo Fisher Scientific) to generate pDonor/scFv-Fc-2A-Puro (
Figure 1a). To generate PB/TRE_scFv-Fc, the scFv-Fc gene fragment was obtained by digesting plasmid R2 [
22] with EcoRI (Nippon gene) and MluI (Nippon gene), and inserted into EcoRI–MluI-digested PB/TRE_aTF/TRE_GFP-2A-Puro. Subsequently, a hygromycin resistance gene expression unit derived from pCEP4 (#V04450; Invitrogen) was inserted into PB/TRE_scFv-Fc to generate PB/TRE_scFv-Fc/Hyg (
Figure 1a).
All plasmid digestion reactions using restriction enzymes were performed at 37 °C for 2–3 h. The constructed plasmids were transformed into E. coli DH5α (#9057; Takara Bio) for propagation and purified using a commercially available DNA purification kit (Qiafilter Plasmid Mid Kit, #12245; Qiagen GmbH, Hilden, Germany) in accordance with the manufacturer’s protocol. Subsequently, the endotoxin-free plasmids were analyzed to measure the concentration using a Nanodrop Spectrophotometer (Model #ND-2000C; Thermo Fisher Scientific) and stored at −20 °C until use for transfection.
2.3. Establishment of scFv-Fc-Producing CHO Cells
CHO cells expressing reporter genes (CHO/aTF_GFP) (
Figure 1b) were established as follows. CHO-K1 cells were seeded in a 24-well plate at a density of 1.2 × 10
5 cells/well. The next day, 640 ng PB/TRE_aTF/TRE_GFP-2A-Puro and 160 ng PiggyBac transposase expression vector plasmids (pPBase, #PB210PA-1; System Biosciences) were transiently transfected into CHO-K1 cells using 2.5 μL transfection reagent, Lipofectamine 2000 (#11668019; Invitrogen), in accordance with the manufacturer’s protocol. After 4 days of transfection, to activate the TRE promoter, the pCMV/aTF plasmid (800 ng) [
18], a vector for constitutively expressing aTF, was introduced into CHO-K1 cells using Lipofectamine 2000 (2.5 μL). Subsequently, GFP-positive cells were sorted using a cell sorter (SH800; Sony, Tokyo, Japan) and selected for 4 days with 50 mg/L puromycin (#A1113803; Invitrogen). Cell cloning was performed from the gene-integrated bulk cells exhibiting strong GFP expression using the limiting dilution method, and the CHO/aTF_GFP cell line was established. GFP expression in CHO/aTF_GFP cells was suppressed after culturing for 8 days in the presence of 1 mg/L doxycycline (Dox, #D9891; Sigma-Aldrich).
Next, to replace the GFP-2A-Puro gene downstream of the TRE promoter in the reporter gene expression unit integrated into CHO/aTF_GFP cells with the scFv-Fc-2A-Puro gene, the pDonor/scFv-Fc-2A-Puro donor vector (800 ng) containing the compatible mutated loxP sites and a Cre expression vector (10 ng) [
22] were co-transfected into CHO/aTF_GFP cells using Lipofectamine 2000 (2.5 μL). The GFP-extinguished cell fraction was sorted using a cell sorter, and cell cloning was performed using the limiting dilution method to establish scFv-Fc-producing CHO cells (CHO/aTF_scFv-Fc1) (
Figure 1b).
To enhance the productivity of scFv-Fc, PB/TRE_scFv-Fc/Hyg and pPBase were co-transfected into CHO/aTF_scFv-Fc1 cells using Lipofectamine 2000 under similar conditions to the establishment of CHO/aTF_GFP cells. After 48 h of transfection, the cells were seeded in 60 mm tissue culture dishes (BioLite #130181; Thermo Fisher Scientific) and selected for 1 week with 4 mg/mL hygromycin (#080-07683; Fujifilm Wako). Subsequently, cell cloning was performed using the limiting dilution method to establish scFv-Fc-producing CHO cells (CHO/aTF_scFv-Fc2) (
Figure 1b) with enhanced scFv-Fc expression.
2.4. Measurement of Transgene Copy Number
To measure the copy number of introduced genes in the recombinant CHO cells, genomic DNA was extracted from each cell line using a commercial genomic DNA extraction kit (#NPK-101, MagExtractor; Toyobo, Osaka, Japan). Quantification of the gene copy number was performed using a quantitative PCR device (AriaMx Real-time PCR System; Agilent Technologies, Santa Clara, CA, USA) via a modified version of a previously published method [
22]. The primers and Taqman probes used are shown in
Table S1. Standard curves were prepared using donor plasmids (PB/TRE_aTF/TRE_GFP-2A-Puro, pDonor/scFv-Fc-2A-Puro, and PB/TRE_scFv-Fc/Hyg). The nuclear genome content of CHO cells was assumed to be 5.4 pg/cell [
23] to calculate the copy number. The copy numbers of GFP and scFv-Fc genes in the cells were measured multiple times (
n = 5) and expressed as mean values with standard deviations.
2.5. scFv-Fc Measurement and Metabolic Analysis
CHO cells and recombinant CHO cells were seeded at a density of 1.0 × 10
5 cells/mL in a culture medium containing serum or 1.0 × 10
6 cells/mL in serum-free medium, with 1 mL per well in 24-well culture plates. The cells were then cultured for 6 days. During this period, the culture media were harvested daily, and cell counting was performed using the trypan blue dye exclusion method with a cell counter (Countess, Model #AMQAF1000; Invitrogen). For the high-cell-density culture, CHO/aTF_scFv-Fc2 cells were seeded in 50 mL bioreactor tubes at a cell density of 0.5–2.0 × 10
7 cells/mL with 10 mL of medium and cultured for 12 days. The culture medium was exchanged for fresh medium every other day, and during each change, the culture medium was harvested and cell counting was performed. The culture conditions were described in
Section 2.1. All harvested medium samples were stored at −80 °C until evaluation for the following assays.
The concentration of scFv-Fc secreted in the culture medium was quantified using an ELISA method [
22]. The rabbit IgG fraction of anti-human IgG F(c) (#609-4103; Rockland Immunochemicals, Philadelphia, PA, USA) and rabbit peroxidase (POD)-conjugated anti-human IgG F(c) antibodies (#609-4303; Rockland Immunochemicals) as primary and secondary antibodies, respectively, were used. Calibration curves were created using a dilution series of purified scFv-Fc or IgG (kindly provided by Dr. Naohiro Noda, Manufacturing Technology Association of Biologics (MAB), Kobe, Japan). From the scFv-Fc concentration in the medium and the number of viable cells, the specific scFv-Fc productivity (pg cell
−1 day
−1) was calculated. Glucose and lactate concentrations were measured using commercially available kits (Glucose Assay Kit-WST, #G264, and Lactate Assay Kit-WST, #L256; both from Dojindo, Kumamoto, Japan) with attached standards according to the manufacturer’s protocols.
Cell cultures were performed using multiple wells or tubes for each condition (n = 3). The measured values obtained from each sample were expressed as the mean value with standard deviation.
2.6. SDS-PAGE
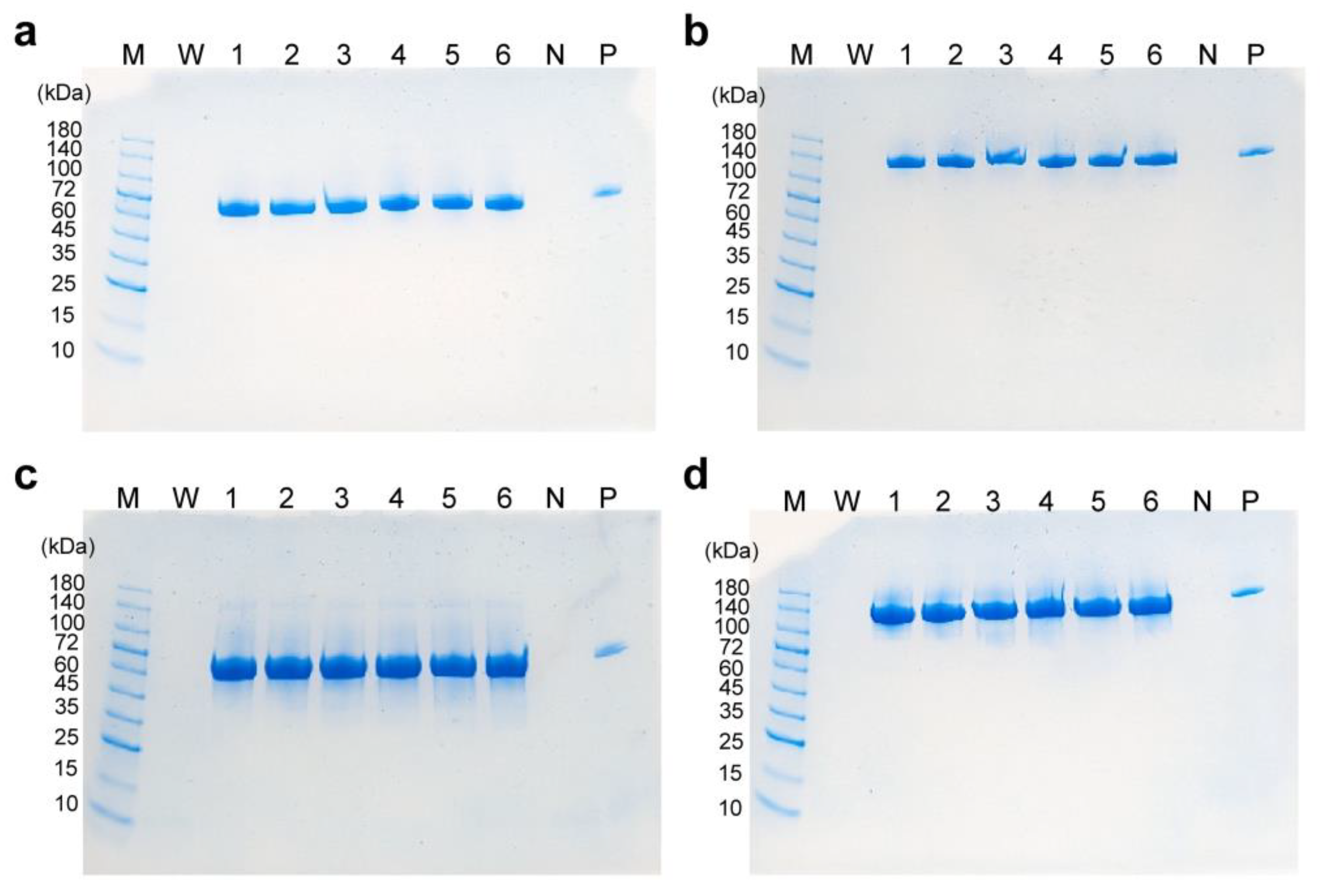
To analyze the structure of scFv-Fc in the culture medium of high-cell-density cultured CHO/aTF_scFv-Fc2 cells, SDS-PAGE was conducted, with the culture supernatants (10 µL) being diluted 100-fold, and the samples were applied to the wells of 4–20% Mini-PROTEAN TGX precast gel (#4561094; Bio-Rad Laboratories, Hercules, CA, USA). The protein bands were stained with SimpleBlue Safe Stain (#LC6060; Invitrogen).
2.7. Glycan Structure Analysis
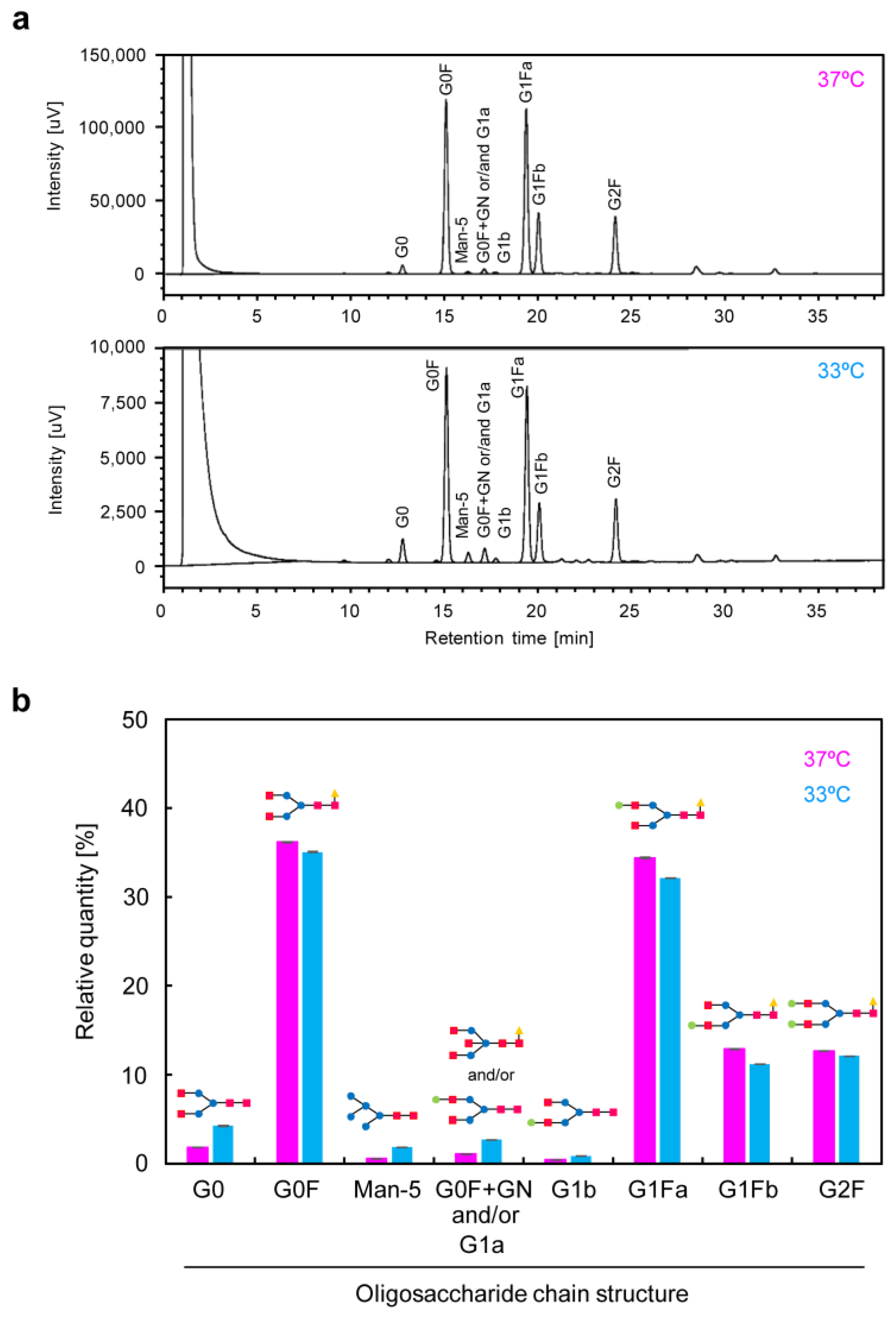
The scFv-Fc contained in the culture medium (days 8–12) of high-cell-density cultured CHO/aTF_scFv-Fc2 cells was purified using a Protein A column (rProtein A Sepharose Fast Flow, #17127901; Cytiva, Marlborough, MA, USA). The purified scFv-Fc samples were treated using a glycan composition analysis kit (EZGlyco mAb-N Kit with 2-AB, #BS-X4410; Sumitomo Bakelite, Tokyo, Japan), and glycan analysis data were obtained using the hydrophilic interaction liquid chromatography–ultra-performance liquid chromatography (HILIC-UPLC) method with a 2-AB labeled human IgG (Waters Glycan Performance Test Standard, #186006349; UVISON Technologies, London, UK) as a standard. The same samples were analyzed three times (n = 3), and the glycan composition distributions were expressed as the mean with standard deviation.
2.8. DNA Microarray Analysis
mRNA was extracted from high-cell-density cultured CHO/aTF_scFv-Fc2 cells (day 12) for transcriptomic analysis. A commercially available DNA chip for Chinese hamster (G4858A#077089 single color 8 × 60K; Agilent Technologies) was used. Microarray analysis was outsourced to Cell Innovator (Fukuoka, Japan). Enrichment analysis for functional clustering was performed using the DAVID (david.ncifcrf.gov) annotation tool. The microarray data were submitted to the ArrayExpress database at EMBL-EBI (
www.ebi.ac.uk/arrayexpress (accessed on 15 November 2023)) under accession number E-MTAB-13440 (available from 1 January 2024).
4. Discussion
In recent years, the production of monoclonal antibodies in recombinant CHO cell cultures with high titers of 5–10 g/L has been reported [
26]. Through the optimization of feed formulation, medium, and process parameters such as pH and temperature, antibody titers have reached up to 10 g/L over a 14 day-period, with a specific production rate of 72 pg cell
−1 day
−1 in a fed-batch process [
26]. Improvements in cell line development and cultivation processes have significantly contributed to high-titer production [
7]. In the cell line construction process, synthetic biology approaches have begun to be adopted for CHO cell engineering to achieve high-titer production of recombinant proteins [
27]. In this study, we applied a gene overexpression system using an aTF based on the Tet-transcription activation system, which can strongly induce the high expression of target genes. To trigger the transient expression of aTF and induce its high-level expression, we incorporated an expression unit that forms a positive feedback loop for the transcription of aTF. We generated recombinant CHO cells in which multiple copies of an expression unit for the target gene (scFv-Fc) were integrated into the cell genome under the control of the TRE promoter driven by aTF. Furthermore, continuous production was carried out using semi-continuous culture at high cell densities of 10 million cells/mL at a low temperature. While maintaining an average specific production rate of approximately 190 pg cell
−1 day
−1 throughout the culture period, we achieved a production concentration of approximately 4.0 g/L of scFv-Fc antibodies. The produced scFv-Fc remained intact, and no non-human glycosylation structures were observed in it.
The PiggyBac transposon vector is a valuable tool for CHO cell line construction because it can be integrated into the CHO cell genome with multiple copies, allowing for the stable expression of a transgene [
24,
28]. Reports indicate that the number of copies introduced by the PiggyBac system in a single transfection is approximately 15 copies [
24,
25], which was also the case in this study. By using the PiggyBac transposon vector, the aTF expression unit with positive feedback of aTF is integrated with multiple copies across the entire genome, leading to the amplification of aTF expression. As a result, it was possible to induce high-level expression of the target gene controlled by the TRE promoter activated by aTF. Using GFP as a reporter gene, after screening for cells with the highest fluorescence intensity, we replaced the reporter gene (GFP-2A-Puro) with the scFv-Fc gene flanked by loxP sites using the Cre/loxP recombination system [
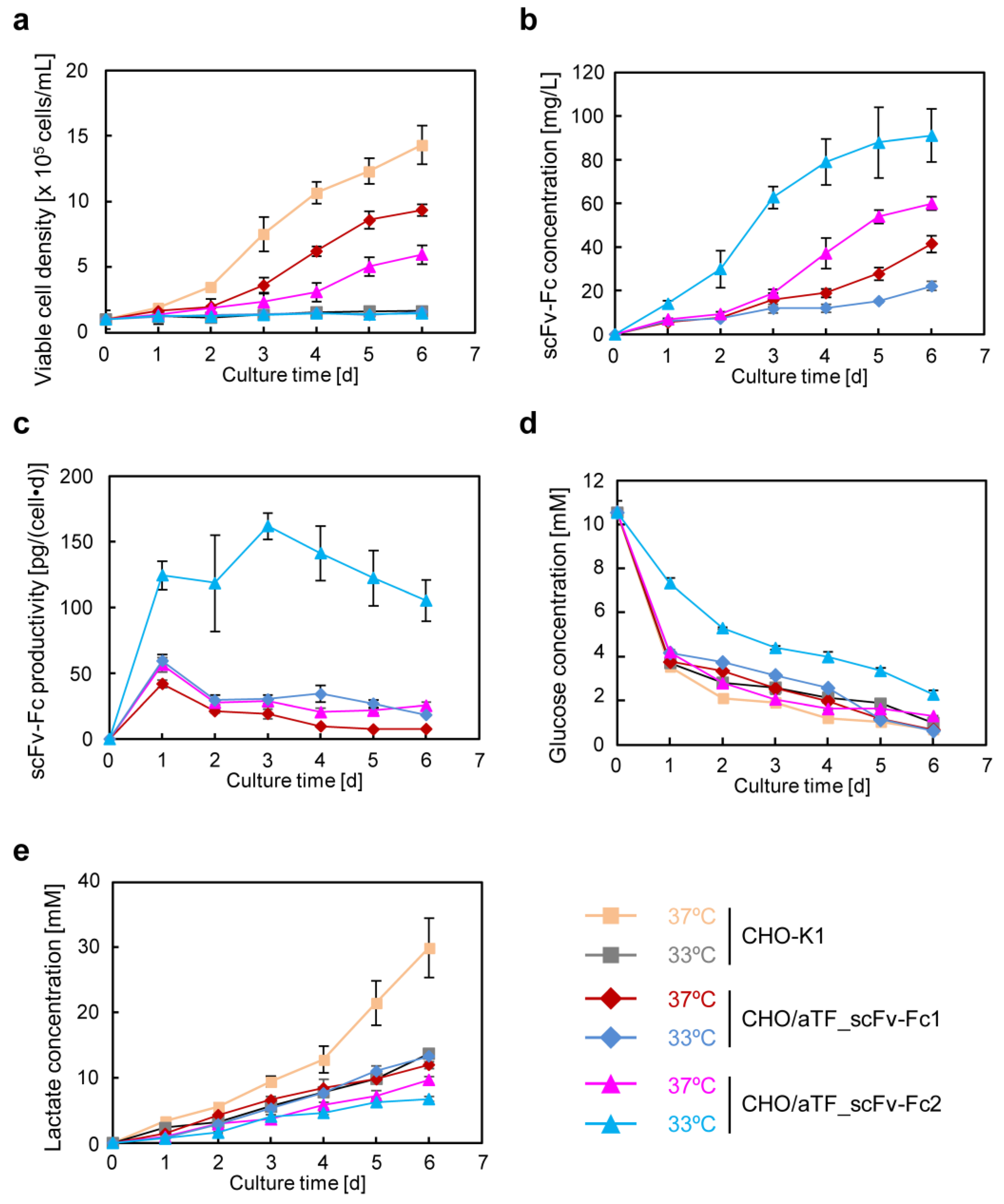
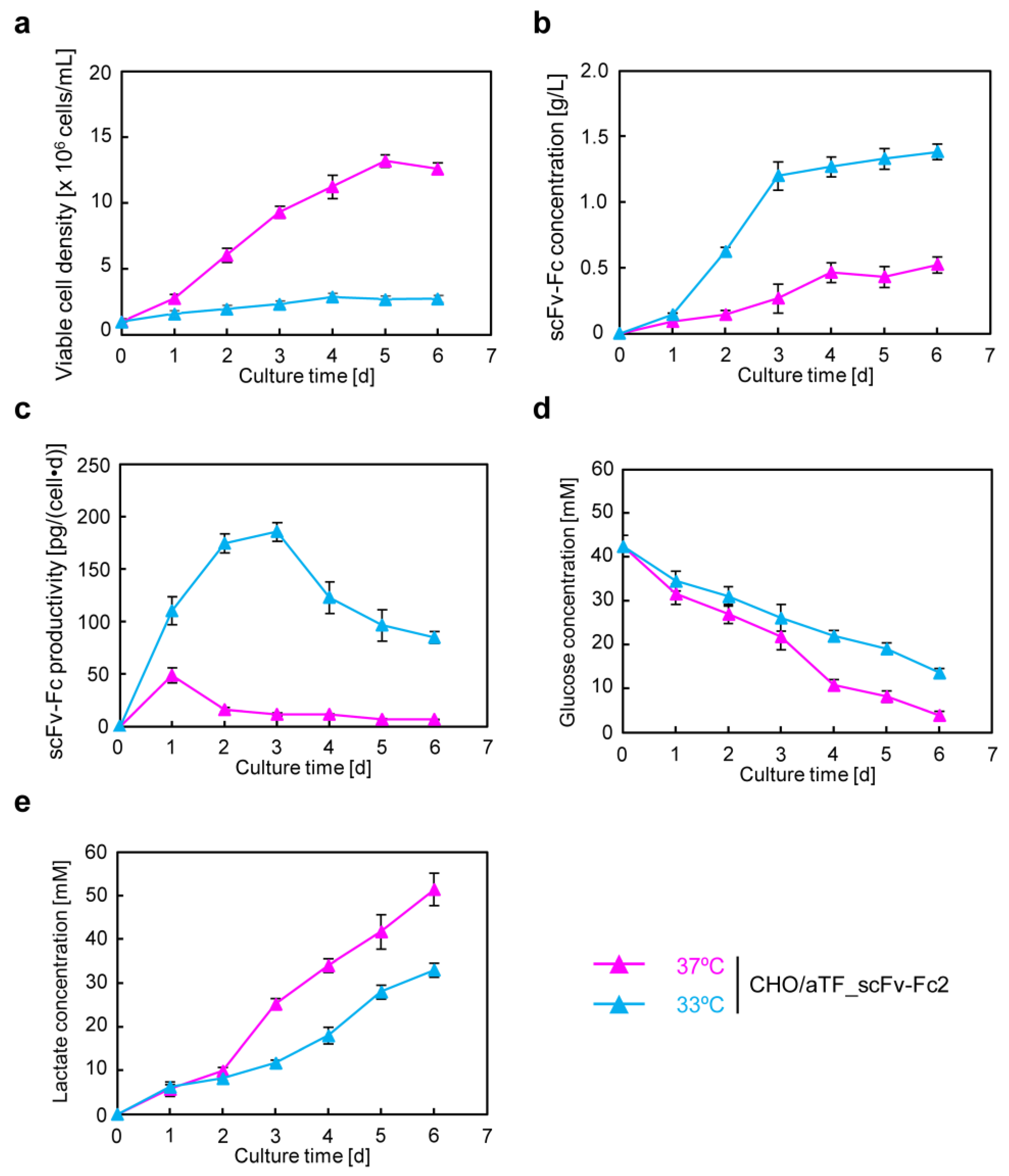
22] to generate CHO/aTF_scFv-Fc1-producing cells. Because aTF is amplified in a positive feedback loop, it is expected that adding response units could further improve expression levels. By introducing additional copies into the cell genome using a PiggyBac transposon vector with expression units containing the TRE promoter as the response sequence to induce expression of the scFv-Fc gene, we generated CHO/aTF_scFv-Fc2 cells. In these cells, on day 6 of culture, scFv-Fc production levels were enhanced by 1.4-fold at 37 °C and 4.1-fold at 33 °C (
Figure 2b). Because aTF expression levels are considered sufficient with positive feedback loop amplification, scFv-Fc expression may be further enhanced by adding more response units.
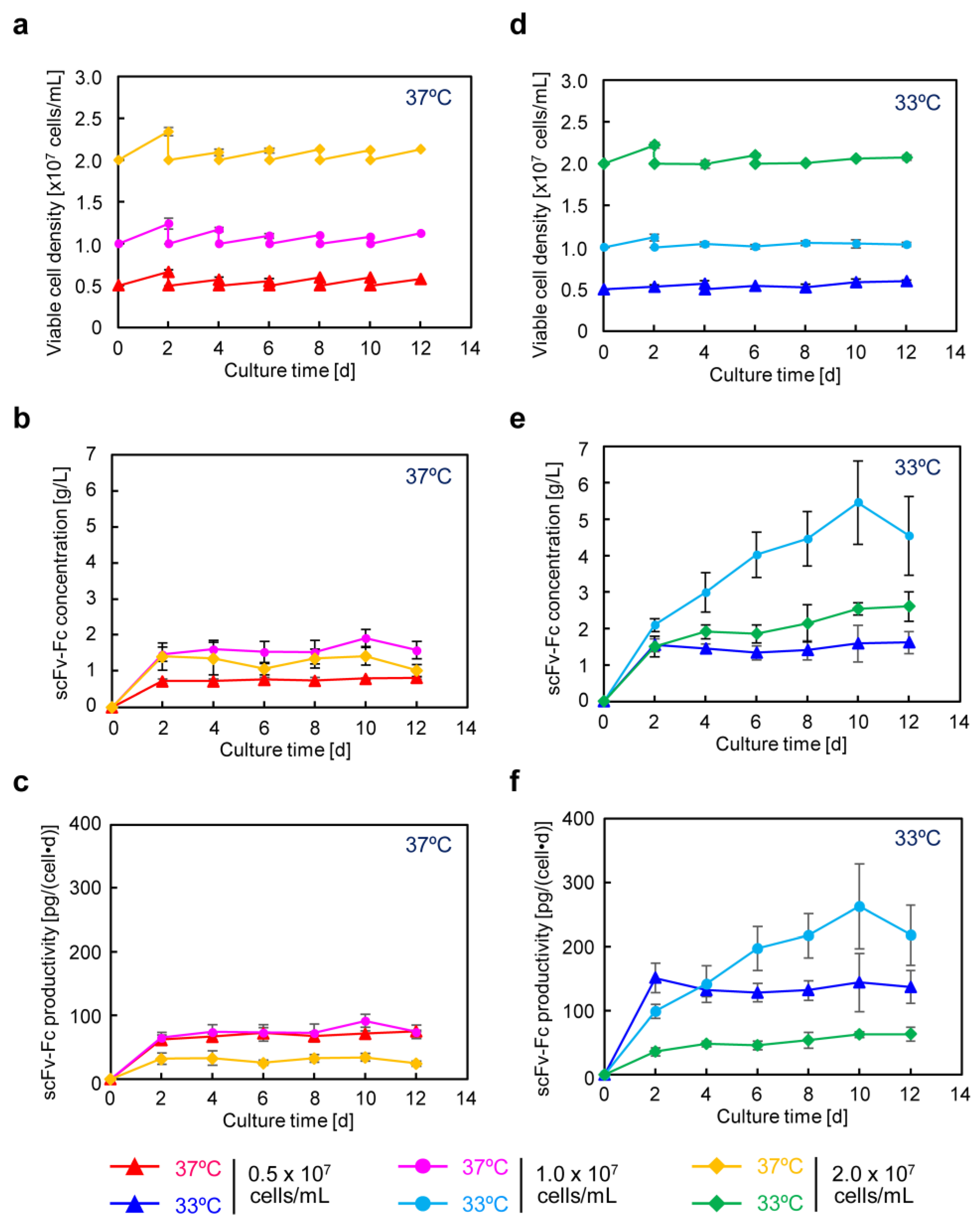
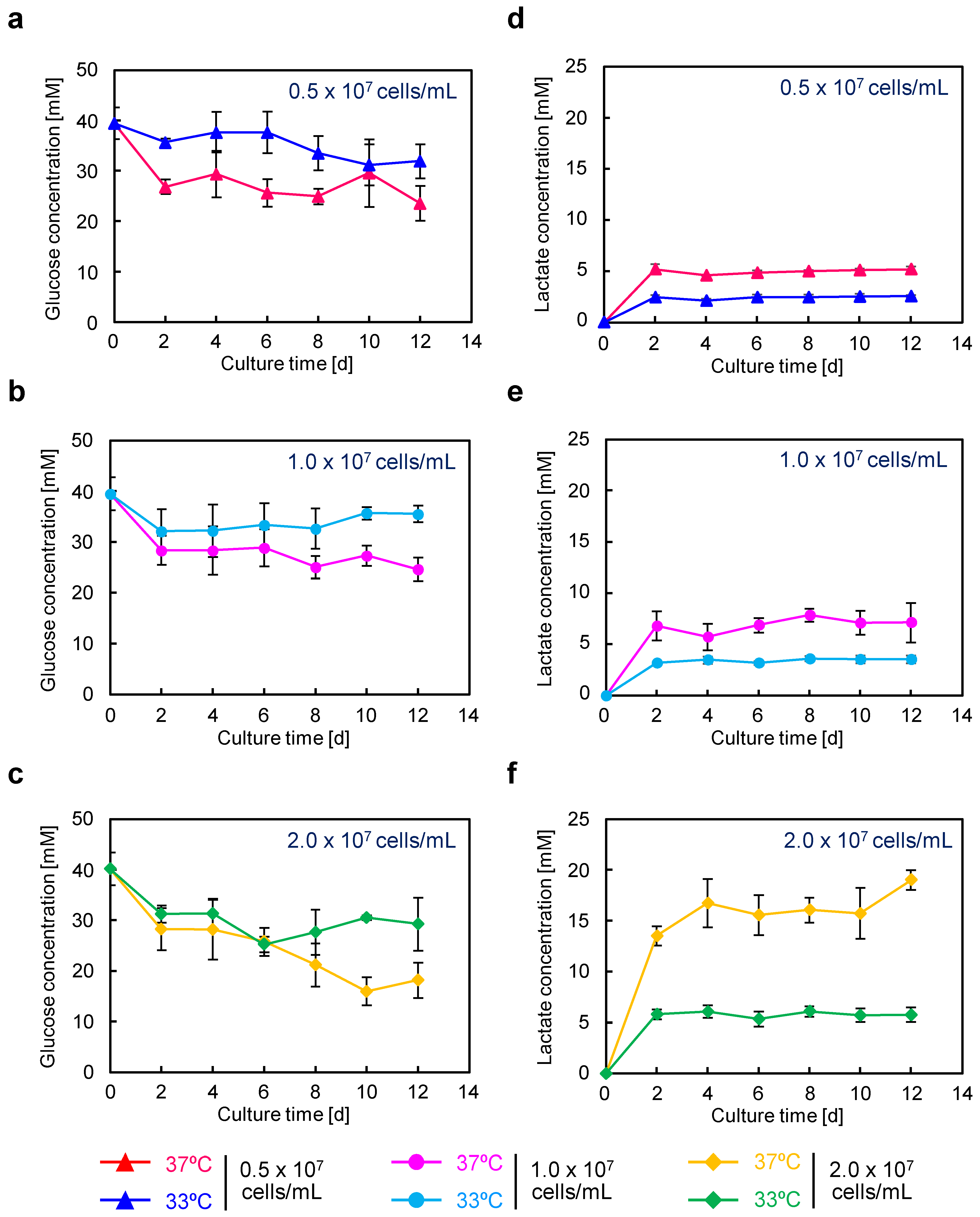
In recombinant protein production using CHO cells, serum-free media are generally employed. After suspension adaptation in serum-free media and subsequent batch culture, the scFv-Fc concentration could be increased by 10-fold while maintaining almost the same specific production rate as in serum-containing medium. The target gene expression system using the positive feedback loop amplification of aTF remained effective even when using serum-free medium, prompting further cultivation at higher seeding cell densities. The results showed that, by seeding at 1.0 × 10
7 cells/mL and conducting low-temperature culture, scFv-Fc production was maximized. In this culture, the observed maximum lactate concentration was 3.6 mM (
Figure 5e), which is less than one-tenth of the harmful lactate concentration (40 mM) often observed in high-cell-density fed-batch culture of CHO cells [
29]. Consequently, under the conditions of seeding at 1.0 × 10
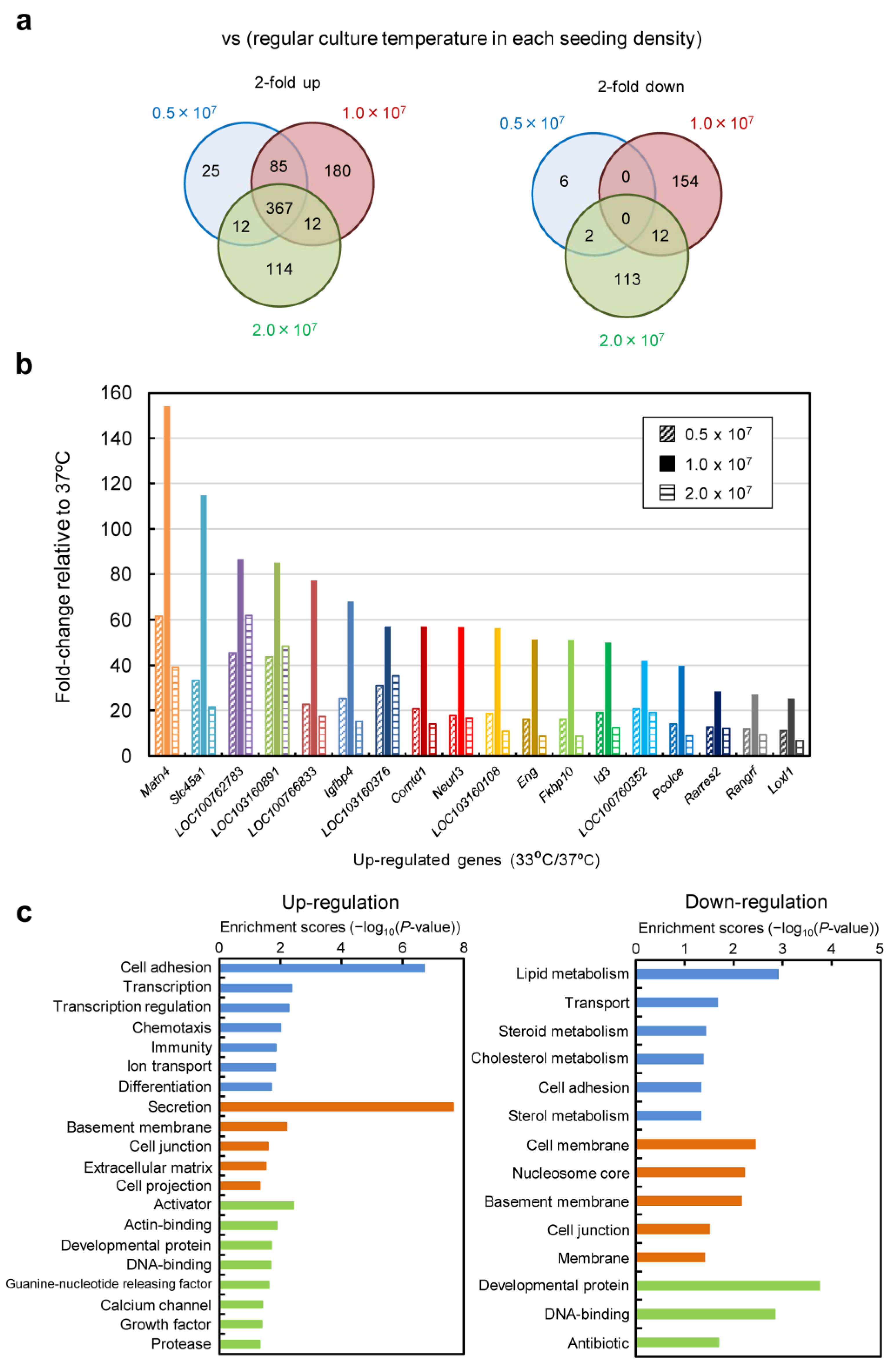
7 cells/mL and low-temperature culture at 33 °C, cell proliferation was suppressed, and the cells were in a favorable state for energy metabolism, allowing for high-level production of scFv-Fc. Furthermore, in low-temperature culture, the low levels of glucose consumption and lactate accumulation suggested the potential for achieving improved production of scFv-Fc with a reduced amount of medium usage by optimizing the timing of medium replacement. Functional clustering analysis based on transcriptomic data obtained using DNA microarrays revealed the upregulation of genes related to cell adhesion and secretion of proteins for culture at 33 °C (
Figure 8 and
Figure S4). Specifically, significant upregulation was observed for genes associated with cell adhesion, including Ncam, Vcam, and Icam, with fold changes of 162.5, 31.0, and 4.6, respectively. It was also found that the factor Sec16, necessary for the formation of COPII-coated vesicles [
30], exhibited a 59.9-fold change in expression. The reduced glucose consumption and lactate accumulation could be related to the downregulation of genes involved in lipid metabolism (
Figure 8c (right)). In recent years, the development of media tailored to production cells and production formats has been actively conducted based on data obtained from transcriptomic and metabolomic analyses. By developing media suitable for the production of target proteins using aTF and optimizing the timing of medium change and feed additions, it may be possible to achieve further increases in the production of target products in the medium and a reduction in the production of inhibitory metabolic byproducts.
We previously developed gene expression systems for the high-level expression of transgenes with transcriptional amplification using an artificial gene circuit that responds to external environmental stimuli, employing the Tet-transcription activation system. To make the entire expression unit compact, we constructed an expression vector that integrates both the induction and the amplification of the target gene expression and integrated it into the cell genome [
19]. In this study, we separated the switch unit, allowing for flexible selection of the trigger for the expression of the aTF. Additionally, because we employed the Tet-transcription activation system, cells incorporating this gene expression system could reset transgene expression in cultures with Dox-supplemented medium and maintain that state or induce re-expression at a later stage. In this study, we induced sustained high-level expression by turning the system “on” through transient aTF expression using pCMV/aTF. By changing the promoter acting as the trigger for aTF expression, the system can be activated at the desired timing. Transcriptomic analysis enabled us to list genes showing more than a 5-fold increase in Z-score of expression at 33 °C cultivation compared with that at 37 °C (
Figure 8b). Using the regulatory regions of these genes as triggers for aTF expression, it is believed that strong gene expression can be induced in low-temperature culture. Furthermore, the potent transcription of the target gene is driven by the transcription activation domain within the aTF [
31]. There have been reports of attempts to select transcription activation domains tailored to artificial transcription factors [
32], structure-based development [
33], and exploration of novel transcription activation domains in human cells using TAD-seq [
34]. The development of aTF suitable for production using CHO cells is expected to enhance the efficiency of target substance production with this system. Recently, a translation activation technology has been reported to further improve the production of recombinant antibodies in production cell lines that have achieved gram-per-liter level production [
35]. The application of this technology may lead to even higher levels of antibody expression in the high-expression system based on the aTF in this study.
In conclusion, we successfully achieved the production of scFv-Fc antibodies at the gram-per-liter level by applying an inducible gene expression system using aTF in CHO cells. In this system, the aTF expression unit was constructed as that capable of amplification through a positive feedback loop. This allowed for the massive expression of scFv-Fc from the multiple copies of the response expression unit containing the scFv-Fc gene, facilitated by a large amount of aTF with potent transcriptional activity. This system remained effective even when the serum-free medium was used, and by combining it with a temperature shift to a lower temperature, a maximum production of 5.5 g/L was achieved. The expression of the CHO/aTF_GFP cells used as the founders in this study can be reset through the addition of Dox. By selecting a trigger promoter for aTF expression, the system can easily adapt to external environmental factors such as heat treatment and hypoxia, and the activation of the expression system can be monitored by observing green fluorescence. The ability to replace the reporter genes with target genes using Cre-loxP recombination means that CHO/aTF_GFP can serve as a universal platform cell for artificial gene expression systems. The artificial gene expression system equipped with the positive feedback loop developed in this study is expected to become a new tool for constructing systems that can produce recombinant proteins at high levels.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







