
Proximity Interactome Analysis of Super Conserved Receptors Expressed in the Brain Identifies EPB41L2, SLC3A2, and LRBA as Main Partners
 , ,
, ,
Abstract
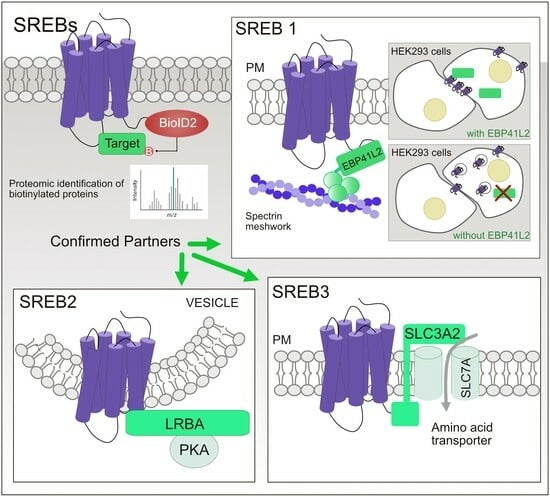
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. DNA Constructs
2.3. Cell Culture and Transfections
2.4. RNA Extraction and qPCR
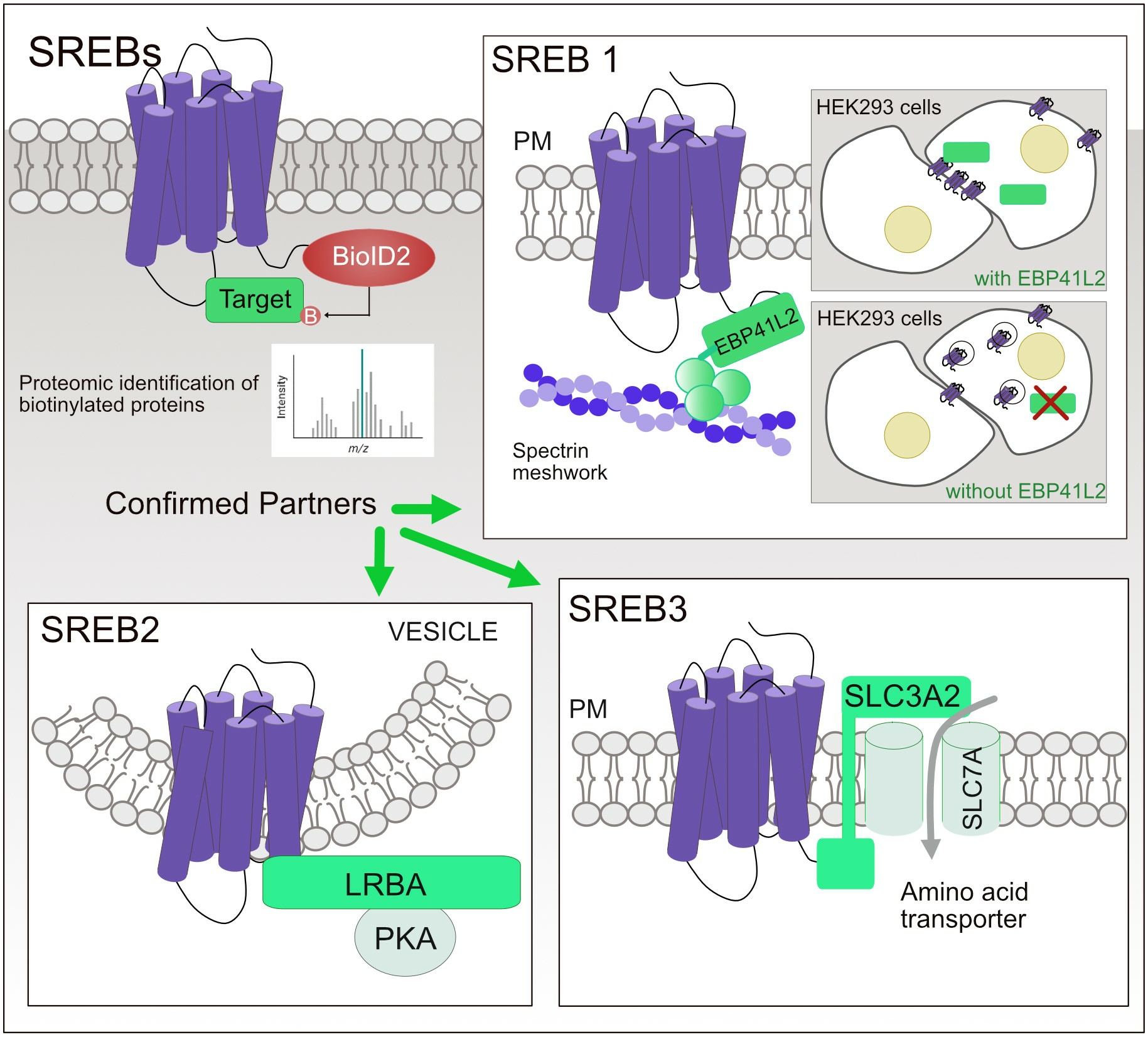
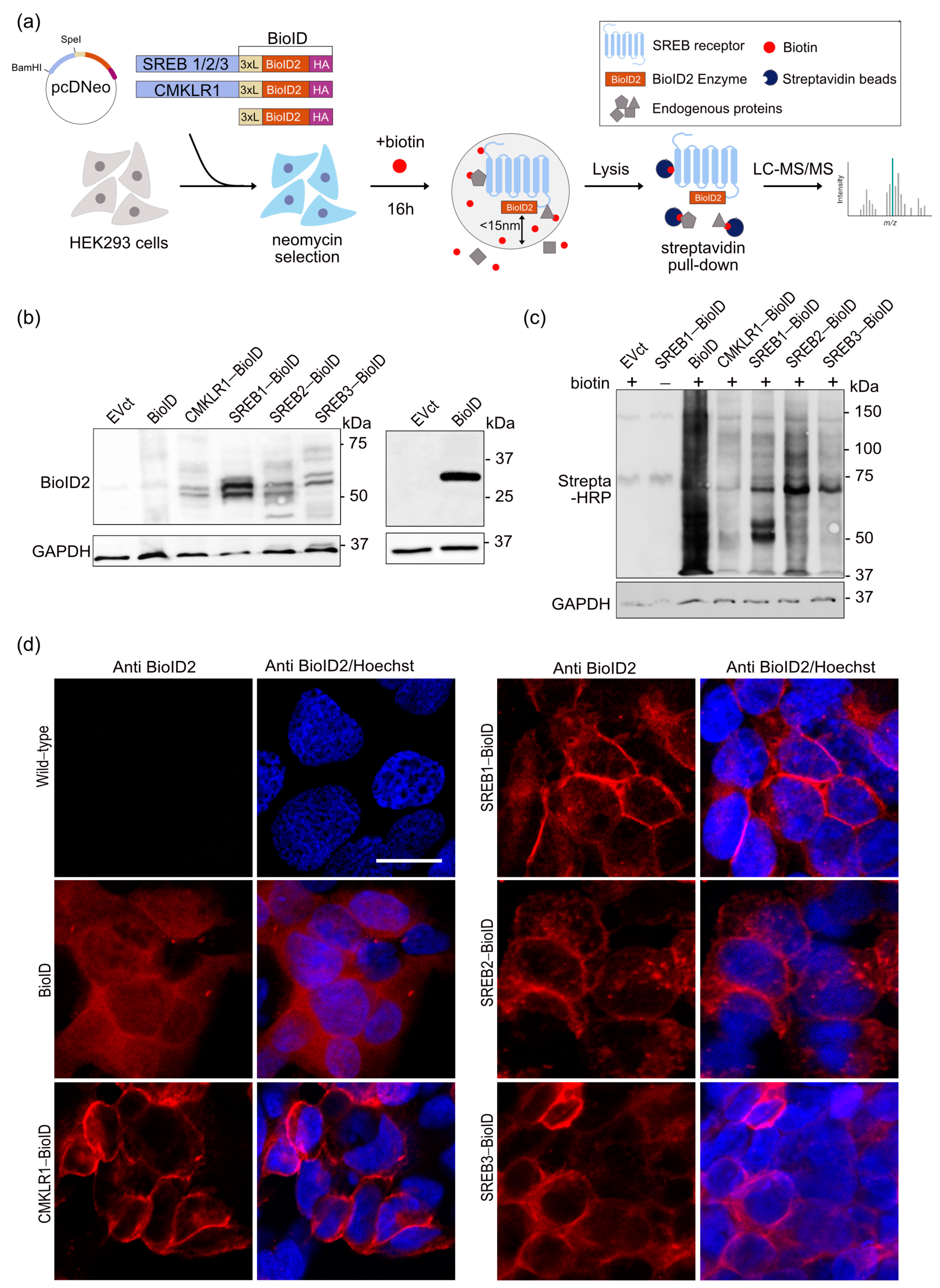
2.5. BioID Assay
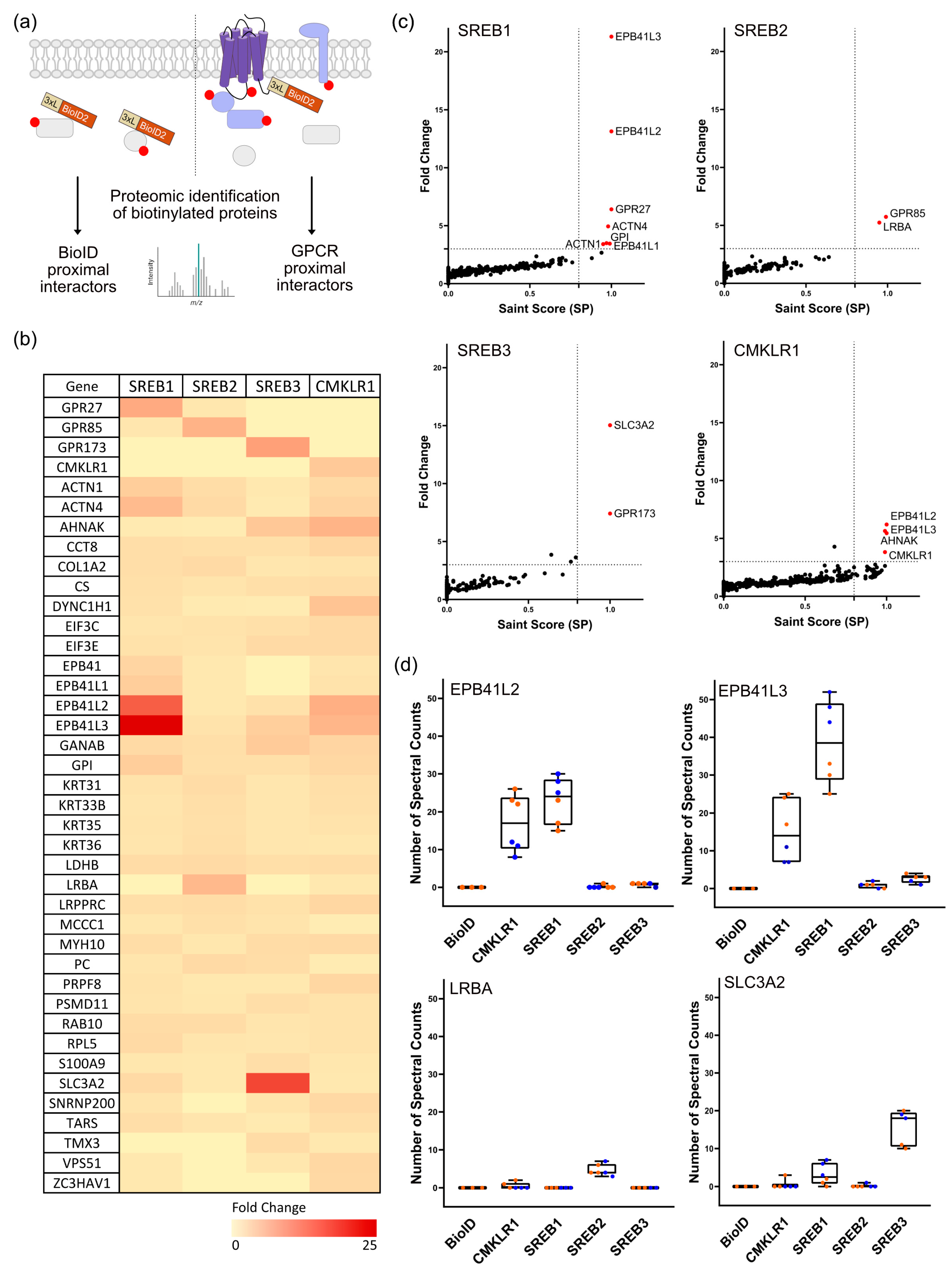
2.6. Mass Spectrometry
2.7. Data Analysis
2.8. Western Blotting, Co-Immunoprecipitation, and Antibodies
2.9. siRNA Knock-Down
2.10. Immunofluorescence
2.11. Solubilization Experiments
2.12. Quantification and Statistic Analysis
3. Results
3.1. BioID2 Fused to SREB Family Members Is Functional and the Fusion Proteins Are Expressed and Localize at the Plasma Membrane
3.2. SREBs Interactome-Mapped using Proximity-Based Biotinylation
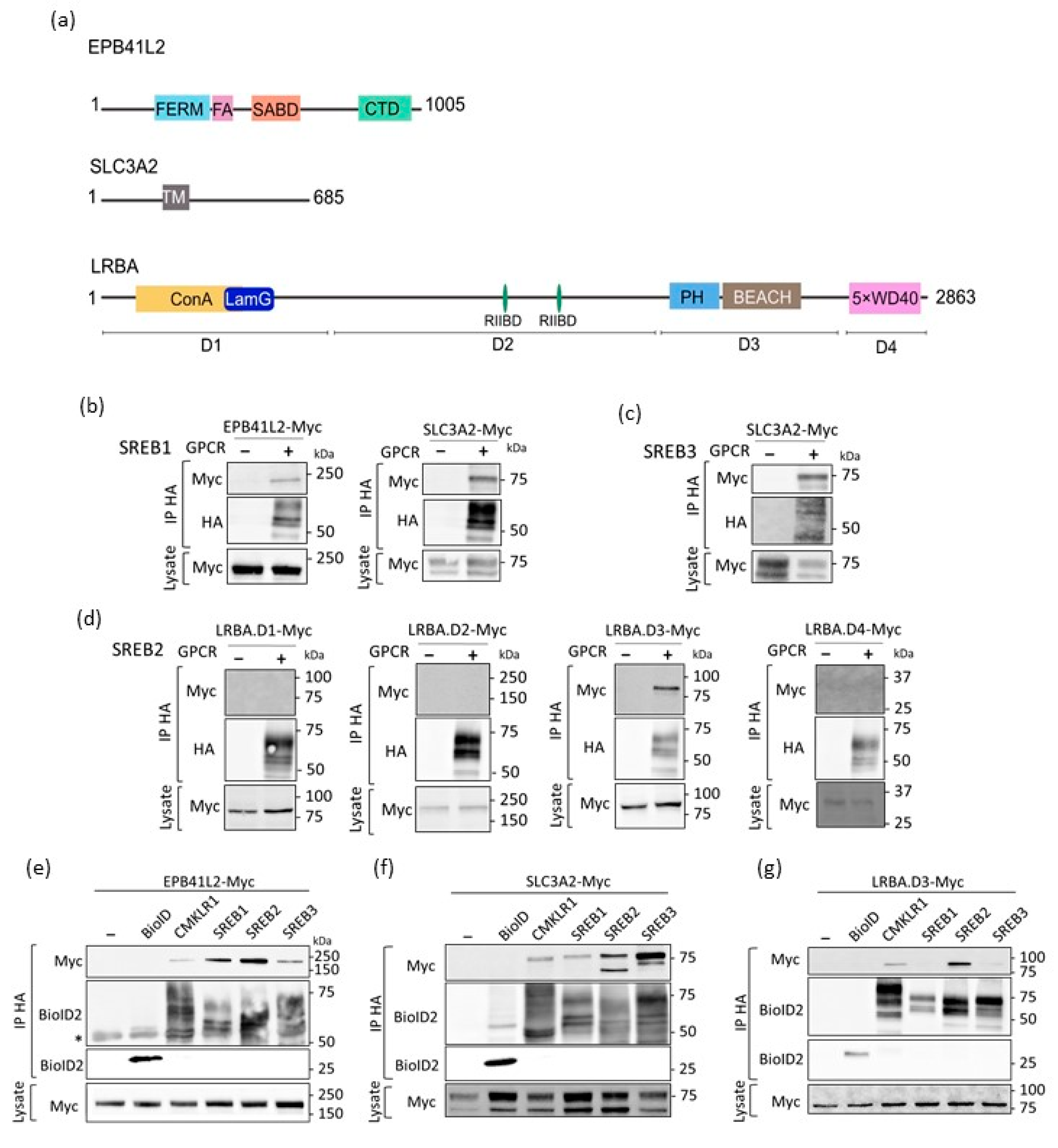
3.3. Confirmation of the Physical Association of SREBs with EPB41L2, SLC3A2, and LRBA via Co-Immunoprecipitation
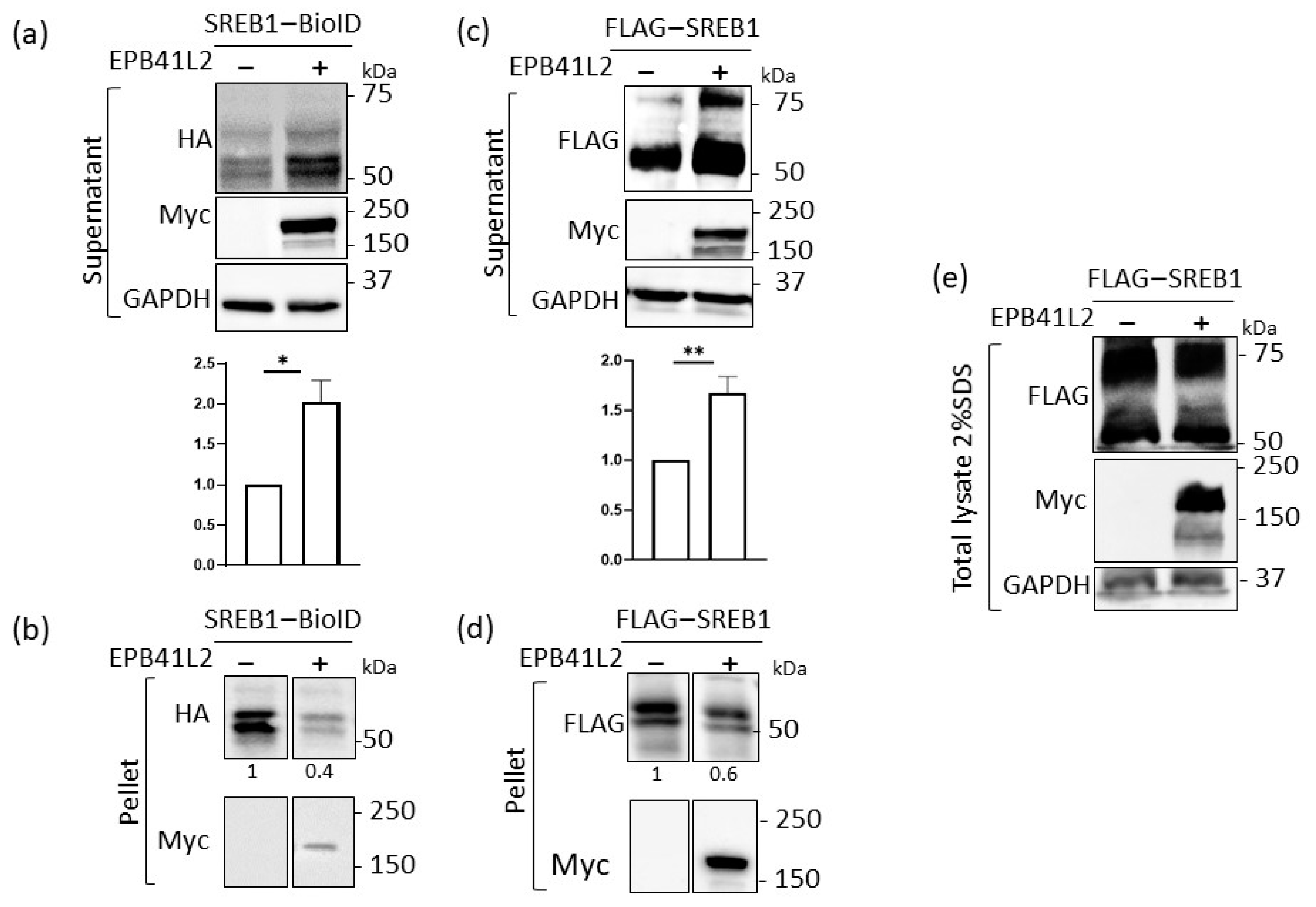
3.4. EPB41L2 Modifies the Solubilization of SREB1 When Using Detergents
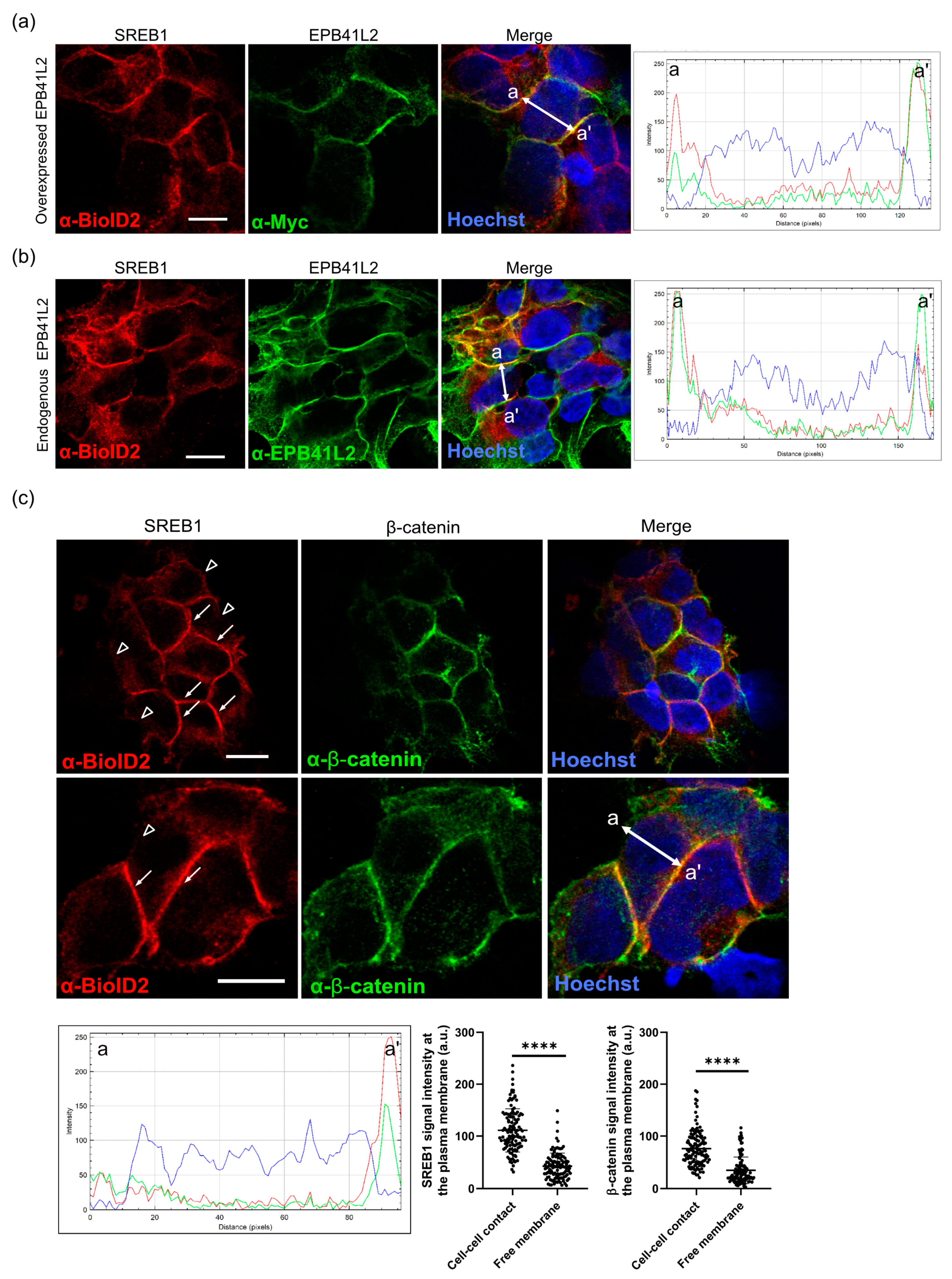
3.5. SREB1 Colocalizes with EPB41L2 at the Plasma Membrane of HEK293 Cells
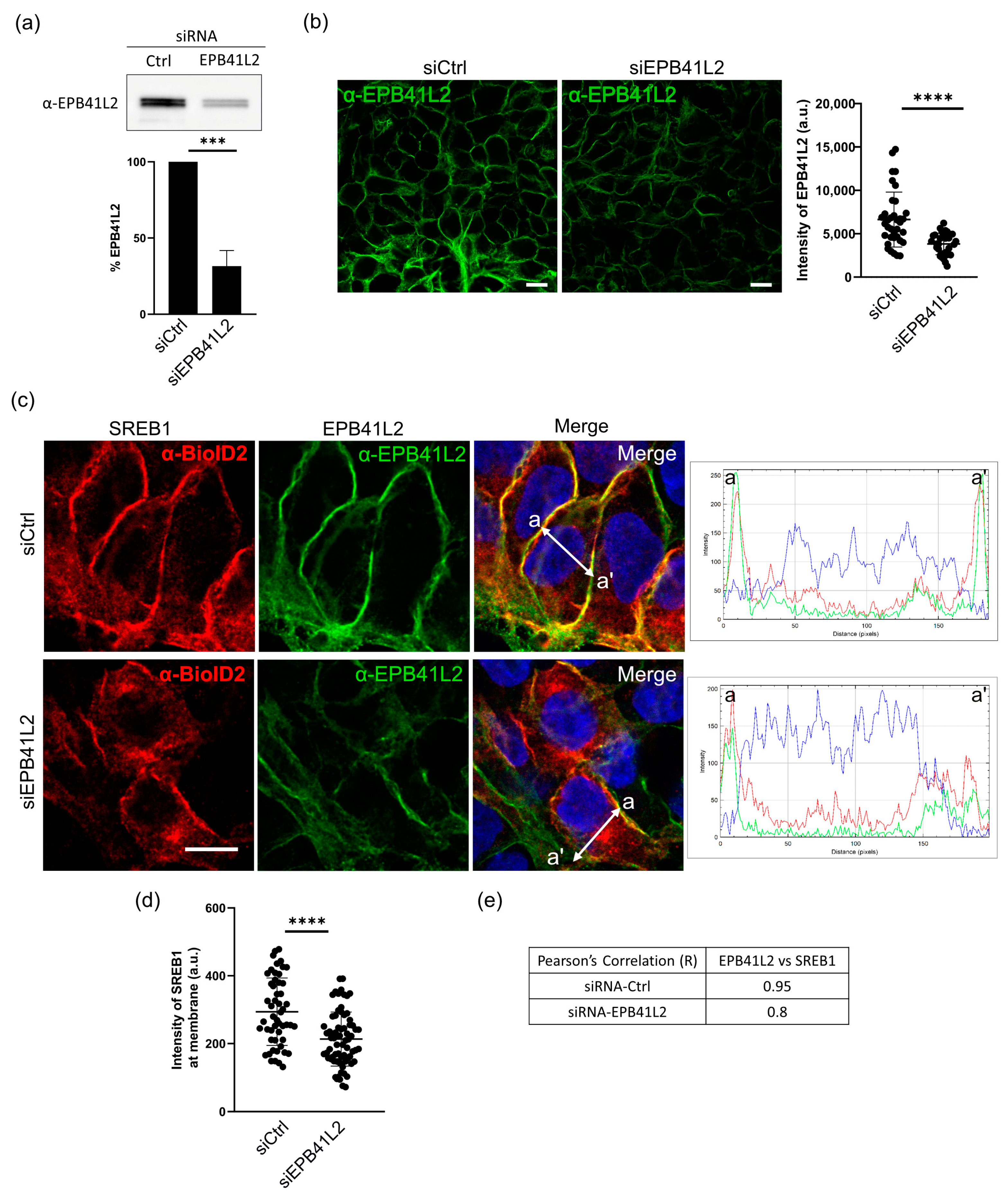
3.6. EPB41L2 Is Important for Proper Localization of SREB1 to the Plasma Membrane
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Laschet, C.; Dupuis, N.; Hanson, J. The G Protein-Coupled Receptors Deorphanization Landscape. Biochem. Pharmacol. 2018, 153, 62–74. [Google Scholar] [CrossRef]
- Hellebrand, S.; Schaller, H.C.; Wittenberger, T. The Brain-Specific G Protein Coupled Receptor GPR85 with Identical Protein Sequence in Man and Mouse Maps to Human Chromosome 7q31. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1493, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Saito, T.; Takasaki, J.; Kamohara, M.; Sugimoto, T.; Kobayashi, M.; Tadokoro, M.; Matsumoto, S.; Ohishi, T.; Furuichi, K. An Evolutionarily Conserved G Protein Coupled Receptor Family, SREB, Expressed in the Central Nervous System. Biochem. Biophys. Res. Commun. 2000, 272, 576–582. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.Q.; Kolakowski, L.F.; George, S.R. Discovery of Three Novel G Protein-Coupled Receptor Genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Regard, J.B.; Sato, I.T.; Coughlin, S.R. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell 2008, 135, 561–571. [Google Scholar] [CrossRef]
- Chopra, D.G.; Yiv, N.; Hennings, T.G.; Zhang, Y.; Ku, G.M. Deletion of Gpr27 in Vivo Reduces Insulin mRNA but Does Not Result in Diabetes. Sci. Rep. 2020, 10, 5629. [Google Scholar] [CrossRef]
- Ku, G.M.; Pappalardo, Z.; Luo, C.C.; German, M.S.; McManus, M.T. An siRNA Screen in Pancreatic Beta Cells Reveals a Role for Gpr27 in Insulin Production. PLoS Genet. 2012, 8, e1002449. [Google Scholar] [CrossRef]
- Nath, A.K.; Ma, J.; Chen, Z.; Li, Z.; Vitery, M.D.C.; Kelley, M.L.; Peterson, R.T.; Gerszten, R.E.; Yeh, J.J. Genetic Deletion of Gpr27 Alters Acylcarnitine Metabolism, Insulin Sensitivity, and Glucose Homeostasis in Zebrafish. FASEB J. 2020, 34, 1546–1557. [Google Scholar] [CrossRef]
- Wang, H.; Du, D.; Huang, J.; Wang, S.; He, X.; Yuan, S.; Xiao, J. GPR27 Regulates Hepatocellular Carcinoma Progression via MAPK/ERK Pathway. Cancer Manag. Res. 2022, 14, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Rijavec, M.; Tomar, S.; Yamani, A.; Ganesan, V.; Krempski, J.; Schuler, C.F.; Bunyavanich, S.; Korosec, P.; Hogan, S.P. Acute Systemic Myeloid Inflammatory and Stress Response in Severe Food Allergic Reactions. Clin. Exp. Allergy 2023, 53, 536–549. [Google Scholar] [CrossRef]
- Matsumoto, M.; Straub, R.E.; Marenco, S.; Nicodemus, K.K.; Matsumoto, S.; Fujikawa, A.; Miyoshi, S.; Shobo, M.; Takahashi, S.; Yarimizu, J.; et al. The Evolutionarily Conserved G Protein-Coupled Receptor SREB2/GPR85 Influences Brain Size, Behavior, and Vulnerability to Schizophrenia. Proc. Natl. Acad. Sci. USA 2008, 105, 6133–6138. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kogan, J.H.; Gross, A.K.; Zhou, Y.; Walton, N.M.; Shin, R.; Heusner, C.L.; Miyake, S.; Tajinda, K.; Tamura, K.; et al. SREB2/GPR85, a Schizophrenia Risk Factor, Negatively Regulates Hippocampal Adult Neurogenesis and Neurogenesis-dependent Learning and Memory. Eur. J. Neurosci. 2012, 36, 2597–2608. [Google Scholar] [CrossRef]
- Fujita-Jimbo, E.; Tanabe, Y.; Yu, Z.; Kojima, K.; Mori, M.; Li, H.; Iwamoto, S.; Yamagata, T.; Momoi, M.Y.; Momoi, T. The Association of GPR85 with PSD-95-Neuroligin Complex and Autism Spectrum Disorder: A Molecular Analysis. Mol. Autism 2015, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yang, H.; Lin, H.; Suo, A.; Wu, S.; Xie, W.; Zhou, N.; Guo, S.; Ding, H.; Zhou, G.; et al. Characterizing microRNA Editing and Mutation Sites in Autism Spectrum Disorder. Front. Mol. Neurosci. 2023, 15, 1105278. [Google Scholar] [CrossRef]
- Larco, D.O.; Cho-Clark, M.; Mani, S.K.; Wu, T.J. The Metabolite GnRH-(1–5) Inhibits the Migration of Immortalized GnRH Neurons. Endocrinology 2013, 154, 783–795. [Google Scholar] [CrossRef]
- Larco, D.O.; Bauman, B.M.; Cho-Clark, M.; Mani, S.K.; Wu, T.J. GnRH-(1–5) Inhibits TGF-β Signaling to Regulate the Migration of Immortalized Gonadotropin-Releasing Hormone Neurons. Front. Endocrinol. 2018, 9, 45. [Google Scholar] [CrossRef]
- Larco, D.O.; Semsarzadeh, N.N.; Cho-Clark, M.; Mani, S.K.; Wu, T.J. β-Arrestin 2 Is a Mediator of GnRH-(1–5) Signaling in Immortalized GnRH Neurons. Endocrinology 2013, 154, 4726–4736. [Google Scholar] [CrossRef]
- Stein, L.M.; Tullock, C.W.; Mathews, S.K.; Garcia-Galiano, D.; Elias, C.F.; Samson, W.K.; Yosten, G.L.C. Hypothalamic Action of Phoenixin to Control Reproductive Hormone Secretion in Females: Importance of the Orphan G Protein-Coupled Receptor Gpr173. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R489–R496. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, N.; Laschet, C.; Franssen, D.; Szpakowska, M.; Gilissen, J.; Geubelle, P.; Soni, A.; Parent, A.-S.; Pirotte, B.; Chevigné, A.; et al. Activation of the Orphan G Protein–Coupled Receptor GPR27 by Surrogate Ligands Promotes β-Arrestin 2 Recruitment. Mol. Pharmacol. 2017, 91, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Weng, X.; Wang, L.; Hao, M.; Li, Y.; Hou, L.; Liang, Y.; Wu, T.; Yao, M.; Lin, G.; et al. HIC1 Deletion Promotes Breast Cancer Progression by Activating Tumor Cell/Fibroblast Crosstalk. J. Clin. Investig. 2018, 128, 5235–5250. [Google Scholar] [CrossRef]
- Civelli, O.; Reinscheid, R.K.; Zhang, Y.; Wang, Z.; Fredriksson, R.; Schiöth, H.B. G Protein–Coupled Receptor Deorphanizations. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 127–146. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef]
- Hauser, A.S.; Kooistra, A.J.; Munk, C.; Heydenreich, F.M.; Veprintsev, D.B.; Bouvier, M.; Babu, M.M.; Gloriam, D.E. GPCR Activation Mechanisms across Classes and Macro/Microscales. Nat. Struct. Mol. Biol. 2021, 28, 879–888. [Google Scholar] [CrossRef]
- Stäubert, C.; Wozniak, M.; Dupuis, N.; Laschet, C.; Pillaiyar, T.; Hanson, J. Superconserved Receptors Expressed in the Brain: Expression, Function, Motifs and Evolution of an Orphan Receptor Family. Pharmacol. Ther. 2022, 240, 108217. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, Y.; Deng, L.; Du, K.; Lu, S.; Chen, T. Structural Understanding of Peptide-Bound G Protein-Coupled Receptors: Peptide–Target Interactions. J. Med. Chem. 2023, 66, 1083–1111. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B.; May, D.G. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2018, 91, 19–23. [Google Scholar] [CrossRef]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An Improved Smaller Biotin Ligase for BioID Proximity-Labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2013, 74, 19.23.1–19.23.14. [Google Scholar] [CrossRef]
- Kong, A.T.; Leprevost, F.V.; Avtonomov, D.M.; Mellacheruvu, D.; Nesvizhskii, A.I. MSFragger: Ultrafast and Comprehensive Peptide Identification in Mass Spectrometry–Based Proteomics. Nat. Methods 2017, 14, 513–520. [Google Scholar] [CrossRef]
- Da Veiga Leprevost, F.; Haynes, S.E.; Avtonomov, D.M.; Chang, H.-Y.; Shanmugam, A.K.; Mellacheruvu, D.; Kong, A.T.; Nesvizhskii, A.I. Philosopher: A Versatile Toolkit for Shotgun Proteomics Data Analysis. Nat. Methods 2020, 17, 869–870. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Haynes, S.E.; Teo, G.C.; Avtonomov, D.M.; Polasky, D.A.; Nesvizhskii, A.I. Fast Quantitative Analysis of timsTOF PASEF Data with MSFragger and IonQuant. Mol. Cell. Proteom. 2020, 19, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Teo, G.C.; Polasky, D.A.; Yu, F.; Nesvizhskii, A.I. Fast Deisotoping Algorithm and Its Implementation in the MSFragger Search Engine. J. Proteome Res. 2021, 20, 498–505. [Google Scholar] [CrossRef]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. SAINT: Probabilistic Scoring of Affinity Purification–Mass Spectrometry Data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Choi, H.; Glatter, T.; Gstaiger, M.; Nesvizhskii, A.I. SAINT-MS1: Protein–Protein Interaction Scoring Using Label-Free Intensity Data in Affinity Purification-Mass Spectrometry Experiments. J. Proteome Res. 2012, 11, 2619–2624. [Google Scholar] [CrossRef]
- Mellacheruvu, D.; Wright, Z.; Couzens, A.L.; Lambert, J.-P.; St-Denis, N.A.; Li, T.; Miteva, Y.V.; Hauri, S.; Sardiu, M.E.; Low, T.Y.; et al. The CRAPome: A Contaminant Repository for Affinity Purification–Mass Spectrometry Data. Nat. Methods 2013, 10, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Baines, A.J.; Lu, H.-C.; Bennett, P.M. The Protein 4.1 Family: Hub Proteins in Animals for Organizing Membrane Proteins. Biochim. Biophys. Acta Biomembr. 2014, 1838, 605–619. [Google Scholar] [CrossRef]
- Martínez Jaramillo, C.; Trujillo Vargas, C.M. LRBA in the Endomembrane System. Colomb. Med. 2018, 49, 236–243. [Google Scholar] [CrossRef]
- Kahlhofer, J.; Teis, D. The Human LAT1–4F2hc (SLC7A5–SLC3A2) Transporter Complex: Physiological and Pathophysiological Implications. Basic Clin. Pharmacol. Toxicol. 2022, 133, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Jiang, X.; Zhao, M.; Su, K.; Tang, M.; Hong, F.; Ye, N.; Zhang, R.; Li, N.; Wang, L.; et al. Identification of the Target Protein and Molecular Mechanism of Honokiol in Anti-Inflammatory Action. Phytomedicine 2023, 109, 154617. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Shi, H.; Zhang, G.; Peng, Y.; Ghosh, A.; Zhang, M.; Hu, X.; Liu, C.; Shao, Y.; Wang, S.; et al. A Novel CCK Receptor GPR173 Mediates Potentiation of GABAergic Inhibition. J. Neurosci. 2023, 43, 2305–2325. [Google Scholar] [CrossRef] [PubMed]
- Treen, A.K.; Luo, V.; Belsham, D.D. Phoenixin Activates Immortalized GnRH and Kisspeptin Neurons Through the Novel Receptor GPR173. Mol. Endocrinol. 2016, 30, 872–888. [Google Scholar] [CrossRef]
- Yanai, T.; Kurosawa, A.; Nikaido, Y.; Nakajima, N.; Saito, T.; Osada, H.; Konno, A.; Hirai, H.; Takeda, S. Identification and Molecular Docking Studies for Novel Inverse Agonists of SREB, Super Conserved Receptor Expressed in Brain. Genes Cells 2016, 21, 717–727. [Google Scholar] [CrossRef]
- Breton, T.S.; Sampson, W.G.B.; Clifford, B.; Phaneuf, A.M.; Smidt, I.; True, T.; Wilcox, A.R.; Lipscomb, T.; Murray, C.; DiMaggio, M.A. Characterization of the G Protein-Coupled Receptor Family SREB across Fish Evolution. Sci. Rep. 2021, 11, 12066. [Google Scholar] [CrossRef]
- Paek, J.; Kalocsay, M.; Staus, D.P.; Wingler, L.; Pascolutti, R.; Paulo, J.A.; Gygi, S.P.; Kruse, A.C. Multidimensional Tracking of GPCR Signaling via Peroxidase-Catalyzed Proximity-Labeling. Cell 2017, 169, 338–349.e11. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.L.; Steurer, M.A.; Aronstam, R.S. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE 2015, 10, e0138463. [Google Scholar] [CrossRef]
- Lefkowitz, R.J.; Shenoy, S.K. Transduction of Receptor Signals by SS-Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Rajagopal, S.; Shenoy, S.K. GPCR Desensitization: Acute and Prolonged Phases. Cell. Signal. 2018, 41, 9–16. [Google Scholar] [CrossRef]
- Cao, W.; Luttrell, L.M.; Medvedev, A.V.; Pierce, K.L.; Daniel, K.W.; Dixon, T.M.; Lefkowitz, R.J.; Collins, S. Direct Binding of Activated C-Src to the Β3-Adrenergic Receptor Is Required for MAP Kinase Activation. J. Biol. Chem. 2000, 275, 38131–38134. [Google Scholar] [CrossRef] [PubMed]
- Liggett, S.B.; Freedman, N.J.; Schwinn, D.A.; Lefkowitz, R.J. Structural Basis for Receptor Subtype-Specific Regulation Revealed by a Chimeric Beta 3/Beta 2-Adrenergic Receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 3665–3669. [Google Scholar] [CrossRef] [PubMed]
- Hislop, J.N.; Caunt, C.J.; Sedgley, K.R.; Kelly, E.; Mundell, S.; Green, L.D.; McArdle, C.A. Internalization of Gonadotropin-Releasing Hormone Receptors (GnRHRs): Does Arrestin Binding to the C-Terminal Tail Target GnRHRs for Dynamin-Dependent Internalization? J. Mol. Endocrinol. 2005, 35, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Wauson, E.M.; Zaganjor, E.; Lee, A.-Y.; Guerra, M.L.; Ghosh, A.B.; Bookout, A.L.; Chambers, C.P.; Jivan, A.; McGlynn, K.; Hutchison, M.R.; et al. The G Protein-Coupled Taste Receptor T1R1/T1R3 Regulates mTORC1 and Autophagy. Mol. Cell 2012, 47, 851–862. [Google Scholar] [CrossRef]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the Cystine/Glutamate Antiporter System x−c in the Brain, Kidney, and Duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type Amino Acid Transporter 1 as a Target for Drug Delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef]
- Reiser, M.; Li, K.; Lockey, R.; Wang, W. Lipopolysaccharide Responsive Beige-Like Anchor Subcellular Localization Involving in Vesicle Trafficking Responsive to Lipopolysaccharide. Austin J. Clin. Immunol. 2014, 1, 8. [Google Scholar]
- Mangodt, T.C.; Vanden Driessche, K.; Norga, K.K.; Moes, N.; De Bruyne, M.; Haerynck, F.; Bordon, V.; Jansen, A.C.; Jonckheere, A.I. Central Nervous System Manifestations of LRBA Deficiency: Case Report of Two Siblings and Literature Review. BMC Pediatr. 2023, 23, 353. [Google Scholar] [CrossRef]
- Pérez-Pérez, D.; Santos-Argumedo, L.; Rodríguez-Alba, J.C.; López-Herrera, G. Role of Protein Kinase A Activation in the Immune System with an Emphasis on Lipopolysaccharide-Responsive and Beige-like Anchor Protein in B Cells. Int. J. Mol. Sci. 2023, 24, 3098. [Google Scholar] [CrossRef]
- Bachmann, V.A.; Mayrhofer, J.E.; Ilouz, R.; Tschaikner, P.; Raffeiner, P.; Röck, R.; Courcelles, M.; Apelt, F.; Lu, T.-W.; Baillie, G.S.; et al. Gpr161 Anchoring of PKA Consolidates GPCR and cAMP Signaling. Proc. Natl. Acad. Sci. USA 2016, 113, 7786–7791. [Google Scholar] [CrossRef]
- Broselid, S.; Berg, K.A.; Chavera, T.A.; Kahn, R.; Clarke, W.P.; Olde, B.; Leeb-Lundberg, L.M.F. G Protein-Coupled Receptor 30 (GPR30) Forms a Plasma Membrane Complex with Membrane-Associated Guanylate Kinases (MAGUKs) and Protein Kinase A-Anchoring Protein 5 (AKAP5) That Constitutively Inhibits cAMP Production. J. Biol. Chem. 2014, 289, 22117–22127. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Jaramillo, C.; Trujillo-Vargas, C.M. Dissecting the Localization of Lipopolysaccharide-Responsive and Beige-like Anchor Protein (LRBA) in the Endomembrane System. Allergol. Immunopathol. 2020, 48, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Binda, A.V.; Kabbani, N.; Lin, R.; Levenson, R. D2 and D3 Dopamine Receptor Cell Surface Localization Mediated by Interaction with Protein 4.1N. Mol. Pharmacol. 2002, 62, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yan, H.; Othman, T.; Turner, C.P.; Woolf, T.; Rivkees, S.A. Cytoskeletal Protein 4.1G Binds to the Third Intracellular Loop of the A1 Adenosine Receptor and Inhibits Receptor Action. Biochem. J. 2004, 377, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Sugai, M.; Katsushima, Y.; Yanagisawa, T.; Sukegawa, J.; Nakahata, N. Increase in Cell-Surface Localization of Parathyroid Hormone Receptor by Cytoskeletal Protein 4.1G. Biochem. J. 2005, 392, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Tateyama, M.; Kubo, Y. Coupling Profile of the Metabotropic Glutamate Receptor 1α Is Regulated by the C-Terminal Domain. Mol. Cell. Neurosci. 2007, 34, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liang, F.; Walensky, L.D.; Huganir, R.L. Regulation of AMPA Receptor GluR1 Subunit Surface Expression by a 4.1N-Linked Actin Cytoskeletal Association. J. Neurosci. 2000, 20, 7932–7940. [Google Scholar] [CrossRef] [PubMed]
- Coleman, S.K.; Cai, C.; Mottershead, D.G.; Haapalahti, J.-P.; Keinänen, K. Surface Expression of GluR-D AMPA Receptor Is Dependent on an Interaction between Its C-Terminal Domain and a 4.1 Protein. J. Neurosci. 2003, 23, 798–806. [Google Scholar] [CrossRef]
- Leonard, A.S.; Davare, M.A.; Horne, M.C.; Garner, C.C.; Hell, J.W. SAP97 Is Associated with the α-Amino-3-Hydroxy-5-Methylisoxazole-4-Propionic Acid Receptor GluR1 Subunit. J. Biol. Chem. 1998, 273, 19518–19524. [Google Scholar] [CrossRef]
- Maguschak, K.A.; Ressler, K.J. A Role for WNT/β-Catenin Signaling in the Neural Mechanisms of Behavior. J. Neuroimmune Pharmacol. 2012, 7, 763–773. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, X.; Su, Y.; Yin, G.; Wang, S.; Yu, B.; Li, H.; Qi, J.; Chen, H.; Zeng, W.; et al. Activation of Wnt/β-Catenin Pathway Mitigates Blood–Brain Barrier Dysfunction in Alzheimer’s Disease. Brain 2022, 145, 4474–4488. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, X.; Yang, S.; Zhang, J. The Wnt /β-Catenin Signaling Pathway in the Adult Neurogenesis: The Wnt /β-Catenin Signaling Pathway. Eur. J. Neurosci. 2011, 33, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Dütting, E.; Enz, R. Band 4.1 Proteins Are Expressed in the Retina and Interact with Both Isoforms of the Metabotropic Glutamate Receptor Type 8: Band 4.1 Proteins Bind to mGluR8. J. Neurochem. 2008, 105, 2375–2387. [Google Scholar] [CrossRef]
- Gibson, A.W.; Li, X.; Wu, J.; Baskin, J.G.; Raman, C.; Edberg, J.C.; Kimberly, R.P. Serine Phosphorylation of FcγRI Cytoplasmic Domain Directs Lipid Raft Localization and Interaction with Protein 4.1G. J. Leukoc. Biol. 2011, 91, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wollscheid, B.; Watts, J.D.; Scheer, B.; Aebersold, R.; DeFranco, A.L. Quantitative Proteomic Analysis of B Cell Lipid Rafts Reveals That Ezrin Regulates Antigen Receptor–Mediated Lipid Raft Dynamics. Nat. Immunol. 2006, 7, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Villar, V.A.M.; Cuevas, S.; Zheng, X.; Jose, P.A. Localization and Signaling of GPCRs in Lipid Rafts. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 132, pp. 3–23. ISBN 978-0-12-803595-5. [Google Scholar]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaafarani, A.; Darche-Gabinaud, R.; Bisteau, X.; Imbault, V.; Wittamer, V.; Parmentier, M.; Pirson, I. Proximity Interactome Analysis of Super Conserved Receptors Expressed in the Brain Identifies EPB41L2, SLC3A2, and LRBA as Main Partners. Cells 2023, 12, 2625. https://doi.org/10.3390/cells12222625
Kaafarani A, Darche-Gabinaud R, Bisteau X, Imbault V, Wittamer V, Parmentier M, Pirson I. Proximity Interactome Analysis of Super Conserved Receptors Expressed in the Brain Identifies EPB41L2, SLC3A2, and LRBA as Main Partners. Cells. 2023; 12(22):2625. https://doi.org/10.3390/cells12222625
Chicago/Turabian StyleKaafarani, Abeer, Romain Darche-Gabinaud, Xavier Bisteau, Virginie Imbault, Valérie Wittamer, Marc Parmentier, and Isabelle Pirson. 2023. "Proximity Interactome Analysis of Super Conserved Receptors Expressed in the Brain Identifies EPB41L2, SLC3A2, and LRBA as Main Partners" Cells 12, no. 22: 2625. https://doi.org/10.3390/cells12222625