
Systemic Inflammatory Disorders, Immunosuppressive Treatment and Increase Risk of Head and Neck Cancers—A Narrative Review of Potential Physiopathological and Biological Mechanisms



Abstract
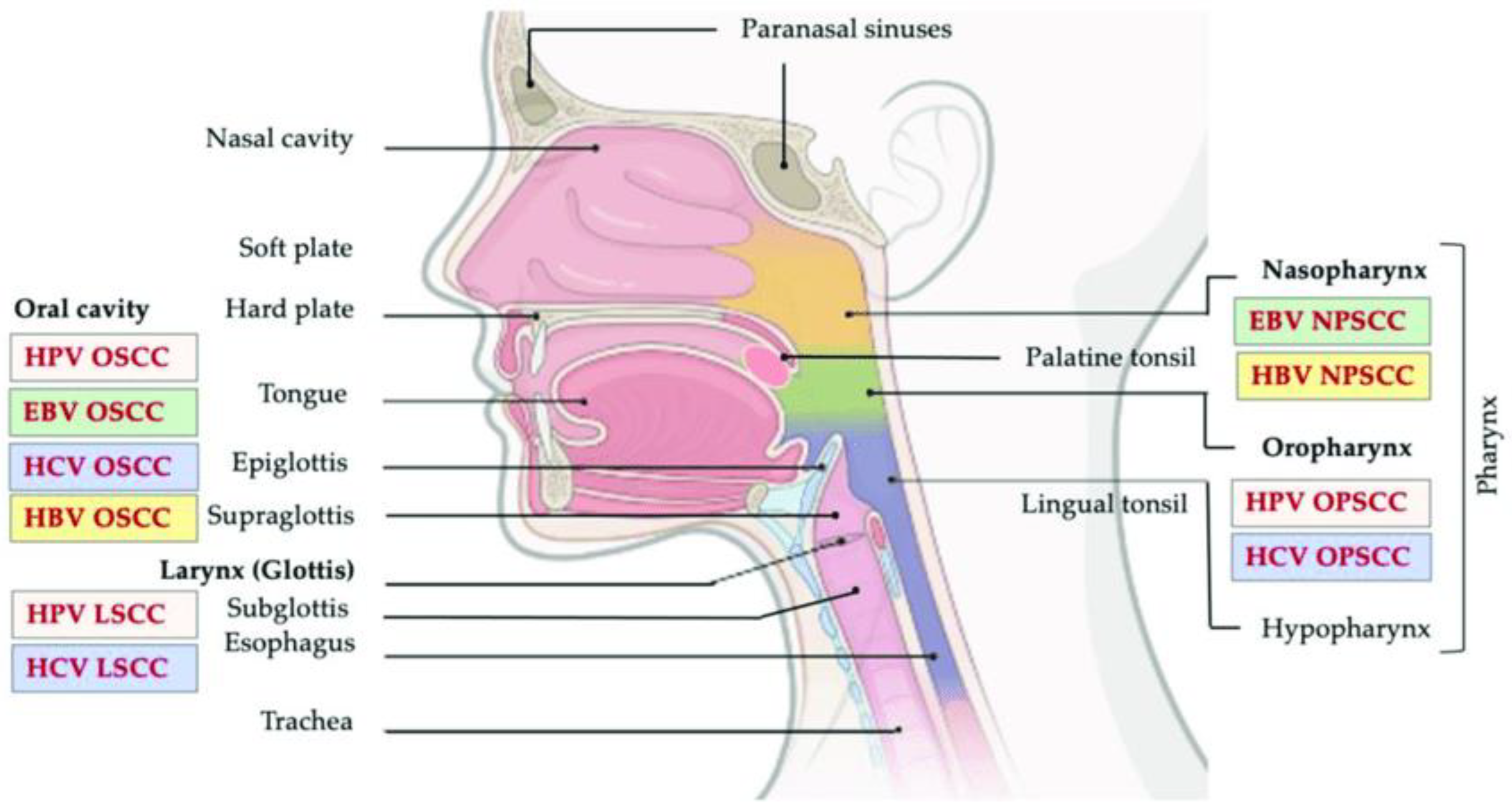
:1. Introduction
2. Materials and Methods
2.1. HNC Incidence in Systemic Inflammatory Diseases
2.2. Immune Pathways Review
3. Results
3.1. HNCs and the Immune System
- Cell-Mediated Inflammatory Diseases:
- Humoral Inflammatory Diseases:
3.1.1. Incidence of HNCs in Systemic Sclerosis
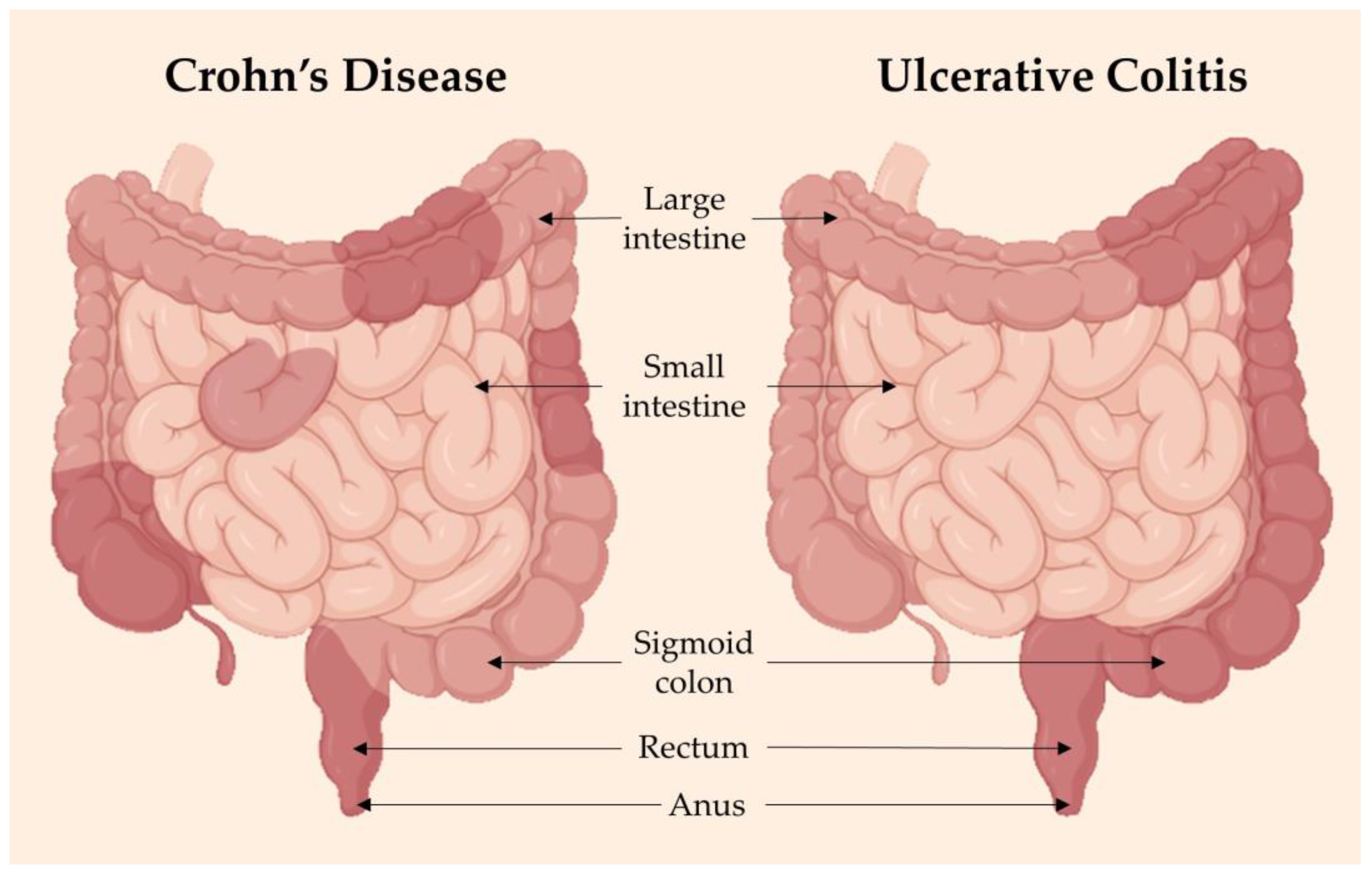
3.1.2. Incidence of HNCs in Inflammatory Bowel Diseases
3.1.3. Incidence of HNCs in Systemic Lupus Erythematous
3.1.4. Incidence of HNCs in Rheumatoid Arthritis
3.1.5. Incidence of HNCs in Dermatomyositis
3.1.6. Incidence of HNCs in Psoriasis
3.2. Pro-Tumor Cytokines and Inflammatory Signaling in Queried Auto-Immune Systemic Diseases
3.2.1. Systemic Sclerosis
3.2.2. Systemic Lupus Erythematous
3.2.3. Rheumatic Arthritis
3.2.4. Inflammatory Bowel Diseases
3.2.5. Psoriasis
3.2.6. Dermatomyositis
3.3. Anti-Tumor Cell-Mediated Immunity Pathways in Queried Auto-Immune Systemic Diseases
3.3.1. Rheumatic Arthritis
3.3.2. Systemic Sclerosis
3.3.3. Systemic Lupus Erythematous
3.3.4. Inflammatory Bowel Disease
3.3.5. Psoriasis
3.3.6. Dermatomyositis
3.4. Effects of Anti-Biological and Immunosuppressive Drugs on Cell Mediated Immunity
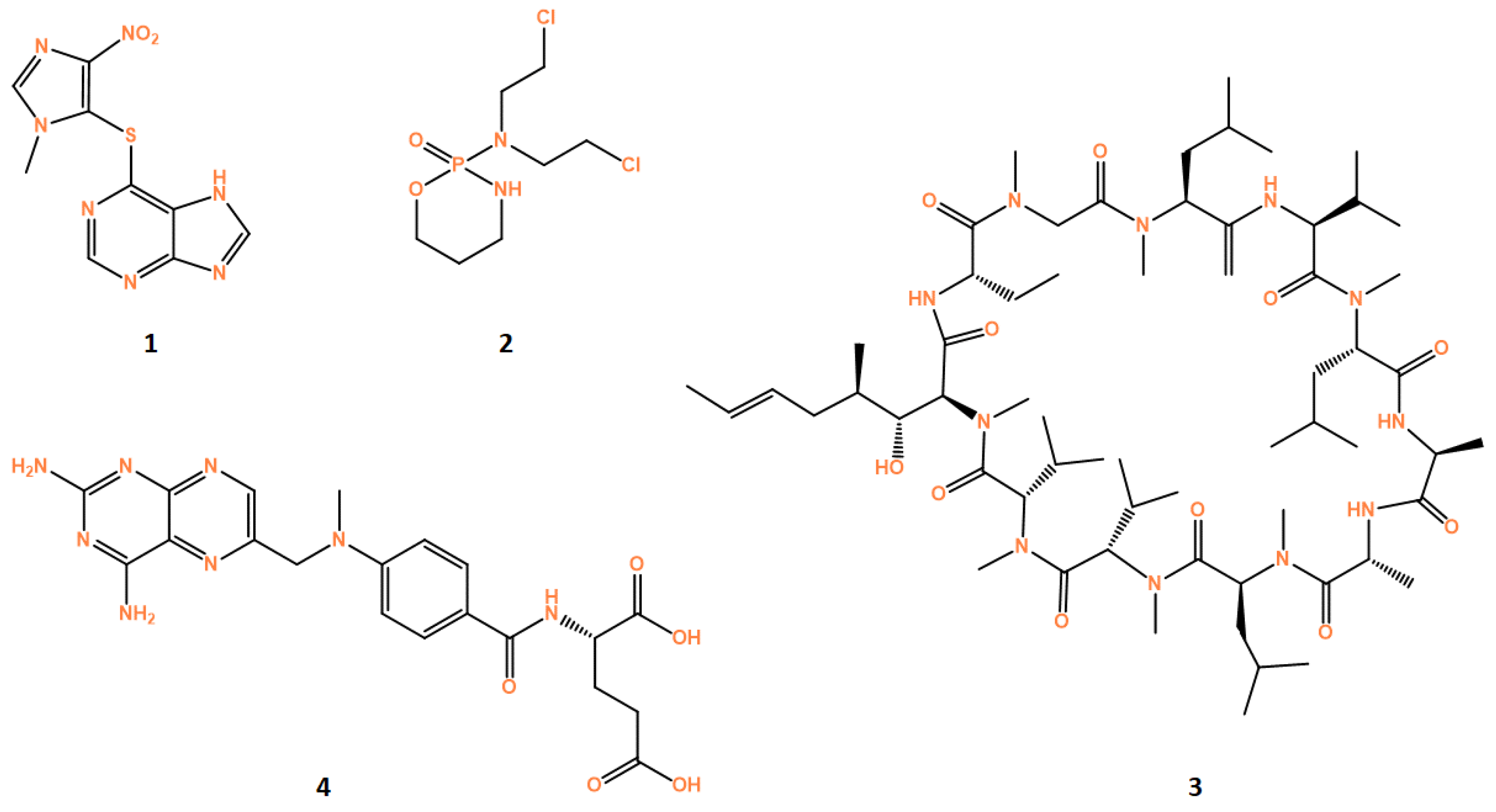
3.4.1. Glucocorticoids
3.4.2. Azathioprine
3.4.3. Cyclophosphamide
3.4.4. Cyclosporine
3.4.5. TNF-α Inhibitors
3.4.6. Methotrexate
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDS | Acquired immunodeficiency syndrome |
| AZA | Azathioprine |
| CCL | chemokine ligands |
| CD | Crohn’s disease |
| CRP | C-reactive protein |
| CsA | Cyclosporine |
| CTLs | Cytolytic T lymphocytes |
| CXCL | Chemokine platelet activating factor |
| CYC | Cyclophosphamide |
| DM | Dermatomyositis |
| DMARD | Disease-modifying antirheumatic drugs |
| EBV | Epstein-Barr virus |
| EGF | Epidermal growth factor |
| ESR | Westerngren erythrocyte sedimentation rate |
| GCS | Glucocorticoids |
| GM-GSF | Granulocyte macrophage colony stimulating factor |
| GRO | growth-related oncogene |
| HCMV-IgC | Human cytomegalovirus-immunoglobulin-G |
| HGF | hepatocyte growth factor |
| HIV | Human immunodeficiency virus |
| HNC | Head and neck cancers |
| HPV | Human papillomavirus |
| IBD | Inflammatory bowel disease |
| ICAM-1 | Intracellular adhesion molecule 1 |
| IFN-α/γ | Interferon alpha/gamma |
| IL | Interleukin |
| IL-2R | Interleukin 2 receptors |
| MAPK-1 | Mitogen-activated protein kinase-activator protein-1 |
| MCP1 | Monocyte chemoattractant protein 1 |
| MDC | Macrophage derived chemokine |
| MIP-1γ | Macrophage inflammatory protein 1 gamma |
| MMP | matrix metalloproteins |
| MTX | Methotrexate |
| NF-κB | Nuclear factor kappa B |
| NKCs | Natural killer cells |
| NPC | Nasopharyngeal carcinoma |
| PGE2 | Prostaglandin E2 |
| PM | Polymyositis |
| PMN | Polymorphonuclear neutrophils |
| RA | Rheumatoid Arthritis |
| SCCs | Squamous cell carcinomas |
| SIR | Standardized incidence ratio |
| SLE | Systemic lupus erythematous |
| SMD | Standardized mean differences |
| SSc | Systemic sclerosis |
| TAA | Tumor-associated antigens |
| TGF-α/β | Transforming growth factor alpha/beta |
| TNF-α | Tumor necrosis factor alpha |
| TNFi | Tumor necrosis factor inhibitors |
| T-regs | Regulatory T-cells |
| UC | Ulcerative colitis |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | vascular endothelial growth factor |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mahmutović, L.; Bilajac, E.; Hromić-Jahjefendić, A. Meet the Insidious Players: Review of Viral Infections in Head and Neck Cancer Etiology with an Update on Clinical Trials. Microorganisms 2021, 9, 1001. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: Linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar] [CrossRef]
- van Kempen, L.C.; de Visser, K.E.; Coussens, L.M. Inflammation, proteases and cancer. Eur. J. Cancer 2006, 42, 728–734. [Google Scholar] [CrossRef]
- Advisory, P.-I. The Link between Periodontal Disease and Cancer: A Review. Available online: https://www.perioimplantadvisory.com/clinical-tips/surgical-techniques/article/16411685/the-link-between-periodontal-disease-and-cancer-a-review (accessed on 23 March 2023).
- Zeng, X.T.; Deng, A.P.; Li, C.; Xia, L.Y.; Niu, Y.M.; Leng, W.D. Periodontal disease and risk of head and neck cancer: A meta-analysis of observational studies. PLoS ONE 2013, 8, e79017. [Google Scholar] [CrossRef]
- Tezal, M.; Sullivan, M.A.; Reid, M.E.; Marshall, J.R.; Hyland, A.; Loree, T.; Lillis, C.; Hauck, L.; Wactawski-Wende, J.; Scannapieco, F.A. Chronic periodontitis and the risk of tongue cancer. Arch. Otolaryngol. Head. Neck Surg. 2007, 133, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Velly, A.M.; Franco, E.L.; Schlecht, N.; Pintos, J.; Kowalski, L.P.; Oliveira, B.V.; Curado, M.P. Relationship between dental factors and risk of upper aerodigestive tract cancer. Oral. Oncol. 1998, 34, 284–291. [Google Scholar] [CrossRef]
- Söder, B.; Yakob, M.; Meurman, J.H.; Andersson, L.C.; Klinge, B.; Söder, P. Periodontal disease may associate with breast cancer. Breast Cancer Res. Treat. 2011, 127, 497–502. [Google Scholar] [CrossRef]
- Gasparoto, T.H.; de Oliveira, C.E.; de Freitas, L.T.; Pinheiro, C.R.; Ramos, R.N.; da Silva, A.L.; Garlet, G.P.; da Silva, J.S.; Campanelli, A.P. Inflammatory events during murine squamous cell carcinoma development. J. Inflamm. 2012, 9, 46. [Google Scholar] [CrossRef]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Zamarron, B.F.; Chen, W. Dual Roles of Immune Cells and Their Factors in Cancer Development and Progression. Int. J. Biol. Sci. 2011, 7, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cytokines in cancer pathogenesis and cancer therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef]
- Purgina, B.; Pantanowitz, L.; Seethala, R.R. A Review of Carcinomas Arising in the Head and Neck Region in HIV-Positive Patients. Pathol. Res. Int. 2011, 2011, 469150. [Google Scholar] [CrossRef] [PubMed]
- Rabinovics, N.; Mizrachi, A.; Hadar, T.; Ad-El, D.; Feinmesser, R.; Guttman, D.; Shpitzer, T.; Bachar, G. Cancer of the head and neck region in solid organ transplant recipients. Head. Neck 2014, 36, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Franks, A.L.; Slansky, J.E. Multiple associations between a broad spectrum of autoimmune diseases, chronic inflammatory diseases and cancer. Anticancer Res. 2012, 32, 1119–1136. [Google Scholar]
- Whiteside, T.L.; Chikamatsu, K.; Nagashima, S.; Okada, K. Antitumor effects of cytolytic T lymphocytes (CTL) and natural killer (NK) cells in head and neck cancer. Anticancer Res. 1996, 16, 2357–2364. [Google Scholar] [PubMed]
- Wang, Y.-T.; Kuo, L.-T.; Weng, H.-H.; Hsu, C.-M.; Tsai, M.-S.; Chang, G.-H.; Lee, Y.-C.; Huang, E.I.; Tsai, Y.-T. Systemic Immun e–Inflammation Index as a Predictor for Head and Neck Cancer Prognosis: A Meta-Analysis. Front. Oncol. 2022, 12, 899518. [Google Scholar] [CrossRef]
- Patini, R.; Cordaro, M.; Marchesini, D.; Scilla, F.; Gioco, G.; Rupe, C.; D’Agostino, M.A.; Lajolo, C. Is Systemic Immunosuppression a Risk Factor for Oral Cancer? A Systematic Review and Meta-Analysis. Cancers 2023, 15, 3077. [Google Scholar] [CrossRef]
- Zajac, A.J.; Harrington, L.E. Immune Response to Viruses: Cell-Mediated Immunity. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Kumari, P.; Debta, P.; Dixit, A. Oral Potentially Malignant Disorders: Etiology, Pathogenesis, and Transformation Into Oral Cancer. Front. Pharmacol. 2022, 13, 825266. [Google Scholar] [CrossRef]
- de Araújo, R.L.; Lyko Kde, F.; Funke, V.A.; Torres-Pereira, C.C. Oral cancer after prolonged immunosuppression for multiorgan chronic graft-versus-host disease. Rev. Bras. Hematol. Hemoter. 2014, 36, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Curtis, R.E.; Metayer, C.; Rizzo, J.D.; Socié, G.; Sobocinski, K.A.; Flowers, M.E.; Travis, W.D.; Travis, L.B.; Horowitz, M.M.; Deeg, H.J. Impact of chronic GVHD therapy on the development of squamous-cell cancers after hematopoietic stem-cell transplantation: An international case-control study. Blood 2005, 105, 3802–3811. [Google Scholar] [CrossRef]
- Leisenring, W.; Friedman, D.L.; Flowers, M.E.; Schwartz, J.L.; Deeg, H.J. Nonmelanoma skin and mucosal cancers after hematopoietic cell transplantation. J. Clin. Oncol. 2006, 24, 1119–1126. [Google Scholar] [CrossRef]
- Friedman, D.L.; Rovo, A.; Leisenring, W.; Locasciulli, A.; Flowers, M.E.; Tichelli, A.; Sanders, J.E.; Deeg, H.J.; Socie, G. Increased risk of breast cancer among survivors of allogeneic hematopoietic cell transplantation: A report from the FHCRC and the EBMT-Late Effect Working Party. Blood 2008, 111, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Atsuta, Y.; Suzuki, R.; Yamashita, T.; Fukuda, T.; Miyamura, K.; Taniguchi, S.; Iida, H.; Uchida, T.; Ikegame, K.; Takahashi, S.; et al. Continuing increased risk of oral/esophageal cancer after allogeneic hematopoietic stem cell transplantation in adults in association with chronic graft-versus-host disease. Ann. Oncol. 2014, 25, 435–441. [Google Scholar] [CrossRef]
- Sonis, S.T.; Amaral Mendes, R. Could the PI3K canonical pathway be a common link between chronic inflammatory conditions and oral carcinogenesis? J. Oral Pathol. Med. 2016, 45, 469–474. [Google Scholar] [CrossRef]
- Janowiak-Majeranowska, A.; Osowski, J.; Mikaszewski, B.; Majeranowski, A. Secondary Oral Cancer after Systemic Treatment of Hematological Malignancies and Oral GVHD: A Systematic Review. Cancers 2022, 14, 2175. [Google Scholar] [CrossRef] [PubMed]
- Zolkind, P.; Uppaluri, R. Checkpoint immunotherapy in head and neck cancers. Cancer Metastasis Rev. 2017, 36, 475–489. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z. The history and advances in cancer immunotherapy: Understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef]
- Mok, C.C.; Lau, C.S. Pathogenesis of systemic lupus erythematosus. J. Clin. Pathol. 2003, 56, 481–490. [Google Scholar] [CrossRef]
- Srivastava, A.; Makarenkova, H.P. Innate Immunity and Biological Therapies for the Treatment of Sjögren’s Syndrome. Int. J. Mol. Sci. 2020, 21, 9172. [Google Scholar] [CrossRef]
- Cooper, L.; Good-Jacobson, K.L. Dysregulation of humoral immunity in chronic infection. Immunol. Cell Biol. 2020, 98, 456–466. [Google Scholar] [CrossRef]
- Pereira, D.; Martins, D.; Mendes, F. Immunotherapy in Head and Neck Cancer When, How, and Why? Biomedicines 2022, 10, 2151. [Google Scholar] [CrossRef]
- Derk, C.T.; Rasheed, M.; Spiegel, J.R.; Jimenez, S.A. Increased incidence of carcinoma of the tongue in patients with systemic sclerosis. J. Rheumatol. 2005, 32, 637–641. [Google Scholar]
- Kuo, C.F.; Luo, S.F.; Yu, K.H.; Chou, I.J.; Tseng, W.Y.; Chang, H.C.; Fang, Y.F.; Chiou, M.J.; See, L.C. Cancer risk among patients with systemic sclerosis: A nationwide population study in Taiwan. Scand. J. Rheumatol. 2012, 41, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Katsanos, K.H.; Roda, G.; Brygo, A.; Delaporte, E.; Colombel, J.F. Oral Cancer and Oral Precancerous Lesions in Inflammatory Bowel Diseases: A Systematic Review. J. Crohns Colitis 2015, 9, 1043–1052. [Google Scholar] [CrossRef]
- Giagkou, E.; Christodoulou, D.K.; Katsanos, K.H. Mouth cancer in inflammatory bowel diseases. Oral Dis. 2016, 22, 260–264. [Google Scholar] [CrossRef]
- Vilas-Boas, F.; Magro, F.; Balhau, R.; Lopes, J.M.; Beça, F.; Eloy, C.; Lopes, S.; Macedo, G. Oral squamous cell carcinoma in a Crohn’s disease patient taking azathioprine: Case report and review of the literature. J. Crohns Colitis 2012, 6, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Engel, S.H.; Hullar, T.E.; Adkins, D.R.; Thorstad, W.L.; Sunwoo, J.B. Temporal relationship between antitumor necrosis factor-alpha antibody therapy and recrudescence of head and neck squamous cell carcinoma. Laryngoscope 2008, 118, 450–452. [Google Scholar] [CrossRef]
- Li, A.C.; Warnakulasuriya, S.; Thompson, R.P. Neoplasia of the tongue in a patient with Crohn’s disease treated with azathioprine: Case report. Eur. J. Gastroenterol. Hepatol. 2003, 15, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Hsu, H.T.; Weng, S.F.; Lin, Y.S. Impact of head and neck malignancies on risk factors and survival in systemic lupus erythematosus. Acta Otolaryngol. 2013, 133, 1088–1095. [Google Scholar] [CrossRef]
- Mao, S.; Shen, H.; Zhang, J. Systemic lupus erythematosus and malignancies risk. J. Cancer Res. Clin. Oncol. 2016, 142, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Tong, H.; Xu, G.; Liu, P.; Meng, H.; Wang, J.; Zhao, X.; Tang, Y.; Jin, J. Systemic lupus erythematous and malignancy risk: A meta-analysis. PLoS ONE 2015, 10, e0122964. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Chang, Y.T.; Wang, C.B.; Wu, C.Y. Malignancy in systemic lupus erythematosus: A nationwide cohort study in Taiwan. Am. J. Med. 2010, 123, 1150.e1–1150.e6. [Google Scholar] [CrossRef]
- Han, J.Y.; Kim, H.; Jung, S.Y.; Jang, E.J.; Cho, S.K.; Sung, Y.K. Increased risk of malignancy in patients with systemic lupus erythematosus: Population-based cohort study in Korea. Arthritis Res. Ther. 2021, 23, 270. [Google Scholar] [CrossRef]
- Grimaldo-Carjevschi, M.; López-Labady, J.; Villarroel-Dorrego, M. Squamous cell carcinoma on the palate in a patient with systemic lupus erythematosus: Case report and review of literature. Lupus 2011, 20, 519–522. [Google Scholar] [CrossRef]
- Unsworth, J.D.; Baldwin, A.; Byrd, L. Systemic lupus erythematosus, pregnancy and carcinoma of the tongue. BMJ Case Rep. 2013, 2013, bcr2013008864. [Google Scholar] [CrossRef]
- Beattie, A.; Stassen, L.F.; Ekanayake, K. Oral Squamous Cell Carcinoma Presenting in a Patient Receiving Adalimumab for Rheumatoid Arthritis. J. Oral Maxillofac. Surg. 2015, 73, 2136–2141. [Google Scholar] [CrossRef] [PubMed]
- Chainani-Wu, N.; Chang, C.; Gross, A.J.; Yom, S.S.; Silverman, S., Jr. Oropharyngeal carcinoma arising after methotrexate and etanercept therapy for rheumatoid arthritis. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, e261–e263. [Google Scholar] [CrossRef]
- Phillips, C.; Zeringue, A.L.; McDonald, J.R.; Eisen, S.A.; Ranganathan, P. Tumor Necrosis Factor Inhibition and Head and Neck Cancer Recurrence and Death in Rheumatoid Arthritis. PLoS ONE 2015, 10, e0143286. [Google Scholar] [CrossRef]
- Sugiyama, T.; Nakagawa, T.; Inui, M.; Tagawa, T. Tongue carcinoma in a young patient with dermatomyositis: A case report and review of the literature. J. Oral Maxillofac. Surg. 2001, 59, 925–928. [Google Scholar] [CrossRef] [PubMed]
- Adi, A.H.; Alturkmani, H.; Spock, T.; Williams Yohannes, P.; Wargo, S.; Szabo, E.; Gutkind, J.S.; Van Waes, C. Dermatomyositis paraneoplastic syndrome before symptomatic tonsillar squamous cell carcinoma: A case report. Head. Neck 2015, 37, E1–E3. [Google Scholar] [CrossRef] [PubMed]
- Botsios, C.; Ostuni, P.; Boscolo-Rizzo, P.; Da Mosto, M.C.; Punzi, L.; Marchiori, C. Dermatomyositis and malignancy of the pharynx in Caucasian patients: Report of two observations. Rheumatol. Int. 2003, 23, 309–311. [Google Scholar] [CrossRef]
- Holden, C.A.; Davis, R.W.; MacDonald, D.M. Dermatomyositis and salivary pleomorphic adenoma. J. R. Soc. Med. 1983, 76, 787–788. [Google Scholar] [CrossRef]
- Kim, S.W.; Shim, J.S.; Eun, Y.G.; Kwon, K.H. Tonsillar squamous cell carcinoma associated with dermatomyositis: The first 2 cases in Korea. Yonsei Med. J. 2010, 51, 605–608. [Google Scholar] [CrossRef]
- Liu, W.; Gao, Z.; Ni, D.; Li, W.; Li, Y.; Wang, H. [Dermatomyositis/polymyositis associated with head and neck malignancy]. Lin. Chuang Er Bi Yan Hou Ke Za Zhi 2005, 19, 340–341, 344. [Google Scholar]
- Song, X.; Peng, J.; Qiu, Q. Nasopharyngeal carcinoma and dermatomyositis (analysis of 12 cases). Lin. Chuang Er Bi Yan Hou Ke Za Zhi 1998, 12, 401–403. [Google Scholar] [PubMed]
- Peng, J.C.; Sheen, T.S.; Hsu, M.M. Nasopharyngeal carcinoma with dermatomyositis. Analysis of 12 cases. Arch. Otolaryngol. Head. Neck Surg. 1995, 121, 1298–1301. [Google Scholar] [CrossRef]
- Mebazâa, A.; Boussen, H.; Nouira, R.; Rokbani, L.; Ben Osman-Dhahri, A.; Bouaouina, N.; Laouani-Kechrid, C.; Louzir, B.; Zahaf, A.; Kamoun, M.R. Dermatomyositis and malignancy in Tunisia: A multicenter national retrospective study of 20 cases. J. Am. Acad. Dermatol. 2003, 48, 530–534. [Google Scholar] [CrossRef]
- Liu, W.C.; Ho, M.; Koh, W.P.; Tan, A.W.; Ng, P.P.; Chua, S.H.; Tan, S.H.; Tang, M.B. An 11-year review of dermatomyositis in Asian patients. Ann. Acad. Med. Singap. 2010, 39, 843–847. [Google Scholar] [CrossRef]
- Teo, P.; Tai, T.H.; Choy, D. Nasopharyngeal carcinoma with dermatomyositis. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 471–474. [Google Scholar] [CrossRef]
- Chen, D.Y.; Chen, Y.M.; Lan, J.L.; Chen, H.H.; Hsieh, C.W.; Wey, S.J.; Lu, J.J. Polymyositis/dermatomyositis and nasopharyngeal carcinoma: The Epstein-Barr virus connection? J. Clin. Virol. 2010, 49, 290–295. [Google Scholar] [CrossRef]
- Abraham, Z.; Rosner, I.; Rozenbaum, M.; Goldenberg, K.; Weller, B. Dermatomyositis and nasopharyngeal carcinoma. J. Dermatol. 1998, 25, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Boffetta, P.; Gridley, G.; Lindelöf, B. Cancer risk in a population-based cohort of patients hospitalized for psoriasis in Sweden. J. Investig. Dermatol. 2001, 117, 1531–1537. [Google Scholar] [CrossRef] [PubMed]
- Frentz, G.; Olsen, J.H. Malignant tumours and psoriasis: A follow-up study. Br. J. Dermatol. 1999, 140, 237–242. [Google Scholar] [CrossRef]
- Hannuksela-Svahn, A.; Pukkala, E.; Läärä, E.; Poikolainen, K.; Karvonen, J. Psoriasis, its treatment, and cancer in a cohort of Finnish patients. J. Investig. Dermatol. 2000, 114, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Seoane, J.; Varela-Centelles, P.; López-Niño, J.; Vázquez, I.; Abdulkader, I.; García-Caballero, T. Gingival mucinous adenocarcinoma of a minor salivary gland. J. Periodontol. 2010, 81, 626–631. [Google Scholar] [CrossRef]
- Carlesimo, M.; Mari, E.; La Pietra, M.; Orsini, D.; Pranteda, G.; Pranteda, G.; Grimaldi, M.; Arcese, A. Occurrence of salivary gland tumours in two patients treated with biological agents. Int. J. Immunopathol. Pharmacol. 2012, 25, 297–300. [Google Scholar] [CrossRef]
- Yamamoto, T.; Katayama, I.; Nishioka, K. Adenocarcinoma of the mouth in a patient with psoriasis under short-term cyclosporine therapy. Dermatology 1996, 193, 72–73. [Google Scholar] [CrossRef]
- Higley, H.; Persichitte, K.; Chu, S.; Waegell, W.; Vancheeswaran, R.; Black, C. Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud’s phenomenon. Arthritis Rheum. 1994, 37, 278–288. [Google Scholar] [CrossRef]
- Muangchant, C.; Pope, J.E. The significance of interleukin-6 and C-reactive protein in systemic sclerosis: A systematic literature review. Clin. Exp. Rheumatol. 2013, 31, 122–134. [Google Scholar]
- Scala, E.; Pallotta, S.; Frezzolini, A.; Abeni, D.; Barbieri, C.; Sampogna, F.; De Pità, O.; Puddu, P.; Paganelli, R.; Russo, G. Cytokine and chemokine levels in systemic sclerosis: Relationship with cutaneous and internal organ involvement. Clin. Exp. Immunol. 2004, 138, 540–546. [Google Scholar] [CrossRef]
- Needleman, B.W.; Wigley, F.M.; Stair, R.W. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. 1992, 35, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Takemura, H.; Suzuki, H.; Yoshizaki, K.; Ogata, A.; Yuhara, T.; Akama, T.; Yamane, K.; Kashiwagi, H. Anti-interleukin-6 autoantibodies in rheumatic diseases. Increased frequency in the sera of patients with systemic sclerosis. Arthritis Rheum. 1992, 35, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Kurasawa, K.; Hirose, K.; Sano, H.; Endo, H.; Shinkai, H.; Nawata, Y.; Takabayashi, K.; Iwamoto, I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000, 43, 2455–2463. [Google Scholar] [CrossRef] [PubMed]
- Furuse, S.; Fujii, H.; Kaburagi, Y.; Fujimoto, M.; Hasegawa, M.; Takehara, K.; Sato, S. Serum concentrations of the CXC chemokines interleukin 8 and growth-regulated oncogene-alpha are elevated in patients with systemic sclerosis. J. Rheumatol. 2003, 30, 1524–1528. [Google Scholar]
- Denton, C.P.; Bickerstaff, M.C.; Shiwen, X.; Carulli, M.T.; Haskard, D.O.; Dubois, R.M.; Black, C.M. Serial circulating adhesion molecule levels reflect disease severity in systemic sclerosis. Br. J. Rheumatol. 1995, 34, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef]
- Vandercappellen, J.; Van Damme, J.; Struyf, S. The role of the CXC chemokines platelet factor-4 (CXCL4/PF-4) and its variant (CXCL4L1/PF-4var) in inflammation, angiogenesis and cancer. Cytokine Growth Factor Rev. 2011, 22, 1–18. [Google Scholar] [CrossRef]
- von Wussow, P.; Jakschies, D.; Hartung, K.; Deicher, H. Presence of interferon and anti-interferon in patients with systemic lupus erythematosus. Rheumatol. Int. 1988, 8, 225–230. [Google Scholar] [CrossRef]
- Yee, A.M.; Yip, Y.K.; Fischer, H.D.; Buyon, J.P. Serum activity that confers acid lability to alpha-interferon in systemic lupus erythematosus: Its association with disease activity and its independence from circulating alpha-interferon. Arthritis Rheum. 1990, 33, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Maury, C.P.; Teppo, A.M. Tumor necrosis factor in the serum of patients with systemic lupus erythematosus. Arthritis Rheum. 1989, 32, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.L.; Chang, K.L.; Chiu, C.C.; Chiang, B.N.; Han, S.H.; Wang, S.R. Defective phagocytosis, decreased tumour necrosis factor-alpha production, and lymphocyte hyporesponsiveness predispose patients with systemic lupus erythematosus to infections. Scand. J. Rheumatol. 1989, 18, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.O.; Fronek, Z.; Lewis, G.D.; Koo, M.; Hansen, J.A.; McDevitt, H.O. Heritable major histocompatibility complex class II-associated differences in production of tumor necrosis factor alpha: Relevance to genetic predisposition to systemic lupus erythematosus. Proc. Natl. Acad. Sci. USA 1990, 87, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Draeger, A.M.; Swaak, A.J.; van den Brink, H.G.; Aarden, L.A. T cell function in systemic lupus erythematosus: Normal production of and responsiveness to interleukin 2. Clin. Exp. Immunol. 1986, 64, 80–87. [Google Scholar]
- Sibbitt, W.L., Jr.; Kenny, C.; Spellman, C.W.; Ley, K.D.; Bankhurst, A.D. Lymphokines in autoimmunity: Relationship between interleukin-2 and interferon-gamma production in systemic lupus erythematosus. Clin. Immunol. Immunopathol. 1984, 32, 166–173. [Google Scholar] [CrossRef]
- Campen, D.H.; Horwitz, D.A.; Quismorio, F.P., Jr.; Ehresmann, G.R.; Martin, W.J. Serum levels of interleukin-2 receptor and activity of rheumatic diseases characterized by immune system activation. Arthritis Rheum. 1988, 31, 1358–1364. [Google Scholar] [CrossRef]
- Tokano, Y.; Murashima, A.; Takasaki, Y.; Hashimoto, H.; Okumura, K.; Hirose, S. Relation between soluble interleukin 2 receptor and clinical findings in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 1989, 48, 803–809. [Google Scholar] [CrossRef]
- Ohtsuka, K.; Gray, J.D.; Stimmler, M.M.; Horwitz, D.A. The relationship between defects in lymphocyte production of transforming growth factor-beta1 in systemic lupus erythematosus and disease activity or severity. Lupus 1999, 8, 90–94. [Google Scholar] [CrossRef]
- Jacob, C.O.; van der Meide, P.H.; McDevitt, H.O. In vivo treatment of (NZB X NZW)F1 lupus-like nephritis with monoclonal antibody to gamma interferon. J. Exp. Med. 1987, 166, 798–803. [Google Scholar] [CrossRef]
- Jacob, C.O.; Hwang, F.; Lewis, G.D.; Stall, A.M. Tumor necrosis factor alpha in murine systemic lupus erythematosus disease models: Implications for genetic predisposition and immune regulation. Cytokine 1991, 3, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Brennan, F.M.; Maini, R.N.; Feldmann, M. TNF alpha--a pivotal role in rheumatoid arthritis? Br. J. Rheumatol. 1992, 31, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Leung, B.P.; Sturrock, R.D.; Field, M.; Liew, F.Y. Interleukin-15 mediates T cell-dependent regulation of tumor necrosis factor-alpha production in rheumatoid arthritis. Nat. Med. 1997, 3, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Alvaro-Gracia, J.M.; Zvaifler, N.J.; Firestein, G.S. Cytokines in chronic inflammatory arthritis. IV. Granulocyte/macrophage colony-stimulating factor-mediated induction of class II MHC antigen on human monocytes: A possible role in rheumatoid arthritis. J. Exp. Med. 1989, 170, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Burmester, G.R.; Weinblatt, M.E.; McInnes, I.B.; Porter, D.; Barbarash, O.; Vatutin, M.; Szombati, I.; Esfandiari, E.; Sleeman, M.A.; Kane, C.D.; et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 1445–1452. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Wei, C.C.; Shieh, D.B.; Chan, C.H.; Chang, M.S. Anti-IL-20 monoclonal antibody alleviates inflammation in oral cancer and suppresses tumor growth. Mol. Cancer Res. 2012, 10, 1430–1439. [Google Scholar] [CrossRef]
- Kragstrup, T.W.; Otkjaer, K.; Holm, C.; Jørgensen, A.; Hokland, M.; Iversen, L.; Deleuran, B. The expression of IL-20 and IL-24 and their shared receptors are increased in rheumatoid arthritis and spondyloarthropathy. Cytokine 2008, 41, 16–23. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Surgeons, T. Inflammatory Bowel Disease (IBD). Available online: https://www.thesurgeons.sg/services/inflammatory-bowel-disease/ (accessed on 5 April 2023).
- Strober, W.; Fuss, I.J. Proinflammatory cytokines in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef]
- Korolkova, O.Y.; Myers, J.N.; Pellom, S.T.; Wang, L.; M’Koma, A.E. Characterization of Serum Cytokine Profile in Predominantly Colonic Inflammatory Bowel Disease to Delineate Ulcerative and Crohn’s Colitides. Clin. Med. Insights Gastroenterol. 2015, 8, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, A.; Boström, L.; Söderberg-Naucler, C. Detection of cytotoxic CD13-specific autoantibodies in sera from patients with ulcerative colitis and Crohn’s disease. J. Autoimmun. 2006, 26, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Sahin, A.; Calhan, T.; Cengiz, M.; Kahraman, R.; Aydin, K.; Ozdil, K.; Korachi, M.; Sokmen, H.M. Serum Interleukin 17 Levels in Patients with Crohn’s Disease: Real Life Data. Dis. Markers 2014, 2014, 690853. [Google Scholar] [CrossRef]
- Gelfand, J.M.; Neimann, A.L.; Shin, D.B.; Wang, X.; Margolis, D.J.; Troxel, A.B. Risk of myocardial infarction in patients with psoriasis. JAMA 2006, 296, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Brauchli, Y.B.; Jick, S.S.; Miret, M.; Meier, C.R. Psoriasis and risk of incident cancer: An inception cohort study with a nested case-control analysis. J. Investig. Dermatol. 2009, 129, 2604–2612. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Choi, H.K.; Setty, A.R.; Curhan, G.C. Psoriasis and the risk of diabetes and hypertension: A prospective study of US female nurses. Arch. Dermatol. 2009, 145, 379–382. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Wajda, A.; Blanchard, J.F. The clustering of other chronic inflammatory diseases in inflammatory bowel disease: A population-based study. Gastroenterology 2005, 129, 827–836. [Google Scholar] [CrossRef]
- Makredes, M.; Robinson, D., Jr.; Bala, M.; Kimball, A.B. The burden of autoimmune disease: A comparison of prevalence ratios in patients with psoriatic arthritis and psoriasis. J. Am. Acad. Dermatol. 2009, 61, 405–410. [Google Scholar] [CrossRef]
- Dowlatshahi, E.A.; van der Voort, E.A.; Arends, L.R.; Nijsten, T. Markers of systemic inflammation in psoriasis: A systematic review and meta-analysis. Br. J. Dermatol. 2013, 169, 266–282. [Google Scholar] [CrossRef]
- Kaji, K.; Fujimoto, M.; Hasegawa, M.; Kondo, M.; Saito, Y.; Komura, K.; Matsushita, T.; Orito, H.; Hamaguchi, Y.; Yanaba, K.; et al. Identification of a novel autoantibody reactive with 155 and 140 kDa nuclear proteins in patients with dermatomyositis: An association with malignancy. Rheumatology 2007, 46, 25–28. [Google Scholar] [CrossRef]
- Targoff, I.N.; Mamyrova, G.; Trieu, E.P.; Perurena, O.; Koneru, B.; O’Hanlon, T.P.; Miller, F.W.; Rider, L.G. A novel autoantibody to a 155-kd protein is associated with dermatomyositis. Arthritis Rheum. 2006, 54, 3682–3689. [Google Scholar] [CrossRef] [PubMed]
- Fiorentino, D.F.; Chung, L.S.; Christopher-Stine, L.; Zaba, L.; Li, S.; Mammen, A.L.; Rosen, A.; Casciola-Rosen, L. Most patients with cancer-associated dermatomyositis have antibodies to nuclear matrix protein NXP-2 or transcription intermediary factor 1γ. Arthritis Rheum. 2013, 65, 2954–2962. [Google Scholar] [CrossRef] [PubMed]
- Trallero-Araguás, E.; Labrador-Horrillo, M.; Selva-O’Callaghan, A.; Martínez, M.A.; Martínez-Gómez, X.; Palou, E.; Rodriguez-Sanchez, J.L.; Vilardell-Tarrés, M. Cancer-associated myositis and anti-p155 autoantibody in a series of 85 patients with idiopathic inflammatory myopathy. Medicine 2010, 89, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Shinjo, S.K.; de Souza, F.H.; de Moraes, J.C. Dermatomyositis and polymyositis: From immunopathology to immunotherapy (immunobiologics). Rev. Bras. Reumatol. 2013, 53, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Bilgic, H.; Ytterberg, S.R.; Amin, S.; McNallan, K.T.; Wilson, J.C.; Koeuth, T.; Ellingson, S.; Newman, B.; Bauer, J.W.; Peterson, E.J.; et al. Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis. Arthritis Rheum. 2009, 60, 3436–3446. [Google Scholar] [CrossRef]
- Kao, L.; Chung, L.; Fiorentino, D.F. Pathogenesis of dermatomyositis: Role of cytokines and interferon. Curr. Rheumatol. Rep. 2011, 13, 225–232. [Google Scholar] [CrossRef]
- Antiga, E.; Kretz, C.C.; Klembt, R.; Massi, D.; Ruland, V.; Stumpf, C.; Baroni, G.; Hartmann, M.; Hartschuh, W.; Volpi, W.; et al. Characterization of regulatory T cells in patients with dermatomyositis. J. Autoimmun. 2010, 35, 342–350. [Google Scholar] [CrossRef]
- Mielnik, P.; Chwalinska-Sadowska, H.; Wiesik-Szewczyk, E.; Maslinski, W.; Olesinska, M. Serum concentration of interleukin 15, interleukin 2 receptor and TNF receptor in patients with polymyositis and dermatomyositis: Correlation to disease activity. Rheumatol. Int. 2012, 32, 639–643. [Google Scholar] [CrossRef]
- Gary S Firestein, M.; Monica Guma, M. Pathogenesis of Rheumatoid Arthritis. Available online: https://www.uptodate.com/contents/pathogenesis-of-rheumatoid-arthritis (accessed on 29 March 2023).
- Andrianakos, A.A.; Sharp, J.T.; Person, D.A.; Lidsky, M.D.; Duffy, J. Cell-mediated immunity in rheumatoid arthritis. Ann. Rheum. Dis. 1977, 36, 13. [Google Scholar] [CrossRef]
- Toh, B.H.; Roberts-Thomson, I.C.; Mathews, J.D.; Whittingham, S.; Mackay, I.R. Depression of cell-mediated immunity in old age and the immunopathic diseases, lupus erythematosus, chronic hepatitis and rheumatoid arthritis. Clin. Exp. Immunol. 1973, 14, 193–202. [Google Scholar]
- Yanagihara, Y.; Shiozawa, K.; Takai, M.; Kyogoku, M.; Shiozawa, S. Natural killer (NK) T cells are significantly decreased in the peripheral blood of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 1999, 118, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Shegarfi, H.; Naddafi, F.; Mirshafiey, A. Natural killer cells and their role in rheumatoid arthritis: Friend or foe? Sci. World J. 2012, 2012, 491974. [Google Scholar] [CrossRef]
- Radstake, T.R.D.J.; van Bon, L.; Broen, J.; Hussiani, A.; Hesselstrand, R.; Wuttge, D.M.; Deng, Y.; Simms, R.; Lubberts, E.; Lafyatis, R. The Pronounced Th17 Profile in Systemic Sclerosis (SSc) Together with Intracellular Expression of TGFβ and IFNγ Distinguishes SSc Phenotypes. PLoS ONE 2009, 4, e5903. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.M.; Harding, B.; Mirick, G.R.; Schneider, J.; Quismorio, F.P.; Friou, G.J. Selective decrease in antibody-dependent cell-mediated cytotoxicity in systemic lupus erythematosus and progressive systemic sclerosis. Clin. Exp. Immunol. 1978, 34, 235–240. [Google Scholar]
- Hughes, P.; Holt, S.; Rowell, N.R.; Allonby, I.D.; Janis, K.; Dodd, J.K. The relationship of defective cell-mediated immunity to visceral disease in systemic sclerosis. Clin. Exp. Immunol. 1977, 28, 233–240. [Google Scholar]
- Mandal, A.; Viswanathan, C. Natural killer cells: In health and disease. Hematol./Oncol. Stem Cell Ther. 2015, 8, 47–55. [Google Scholar] [CrossRef]
- Funderburg, N.T.; Stubblefield Park, S.R.; Sung, H.C.; Hardy, G.; Clagett, B.; Ignatz-Hoover, J.; Harding, C.V.; Fu, P.; Katz, J.A.; Lederman, M.M.; et al. Circulating CD4(+) and CD8(+) T cells are activated in inflammatory bowel disease and are associated with plasma markers of inflammation. Immunology 2013, 140, 87–97. [Google Scholar] [CrossRef]
- Beeken, W.L.; Macpherson, B.R.; Gundel, R.M.; St Andre-Ukena, S.; Wood, S.G.; Sylwester, D.L. Depressed spontaneous cell-mediated cytotoxicity in Crohn’s disease. Clin. Exp. Immunol. 1983, 51, 351–358. [Google Scholar]
- Egawa, S.; Hiwatashi, N. Natural killer cell activity in patients with inflammatory bowel disease. J. Clin. Lab. Immunol. 1986, 20, 187–192. [Google Scholar]
- Ventura, M.; Colizzi, M.; Ottolenghi, A.; Polignano, A.; Sinisi, D.A.; Rantuccio, F.; Antonaci, S.; Bonomo, L. Cell-mediated immune response in psoriasis and psoriatic arthritis. Recent. Prog. Med. 1989, 80, 449–454. [Google Scholar]
- Cameron, A.L.; Kirby, B.; Griffiths, C.E. Circulating natural killer cells in psoriasis. Br. J. Dermatol. 2003, 149, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Tobin, A.M.; Lynch, L.; Kirby, B.; O’Farrelly, C. Natural Killer Cells in Psoriasis. J. Innate Immun. 2011, 3, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Majewski, S.; Wasik, M.; Jabłońska, S.; Kamiński, M.; Fraczykowska, M. Defective natural-killer- and killer-cell activity associated with increased polymorphonuclear leukocyte adherence in psoriasis. Arch. Dermatol. Res. 1986, 278, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Dunphy, S.; Gardiner, C.M. NK Cells and Psoriasis. J. Biomed. Biotechnol. 2011, 2011, 248317. [Google Scholar] [CrossRef]
- Greenberg, S.A. Type 1 interferons and myositis. Arthritis Res. Ther. 2010, 12 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Walsh, R.J.; Kong, S.W.; Yao, Y.; Jallal, B.; Kiener, P.A.; Pinkus, J.L.; Beggs, A.H.; Amato, A.A.; Greenberg, S.A. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007, 56, 3784–3792. [Google Scholar] [CrossRef]
- Aleksza, M.; Szegedi, A.; Antal-Szalmás, P.; Irinyi, B.; Gergely, L.; Ponyi, A.; Hunyadi, J.; Sipka, S.; Zeher, M.; Szegedi, G.; et al. Altered cytokine expression of peripheral blood lymphocytes in polymyositis and dermatomyositis. Ann. Rheum. Dis. 2005, 64, 1485. [Google Scholar] [CrossRef]
- O’Gorman, M.R.; Bianchi, L.; Zaas, D.; Corrochano, V.; Pachman, L.M. Decreased levels of CD54 (ICAM-1)-positive lymphocytes in the peripheral blood in untreated patients with active juvenile dermatomyositis. Clin. Diagn. Lab. Immunol. 2000, 7, 693–697. [Google Scholar] [CrossRef]
- Mitsuo, A.; Morimoto, S.; Nakiri, Y.; Suzuki, J.; Kaneko, H.; Tokano, Y.; Tsuda, H.; Takasaki, Y.; Hashimoto, H. Decreased CD161+CD8+ T cells in the peripheral blood of patients suffering from rheumatic diseases. Rheumatology 2006, 45, 1477–1484. [Google Scholar] [CrossRef]
- Viguier, M.; Fouéré, S.; de la Salmonière, P.; Rabian, C.; Lebbe, C.; Dubertret, L.; Morel, P.; Bachelez, H. Peripheral blood lymphocyte subset counts in patients with dermatomyositis: Clinical correlations and changes following therapy. Medicine 2003, 82, 82–86. [Google Scholar] [CrossRef]
- O’Gorman, M.R.; Corrochano, V.; Roleck, J.; Donovan, M.; Pachman, L.M. Flow cytometric analyses of the lymphocyte subsets in peripheral blood of children with untreated active juvenile dermatomyositis. Clin. Diagn. Lab. Immunol. 1995, 2, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Matsumoto, Y.; Ohashi, M.; Sasaki, R. Analysis of lymphocyte subpopulations in peripheral blood in adult and juvenile cases of dermatomyositis. J. Dermatol. 1993, 20, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.X.; Lu, X.; Zu, N.; Lin, B.; Wang, L.Y.; Shu, X.M.; Ma, L.; Wang, G.C. Clinical significance of peripheral blood lymphocyte subsets in patients with polymyositis and dermatomyositis. Clin. Rheumatol. 2012, 31, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Brattsand, R.; Linden, M. Cytokine modulation by glucocorticoids: Mechanisms and actions in cellular studies. Aliment. Pharmacol. Ther. 1996, 10 (Suppl. S2), 81–90; discussion 91–92. [Google Scholar] [CrossRef]
- Boumpas, D.T.; Chrousos, G.P.; Wilder, R.L.; Cupps, T.R.; Balow, J.E. Glucocorticoid therapy for immune-mediated diseases: Basic and clinical correlates. Ann. Intern. Med. 1993, 119, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, J.D.; Lu, F.W.; Vacchio, M.S. Glucocorticoids in T cell development and function*. Annu. Rev. Immunol. 2000, 18, 309–345. [Google Scholar] [CrossRef]
- Lanza, L.; Scudeletti, M.; Puppo, F.; Bosco, O.; Peirano, L.; Filaci, G.; Fecarotta, E.; Vidali, G.; Indiveri, F. Prednisone increases apoptosis in in vitro activated human peripheral blood T lymphocytes. Clin. Exp. Immunol. 1996, 103, 482–490. [Google Scholar] [CrossRef]
- Franchimont, D.; Louis, E.; Dewe, W.; Martens, H.; Vrindts-Gevaert, Y.; De Groote, D.; Belaiche, J.; Geenen, V. Effects of dexamethasone on the profile of cytokine secretion in human whole blood cell cultures. Regul. Pept. 1998, 73, 59–65. [Google Scholar] [CrossRef]
- Visser, J.; van Boxel-Dezaire, A.; Methorst, D.; Brunt, T.; de Kloet, E.R.; Nagelkerken, L. Differential regulation of interleukin-10 (IL-10) and IL-12 by glucocorticoids in vitro. Blood 1998, 91, 4255–4264. [Google Scholar] [CrossRef]
- Franchimont, D.; Galon, J.; Gadina, M.; Visconti, R.; Zhou, Y.; Aringer, M.; Frucht, D.M.; Chrousos, G.P.; O’Shea, J.J. Inhibition of Th1 immune response by glucocorticoids: Dexamethasone selectively inhibits IL-12-induced Stat4 phosphorylation in T lymphocytes. J. Immunol. 2000, 164, 1768–1774. [Google Scholar] [CrossRef]
- Weaver, C.T.; Harrington, L.E.; Mangan, P.R.; Gavrieli, M.; Murphy, K.M. Th17: An effector CD4 T cell lineage with regulatory T cell ties. Immunity 2006, 24, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Kozhaya, L.; McKevitt, K.; Djuretic, I.M.; Carlson, T.J.; Quintero, M.A.; McCauley, J.L.; Abreu, M.T.; Unutmaz, D.; Sundrud, M.S. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J. Exp. Med. 2014, 211, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, A. The effects of azathioprine and 6 MP on immunity. J. Immunopharmacol. 1979, 1, 429–454. [Google Scholar] [CrossRef]
- Leitner, J.; Drobits, K.; Pickl, W.F.; Majdic, O.; Zlabinger, G.; Steinberger, P. The effects of Cyclosporine A and azathioprine on human T cells activated by different costimulatory signals. Immunol. Lett. 2011, 140, 74–80. [Google Scholar] [CrossRef]
- Tiede, I.; Fritz, G.; Strand, S.; Poppe, D.; Dvorsky, R.; Strand, D.; Lehr, H.A.; Wirtz, S.; Becker, C.; Atreya, R.; et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Investig. 2003, 111, 1133–1145. [Google Scholar] [CrossRef]
- Cossu, A.; Biancone, L.; Ascolani, M.; Pallone, F.; Boirivant, M. “In vitro” azathioprine-induced changes in peripheral T cell apoptosis and IFN-γ production associate with drug response in patients with Crohn’s Disease. J. Crohn’s Colitis 2013, 7, 441–450. [Google Scholar] [CrossRef]
- Hildner, K.; Märker-Hermann, E.; Schlaak, J.F.; Becker, C.; Germann, T.; Schmitt, E.; Zum Büschenfelde, K.-H.M.; Neurath, M.F. Azathioprine, Mycophenolate Mofetil, and Methotrexate Specifically Modulate Cytokine Production by T Cells. Ann. N. Y. Acad. Sci. 1998, 859, 204–207. [Google Scholar] [CrossRef]
- Gómez-Martín, D.; Díaz-Zamudio, M.; Vanoye, G.; Crispín, J.C.; Alcocer-Varela, J. Quantitative and functional profiles of CD4+ lymphocyte subsets in systemic lupus erythematosus patients with lymphopenia. Clin. Exp. Immunol. 2011, 164, 17–25. [Google Scholar] [CrossRef]
- Al Rifai, A.; Prasad, N.; Shuttleworth, E.; McBurney, H.; Pushpakom, S.; Robinson, A.; Newman, W.; Campbell, S. Natural history of azathioprine-associated lymphopenia in inflammatory bowel disease patients: A prospective observational study. Eur. J. Gastroenterol. Hepatol. 2011, 23, 153–158. [Google Scholar] [CrossRef]
- Stocco, G.; Martelossi, S.; Barabino, A.; Decorti, G.; Bartoli, F.; Montico, M.; Gotti, A.; Ventura, A. Glutathione-S-transferase genotypes and the adverse effects of azathioprine in young patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 57–64. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Naito, T.; Baba, T.; Takeda, K.; Sasaki, S.; Nakamoto, Y.; Mukaida, N. High-dose cyclophosphamide induces specific tumor immunity with concomitant recruitment of LAMP1/CD107a-expressing CD4-positive T cells into tumor sites. Cancer Lett. 2015, 366, 93–99. [Google Scholar] [CrossRef]
- Madondo, M.T.; Quinn, M.; Plebanski, M. Low dose cyclophosphamide: Mechanisms of T cell modulation. Cancer Treat. Rev. 2016, 42, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Turk, J.L.; Parker, D. Effect of cyclophosphamide on immunological control mechanisms. Immunol. Rev. 1982, 65, 99–113. [Google Scholar] [CrossRef]
- Balow, J.E.; Hurley, D.L.; Fauci, A.S. Cyclophosphamide suppression of established cell-mediated immunity. Quantitative vs. qualitative changes in lymphocyte populations. J. Clin. Investig. 1975, 56, 65–70. [Google Scholar] [CrossRef]
- Motoyoshi, Y.; Kaminoda, K.; Saitoh, O.; Hamasaki, K.; Nakao, K.; Ishii, N.; Nagayama, Y.; Eguchi, K. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol. Rep. 2006, 16, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Hardinger, K.; Magee, C.C. Pharmacology of Cyclosporine and Tacrolimus. Available online: https://www.uptodate.com/contents/pharmacology-of-cyclosporine-and-tacrolimus (accessed on 30 March 2023).
- Schreiber, S.L.; Crabtree, G.R. The mechanism of action of cyclosporin A and FK506. Immunol. Today 1992, 13, 136–142. [Google Scholar] [CrossRef]
- Suzuki, N.; Kaneko, S.; Ichino, M.; Mihara, S.; Wakisaka, S.; Sakane, T. In vivo mechanisms for the inhibition of T lymphocyte activation by long-term therapy with tacrolimus (FK-506): Experience in patients with Behçet’s disease. Arthritis Rheum. 1997, 40, 1157–1167. [Google Scholar] [CrossRef]
- Wiederrecht, G.; Lam, E.; Hung, S.; Martin, M.; Sigal, N. The mechanism of action of FK-506 and cyclosporin A. Ann. N. Y Acad. Sci. 1993, 696, 9–19. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Clipstone, N.A.; Ho, S.N.; Northrop, J.P.; Crabtree, G.R. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature 1996, 383, 837–840. [Google Scholar] [CrossRef]
- Kim, W.U.; Cho, M.L.; Kim, S.I.; Yoo, W.H.; Lee, S.S.; Joo, Y.S.; Min, J.K.; Hong, Y.S.; Lee, S.H.; Park, S.H.; et al. Divergent effect of cyclosporine on Th1/Th2 type cytokines in patients with severe, refractory rheumatoid arthritis. J. Rheumatol. 2000, 27, 324–331. [Google Scholar]
- Rentenaar, R.J.; Heydendael, V.M.R.; Diepen, F.N.J.V.; Rie, M.A.d.; Berge, I.J.M.T. Systemic Treatment with Either Cyclosporin A or Methotrexate Does Not Influence the T Helper 1/T Helper 2 Balance in Psoriatic Patients. J. Clin. Immunol. 2004, 24, 361–369. [Google Scholar] [CrossRef]
- Shin, G.T.; Khanna, A.; Ding, R.; Sharma, V.K.; Lagman, M.; Li, B.; Suthanthiran, M. In vivo expression of transforming growth factor-beta1 in humans: Stimulation by cyclosporine. Transplantation 1998, 65, 313–318. [Google Scholar] [CrossRef]
- Zeevi, A.; Eiras, G.; Bach, F.H.; Fung, J.J.; Todo, S.; Starzl, T.; Duquesnoy, R.J. Functional differentiation of human cytotoxic T lymphocytes in the presence of FK 506 and CyA. Transplant. Proc. 1990, 22, 106–109. [Google Scholar] [PubMed]
- Bunjes, D.; Hardt, C.; Röllinghoff, M.; Wagner, H. Cyclosporin A mediates immunosuppression of primary cytotoxic T cell responses by impairing the release of interleukin 1 and interleukin 2. Eur. J. Immunol. 1981, 11, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Fahrbach, K.; Nordstrom, B.; Ross, S.; Schmid, C.H.; Symmons, D. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: Meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol. Drug Saf. 2011, 20, 119–130. [Google Scholar] [CrossRef]
- Askling, J.; van Vollenhoven, R.F.; Granath, F.; Raaschou, P.; Fored, C.M.; Baecklund, E.; Dackhammar, C.; Feltelius, N.; Cöster, L.; Geborek, P.; et al. Cancer risk in patients with rheumatoid arthritis treated with anti-tumor necrosis factor alpha therapies: Does the risk change with the time since start of treatment? Arthritis Rheum. 2009, 60, 3180–3189. [Google Scholar] [CrossRef]
- Dommasch, E.D.; Abuabara, K.; Shin, D.B.; Nguyen, J.; Troxel, A.B.; Gelfand, J.M. The risk of infection and malignancy with tumor necrosis factor antagonists in adults with psoriatic disease: A systematic review and meta-analysis of randomized controlled trials. J. Am. Acad. Dermatol. 2011, 64, 1035–1050. [Google Scholar] [CrossRef]
- Mariette, X.; Matucci-Cerinic, M.; Pavelka, K.; Taylor, P.; van Vollenhoven, R.; Heatley, R.; Walsh, C.; Lawson, R.; Reynolds, A.; Emery, P. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: A systematic review and meta-analysis. Ann. Rheum. Dis. 2011, 70, 1895–1904. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Askling, J.; Fored, C.M.; Baecklund, E.; Brandt, L.; Backlin, C.; Ekbom, A.; Sundström, C.; Bertilsson, L.; Cöster, L.; Geborek, P.; et al. Haematopoietic malignancies in rheumatoid arthritis: Lymphoma risk and characteristics after exposure to tumour necrosis factor antagonists. Ann. Rheum. Dis. 2005, 64, 1414–1420. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Michaud, K. The effect of methotrexate and anti-tumor necrosis factor therapy on the risk of lymphoma in rheumatoid arthritis in 19,562 patients during 89,710 person-years of observation. Arthritis Rheum. 2007, 56, 1433–1439. [Google Scholar] [CrossRef]
- Thompson, A.E.; Rieder, S.W.; Pope, J.E. Tumor necrosis factor therapy and the risk of serious infection and malignancy in patients with early rheumatoid arthritis: A meta-analysis of randomized controlled trials. Arthritis Rheum. 2011, 63, 1479–1485. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011, 585, 3611–3618. [Google Scholar] [CrossRef]
- Aravena, O.; Pesce, B.; Soto, L.; Orrego, N.; Sabugo, F.; Wurmann, P.; Molina, M.C.; Alfaro, J.; Cuchacovich, M.; Aguillón, J.C.; et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology 2011, 216, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Luan, L.; Han, S.; Wang, H.; Liu, X. Down-regulation of the Th1, Th17, and Th22 pathways due to anti-TNF-α treatment in psoriasis. Int. Immunopharmacol. 2015, 29, 278–284. [Google Scholar] [CrossRef]
- Ross, S.E.; Williams, R.O.; Mason, L.J.; Mauri, C.; Marinova-Mutafchieva, L.; Malfait, A.M.; Maini, R.N.; Feldmann, M. Suppression of TNF-alpha expression, inhibition of Th1 activity, and amelioration of collagen-induced arthritis by rolipram. J. Immunol. 1997, 159, 6253–6259. [Google Scholar] [CrossRef]
- Eriksson, C.; Rantapää-Dahlqvist, S.; Sundqvist, K.G. Changes in chemokines and their receptors in blood during treatment with the TNF inhibitor infliximab in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2013, 42, 260–265. [Google Scholar] [CrossRef]
- Herman, S.; Zurgil, N.; Machlav, S.; Shinberg, A.; Langevitz, P.; Ehrenfeld, M.; Deutsch, M. Distinct effects of anti-tumor necrosis factor combined therapy on TH1/TH2 balance in rheumatoid arthritis patients. Clin. Vaccine Immunol. 2011, 18, 1077–1082. [Google Scholar] [CrossRef]
- McGovern, J.L.; Nguyen, D.X.; Notley, C.A.; Mauri, C.; Isenberg, D.A.; Ehrenstein, M.R. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum. 2012, 64, 3129–3138. [Google Scholar] [CrossRef]
- Bosè, F.; Raeli, L.; Garutti, C.; Frigerio, E.; Cozzi, A.; Crimi, M.; Caprioli, F.; Scavelli, R.; Altomare, G.; Geginat, J.; et al. Dual role of anti-TNF therapy: Enhancement of TCR-mediated T cell activation in peripheral blood and inhibition of inflammation in target tissues. Clin. Immunol. 2011, 139, 164–176. [Google Scholar] [CrossRef]
- Talotta, R.; Berzi, A.; Atzeni, F.; Batticciotto, A.; Clerici, M.; Sarzi-Puttini, P.; Trabattoni, D. Paradoxical Expansion of Th1 and Th17 Lymphocytes in Rheumatoid Arthritis Following Infliximab Treatment: A Possible Explanation for a Lack of Clinical Response. J. Clin. Immunol. 2015, 35, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Szalay, B.; Vásárhelyi, B.; Cseh, A.; Tulassay, T.; Deák, M.; Kovács, L.; Balog, A. The impact of conventional DMARD and biological therapies on CD4+ cell subsets in rheumatoid arthritis: A follow-up study. Clin. Rheumatol. 2014, 33, 175–185. [Google Scholar] [CrossRef]
- Dahlén, R.; Magnusson, M.K.; Bajor, A.; Lasson, A.; Ung, K.A.; Strid, H.; Öhman, L. Global mucosal and serum cytokine profile in patients with ulcerative colitis undergoing anti-TNF therapy. Scand. J. Gastroenterol. 2015, 50, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Campanati, A.; Ganzetti, G.; Giuliodori, K.; Marra, M.; Bonfigli, A.; Testa, R.; Offidani, A. Serum levels of adipocytokines in psoriasis patients receiving tumor necrosis factor-α inhibitors: Results of a retrospective analysis. Int. J. Dermatol. 2015, 54, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M. Molecular and cellular effects of methotrexate. Curr. Opin. Rheumatol. 1999, 11, 226–232. [Google Scholar] [CrossRef]
- Schröder, O.; Stein, J. Low dose methotrexate in inflammatory bowel disease: Current status and future directions. Am. J. Gastroenterol. 2003, 98, 530–537. [Google Scholar] [CrossRef]
- Cipriani, P.; Ruscitti, P.; Carubbi, F.; Liakouli, V.; Giacomelli, R. Methotrexate: An old new drug in autoimmune disease. Expert. Rev. Clin. Immunol. 2014, 10, 1519–1530. [Google Scholar] [CrossRef]
- Yélamos, O.; Puig, L. Systemic methotrexate for the treatment of psoriasis. Expert. Rev. Clin. Immunol. 2015, 11, 553–563. [Google Scholar] [CrossRef]
- Xinqiang, S.; Fei, L.; Nan, L.; Yuan, L.; Fang, Y.; Hong, X.; Lixin, T.; Juan, L.; Xiao, Z.; Yuying, S.; et al. Therapeutic efficacy of experimental rheumatoid arthritis with low-dose methotrexate by increasing partially CD4+ CD25+ Treg cells and inducing Th1 to Th2 shift in both cells and cytokines. Biomed. Pharmacother. 2010, 64, 463–471. [Google Scholar] [CrossRef]
- Yamaki, K.; Uchida, H.; Harada, Y.; Li, X.; Yanagisawa, R.; Takano, H.; Hayashi, H.; Taneda, S.; Mori, Y.; Yoshino, S. Effect of methotrexate on Th1 and Th2 immune responses in mice. J. Pharm. Pharmacol. 2003, 55, 1661–1666. [Google Scholar] [CrossRef]
- Herman, S.; Zurgil, N.; Langevitz, P.; Ehrenfeld, M.; Deutsch, M. Methotrexate selectively modulates TH1/TH2 balance in active rheumatoid arthritis patients. Clin. Exp. Rheumatol. 2008, 26, 317–323. [Google Scholar] [PubMed]
- Ghoreschi, K.; Mrowietz, U.; Röcken, M. A molecule solves psoriasis? Systemic therapies for psoriasis inducing interleukin 4 and Th2 responses. J. Mol. Med. 2003, 81, 471–480. [Google Scholar] [CrossRef]
- Dayan, M.; Segal, R.; Mozes, E. Cytokine manipulation by methotrexate treatment in murine experimental systemic lupus erythematosus. J. Rheumatol. 1997, 24, 1075–1082. [Google Scholar] [PubMed]
- Solomon, D.H.; Kremer, J.M.; Fisher, M.; Curtis, J.R.; Furer, V.; Harrold, L.R.; Hochberg, M.C.; Reed, G.; Tsao, P.; Greenberg, J.D. Comparative cancer risk associated with methotrexate, other non-biologic and biologic disease-modifying anti-rheumatic drugs. Semin. Arthritis Rheum. 2014, 43, 489–497. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | Risk of HNCs | SIR | Related Cytokines |
|---|---|---|---|
| SSc | Oral cavity and pharyngeal | 3.67 | IL-1, 1α, 2, 2R, 4, 6, 8, 10, 17, 18, 23 (↑) IFN-γ (↓) TNF-α (↑) TGF-β (↑) CXCL4 (↑)) CRP, MCP1, MIP-1γ, VCAM-1 (↑) MDC (↓) |
| Oropharyngeal | 9.63 | ||
| Tongue | 25 | ||
| SLE | Oropharyngeal | 1.54–2.02 | IL-2, 2R (↑) IFN-α, γ (↑) TNF-α (↓) TGF-β (↓) |
| HNCs | 2.02–2.16 | ||
| Nasopharyngeal | 2.02–4.18 | ||
| Thyroid | 2.31 | ||
| Laryngeal | 4.19 | ||
| RA | Oropharyngeal | – | IL-15, 20 (↑) TNF-α (↑) GM-CSF (↑) |
| UC | None | – | IL-2, 6, 8 (↑) TNF-α (↑) GRO (CXCL-1) (↑) Eotaxin (↑) |
| CD | Oral | – | Il-5, 6, 7, 8, 13 (↑) IFN-γ CRP (↑) |
| Neck cavity | |||
| Tongue | |||
| Psoriasis | Oral | 0.7–2.1 | IL-6 (↑) TNF-α (↑) CRP, ICAM-1 (↑) |
| Pharyngeal | 1.3–4.1 | ||
| Laryngeal | 1.43–2.9 | ||
| Oropharyngeal | 2.8 | ||
| DM | Nasopharyngeal | – | IL-1, 2R, 6, 15, 17, 18 (↑) IFN-signature (↑) TNF-R1 (↑) TGF- β (↓) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vale, N.; Pereira, M.; Mendes, R.A. Systemic Inflammatory Disorders, Immunosuppressive Treatment and Increase Risk of Head and Neck Cancers—A Narrative Review of Potential Physiopathological and Biological Mechanisms. Cells 2023, 12, 2192. https://doi.org/10.3390/cells12172192
Vale N, Pereira M, Mendes RA. Systemic Inflammatory Disorders, Immunosuppressive Treatment and Increase Risk of Head and Neck Cancers—A Narrative Review of Potential Physiopathological and Biological Mechanisms. Cells. 2023; 12(17):2192. https://doi.org/10.3390/cells12172192
Chicago/Turabian StyleVale, Nuno, Mariana Pereira, and Rui Amaral Mendes. 2023. "Systemic Inflammatory Disorders, Immunosuppressive Treatment and Increase Risk of Head and Neck Cancers—A Narrative Review of Potential Physiopathological and Biological Mechanisms" Cells 12, no. 17: 2192. https://doi.org/10.3390/cells12172192