
Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview
by
 , ,
, ,
Snehal Raut
1,
Aditya Bhalerao
1,
Michael Powers
2,
Minelly Gonzalez
1,
Salvatore Mancuso
1 and
Luca Cucullo
1,* 1
Department of Foundational Medical Studies, Oakland University William Beaumont School of Medicine, Rochester, MI 48309, USA
2
Department of Biological and Biomedical Sciences, Oakland University, Rochester, MI 48309, USA
*
Author to whom correspondence should be addressed.
Cells 2023, 12(16), 2019; https://doi.org/10.3390/cells12162019
Submission received: 17 May 2023
/
Revised: 19 July 2023
/
Accepted: 6 August 2023
/
Published: 8 August 2023
(This article belongs to the Special Issue Therapeutic Mechanism of Nervous System Inflammation)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The brain is a highly dynamic organ that requires a constant energy source to function normally. This energy is mostly supplied by glucose, a simple sugar that serves as the brain’s principal fuel source. Glucose transport across the blood–brain barrier (BBB) is primarily controlled via sodium-independent facilitated glucose transport, such as by glucose transporter 1 (GLUT1) and 3 (GLUT3). However, other glucose transporters, including GLUT4 and the sodium-dependent transporters SGLT1 and SGLT6, have been reported in vitro and in vivo. When the BBB endothelial layer is crossed, neurons and astrocytes can absorb the glucose using their GLUT1 and GLUT3 transporters. Glucose then enters the glycolytic pathway and is metabolized into adenosine triphosphate (ATP), which supplies the energy to support cellular functions. The transport and metabolism of glucose in the brain are impacted by several medical conditions, which can cause neurological and neuropsychiatric symptoms. Alzheimer’s disease (AD), Parkinson’s disease (PD), epilepsy, traumatic brain injury (TBI), schizophrenia, etc., are a few of the most prevalent disorders, characterized by a decline in brain metabolism or hypometabolism early in the course of the disease. Indeed, AD is considered a metabolic disorder related to decreased brain glucose metabolism, involving brain insulin resistance and age-dependent mitochondrial dysfunction. Although the conventional view is that reduced cerebral metabolism is an effect of neuronal loss and consequent brain atrophy, a growing body of evidence points to the opposite, where hypometabolism is prodromal or at least precedes the onset of brain atrophy and the manifestation of clinical symptoms. The underlying processes responsible for these glucose transport and metabolic abnormalities are complicated and remain poorly understood. This review article provides a comprehensive overview of the current understanding of hypometabolism in AD and potential therapeutic targets.
1. Brain Energetics
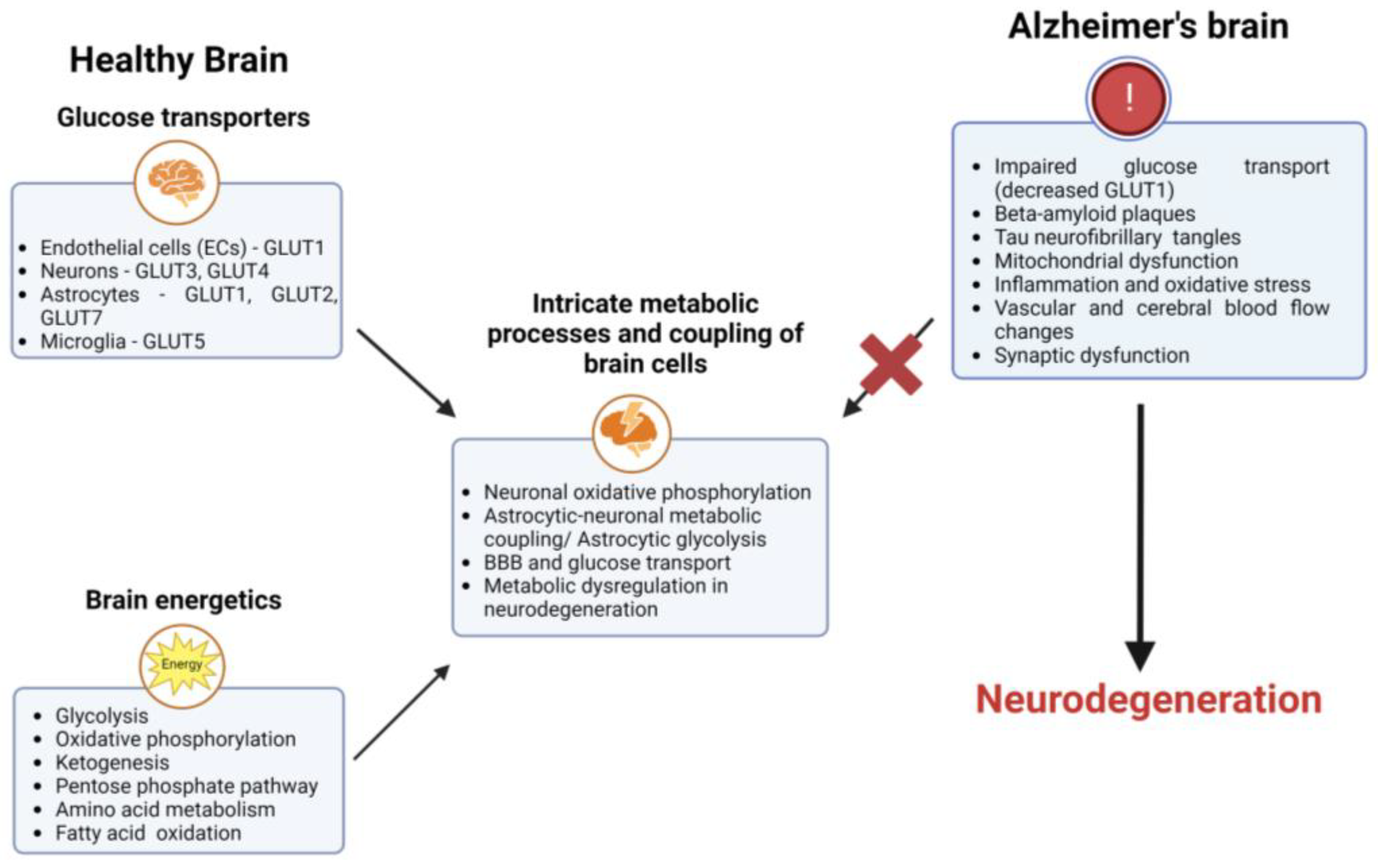
The brain requires a constant supply of energy to carry out essential activities such as neurotransmission, protein synthesis, and maintenance of membrane potentials [1,2]. Most of the energy consumed by the brain is derived from glucose metabolism, which is tightly regulated to ensure adequate energy delivery to the brain [3]. Glucose is transported into the brain across the BBB and taken up by neurons and astrocytes. It is converted into ATP through biochemical reactions involving glycolysis, the tricarboxylic acid cycle (TCA), and oxidative phosphorylation [4], which is used by the brain to support a variety of cellular processes. The brain produces 95% or more of its ATP through glucose metabolism. The neurovascular unit, which includes the brain microvascular endothelial cells, pericytes [5], astrocytes, oligodendrocytes [6], microglia [7], and neurons [8] as the final recipient of glucose absorption, coordinates glucose uptake among a variety of cell types inside the brain [9]. The brain can utilize other energy sources, such as ketones, and lactate, in the absence of glucose [10,11,12].
The energy requirements of active neurons, not the blood glucose level, determine how much glucose is taken up by the brain from the bloodstream. Due to the increased energy requirements for neuronal signaling, neurotransmitter release, ionic balance, and synaptic plasticity [13,14,15,16], glucose is actively “drawn” into the brain [17,18]. Glucose transport is accomplished by the coordinated activation of glucose transporters on the capillary endothelium (GLUT1) [19,20,21,22,23,24,25], astrocytes (GLUT1 [26], GLUT2 [27], and GLUT7 [28]), oligodendrocytes (GLUT1) [29], and neurons (GLUT3 [28,30] and GLUT4) [26,31,32,33,34]. Blood glucose diffuses either directly from capillaries to neurons via the extracellular space, or is mediated by the astrocyte end-feet surrounding the capillary walls [25,35,36,37]. The distribution of energy and metabolic support inside the brain is greatly aided by astrocytes. They have extensive processes that wrap around blood vessels and synapses, allowing them to take up nutrients such as glucose and lactate from the bloodstream and deliver them to neurons.
The astrocyte–neuron lactate shuttle is a metabolic link between astrocytes and neurons. Astrocytes absorb glucose and use it to fuel glycolysis, which produces lactate as a byproduct. Subsequently, monocarboxylate transporter 1 (MCT1) transports lactate to the extracellular environment, where it is taken up by MCT2 and delivered to neurons [38]. Neurons can use lactate as a source of energy once astrocytes have released it and taken it up. This metabolic coupling supports neuronal function and plasticity by providing neurons with an extra energy source.
Unlike astrocytes and oligodendrocytes, microglia do not directly fuel neurons, although the large quantities of lactate generated by active microglia are very likely to be absorbed by nearby neurons [39]. The high energy requirements of activated microglia significantly restrict the energy available to neurons when the brain glucose supply remains limited for an extended time [40,41]. One example is during episodes of hypoglycemia [42], which can occur due to various reasons, including diabetes [43], starvation/fasting [44], critical illness [45], alcohol consumption [46], etc. When blood glucose levels drop significantly the brain’s glucose supply becomes limited, potentially leading to confusion, dizziness, seizures, loss of consciousness, and, in severe cases, brain damage. Therefore, the opportunistic use of alternate fuels derived from sources outside the brain plays a significant role in the brain’s resilience in the face of energetic strain. The primary substitute fuels for glucose are ketones and lactate, which are transported to the brain via monocarboxylate transporters on astrocytes and the capillary endothelium [47,48,49,50,51].
The brain can use ketone bodies for energy when blood levels of ketones rise sufficiently, typically after several days of fasting or after several weeks of adherence to a low-carbohydrate and high-fat (ketogenic) diet [52,53]. Fatty acid oxidation (FAO) can provide an alternative fuel source in the brain when glucose metabolism is impaired or in hypometabolic conditions [54,55]. During FAO, long-chain fatty acids are transported into the mitochondria, undergoing a series of enzymatic reactions. These reactions involve sequential cleavage of the fatty acid chain, producing acetyl-CoA units. Acetyl-CoA is then further metabolized through the TCA cycle [56,57].
2. Hypometabolism in AD
Amyloid and tau are well-established AD risk factors that affect millions of people globally [62,63]. One of the main findings in the brains of AD patients is hypometabolism, a reduction in the brain’s metabolic rate [64,65]. This hypometabolism is seen in specific brain regions, including the temporal and parietal lobes, which are involved in memory and cognition [66,67,68,69,70]. The data from animal models and AD patients are consistent with the findings that brain hypometabolism is a major feature of AD [64,71,72]. In animal models of AD, there is a widespread decrease in the cerebral metabolic rate of glucose (CMRglu) in the hippocampus, cortex, and other brain regions [73,74,75]. This decline in glucose metabolism occurs progressively, worsening over time. Decrease in CMRglu can lead to neuronal death and cognitive decline [76,77]. While the exact reason for this hypometabolism is not well understood, it may be related to the accumulation of amyloid plaques and neurofibrillary tangles [78]. Clinical fluorodeoxyglucose (FDG) positron emission tomography (PET) studies have repeatedly demonstrated impaired glucose uptake in individuals with mild cognitive impairment (MCI), early AD, and genetic risk/family history of AD [79,80,81,82]. In AD, studies have shown that reductions in brain glucose metabolism occur early in the disease process before significant memory impairment is observed [78,83,84,85]. Additionally, neurodegeneration and loss of neurons may contribute to the decline in glucose uptake and utilization found in AD [86,87].
The exact relationship between glucose metabolism and AD is not yet fully understood. One theory is that the reduction in glucose metabolism may lead to decreased brain energy production, affecting nerve cells’ normal functioning and contributing to brain damage [88]. There is evidence that changes in insulin signaling may play a role in reducing glucose metabolism in AD [89,90]. However, research shows that glucose uptake in the brain is largely independent of insulin and that only a small percentage of glucose uptake in the brain is insulin-dependent [91,92,93], as glucose transport into the brain is primarily mediated by glucose transporters (GLUTs), which are insulin-independent [94]. While insulin can regulate glucose metabolism and uptake in other tissues in the body, it appears to have a limited role in the brain [95,96]. Research suggests that insulin may inhibit glucose uptake in the brain under certain conditions. For example, in individuals with type 2 diabetes, where insulin resistance is present, glucose uptake in the brain is not significantly impaired [97,98]. However, the reduction in glucose metabolism in certain brain regions in AD suggests that it may play a role in the disease’s development; thus, targeting glucose metabolism may provide therapeutic benefits in AD [99].
3. Glucose Transporters in AD
Glucose transporters play a critical role in regulating glucose uptake into the brain, and their expression levels have been investigated in AD. Here are several key findings related to glucose transporters expression in AD:
- GLUT4 is typically found in muscle and adipose tissue; however, recent studies have suggested that it may be expressed in the brain as well [108,109,110]. One study found that in AD patients GLUT4 expression was increased in the hippocampus, suggesting that it may be upregulated in response to the impaired glucose metabolism in this region [111,112,113].
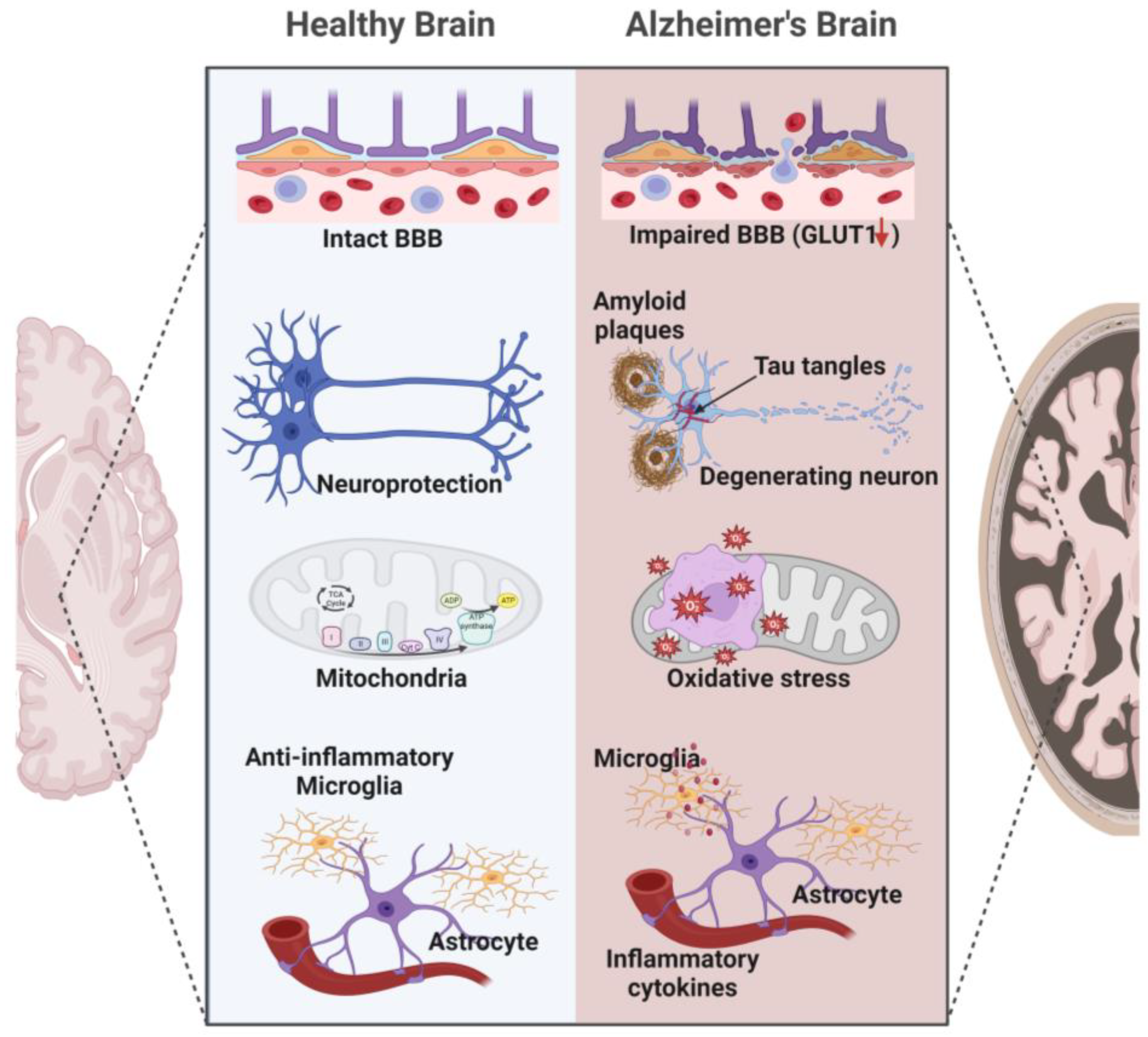
These studies suggest that alterations in these glucose transporters’ expression may impair glucose metabolism in AD. However, this reduced expression/activity of glucose transporters in AD is likely multifactorial, resulting from a complex interplay of beta-amyloid toxicity, inflammation, and oxidative stress in the brain. Indeed, one possible explanation is that beta-amyloid plaques can directly impair the function of glucose transporters. Studies have shown that beta-amyloid can bind to these transporters, promoting their internalization and reducing glucose transport activity [101,114,115,116], thereby leading to energy deficits that contribute to the cognitive decline in AD. Another possible explanation is that inflammation, a common AD feature, can reduce expression of glucose transporters.
Inflammatory cytokines such as interleukin-1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-α) have been shown to decrease glucose transporter expression in brain cells [117,118]. Finally, oxidative stress, which is increased in AD, can severely impact the membranes of brain cells, and promote irreversible covalent modifications that destabilize and inactivate the embedded proteins, including glucose transporters [119,120,121] (Figure 2).
4. The Potential Therapeutic Targets for Hypometabolism in AD
Studies on AD have revealed that changes in brain glucose metabolism early in the disease process lead to the loss of synapses and neuronal death. Insufficient neuronal glucose and mitochondrial energy production compromise the clearance of amyloid and tau proteins from the brain [122]. On the other hand, the buildup of amyloid plaques and tau tangles exacerbates brain glucose hypometabolism and mitochondrial impairment, reduces energy output, and elevates oxidative stress [123,124]. Hypometabolism then causes cellular injury and neuroinflammation. This destructive cycle caused by energy failure in AD is comparable to the brain circuit disruption found in other neurodegenerative illnesses; it worsens memory and cognition and leads to unusual behavior in those affected.
While beta-amyloid is undoubtedly a major factor in the progression of AD, it is becoming increasingly obvious that a variety of other factors, such as inflammation, oxidative stress, dysfunction of the innate immune system in the brain, hereditary factors, and lifestyle choices all play a role [122,123,124]. Studies have shown that AD is associated with decreased glucose uptake and utilization in the brain, which is thought to contribute to the decline in brain function observed in this condition [125,126,127,128].
As mentioned, the conventional thinking that reduced brain glucose metabolism in AD is just a byproduct of neuronal dysfunction is now being challenged; particularly in AD, a continuous energy gap in the brain results from the gradual loss of brain glucose uptake and metabolism, resulting in brain cell malfunction and the buildup of neurotoxic proteins long before clinical symptoms become apparent. The brain’s energy demands cannot be resolved only by raising blood glucose concentration if glycolysis is compromised and neuronal function deteriorates. Furthermore, neuronal activities, not blood glucose levels, determine brain glucose absorption. Conversely, the availability of ketones and lactate in the bloodstream influences their absorption by the brain, making them an alternate source of brain energy. Brain energy rescue strategies may need to target several metabolic pathways and activities depending on the condition, because AD does not have a single common mechanism that results in hypometabolism.
Rescue strategies could focus on a single enzyme, transporter, or metabolite. One approach involves targeting the mitochondrial dysfunction associated with hypometabolism in AD. Several potential drugs that target mitochondrial function, such as coenzyme Q10, have been investigated as potential treatments for AD [129,130]. Mixed results have been found in research examining the possible therapeutic benefits of coenzyme Q10 in AD. Studies have found that CoQ10 supplementation can improve cognitive function, slow down the progression of AD, and reduce oxidative stress in individuals with the disease. Additionally, research has shown that CoQ10 can enhance mitochondrial function [130], which plays a crucial role in energy production and neuronal health. While several studies have reported positive results, others have not seen significant changes [131,132]. These studies have failed to demonstrate improvements in cognitive function or disease progression compared to a placebo or standard treatment. For instance, a large randomized controlled trial called the IDEAL Study did not find any significant differences in cognitive decline between the CoQ10-treated group and the placebo group over a two-year period [130,133]. Another approach involves targeting oxidative stress, which is believed to contribute to hypometabolism in AD [134]. Antioxidant therapies such as vitamin E and curcumin have been investigated in clinical trials as a potential treatment to reduce oxidative stress and improve cognitive function in patients with AD [135,136,137]. Vitamin E is a fat-soluble antioxidant shown to have neuroprotective effects in animal models of AD. Clinical trials have suggested that vitamin E may be beneficial in slowing cognitive decline in patients with mild to moderate AD [138,139,140]. In addition, vitamin E is a widely available and inexpensive supplement that has relatively few side effects. However, high doses of vitamin E have been associated with an increased risk of mortality and bleeding, and may have negative interactions with other medications [139]. In addition, clinical trials of vitamin E have failed to show significant cognitive benefits in patients with AD [139]. Curcumin is a naturally occurring turmeric compound with antioxidant, anti-inflammatory, and neuroprotective properties. Studies have suggested that curcumin may improve cognitive function and reduce amyloid beta and tau pathology in animal models of AD [141,142,143,144]. In addition, curcumin is a relatively safe supplement that has few side effects and low bioavailability, meaning that it is poorly absorbed by the body and quickly metabolized [145,146]. This has limited its effectiveness in clinical trials, as it is difficult to achieve therapeutic levels of curcumin in the brain. Curcumin may interact with certain medications and may negatively affect the liver in high doses [147,148,149].
In addition, growing evidence suggests that targeting the BBB can improve glucose transport and metabolism in the brain [150,151]. Disruption of the BBB is a common feature of AD, and targeting the BBB to improve glucose delivery to the brain could have therapeutic implications for AD [20,152,153]. Studies have shown a reduction of glucose transporter expression at the BBB endothelium [102,154,155]. In AD, the reduction in glucose transporter expression and distribution primarily affects the GLUT1 and GLUT3 transporters. These transporters facilitate the movement of glucose from the bloodstream into brain cells. They are integral membrane proteins that undergo conformational changes to transport glucose in a concentration-dependent manner. The expression and activity of these transporters are tightly regulated to maintain glucose homeostasis in the brain. Disruptions in the expression or function of GLUT1 and GLUT3 can lead to impaired glucose transport across the BBB, resulting in reduced glucose availability and compromised energy metabolism in the brain.
As discussed previously, a reduction in expression may result from factors such as beta-amyloid toxicity, inflammation, and oxidative stress. Targeting the disrupted BBB can help to normalize the expression and function of glucose transporters, improving the transport of glucose across the barrier and into the brain, thereby clearing amyloid-beta, modulating inflammatory responses, and preserving the integrity of the neurovascular unit [20,154,156]. Several of the cited studies discuss strategies to restore BBB integrity, such as by modulating neuroinflammation, enhancing pericyte function, and promoting tight junction integrity. Additionally, altered distribution or localization of glucose transporters within the cell membrane may contribute to impaired glucose uptake in AD. In AD, GLUT1 expression is reduced in regions such as the hippocampus and frontal cortex [28,100,157,158]. This reduction in expression may impair glucose uptake from the bloodstream into the brain. In addition, studies have reported reduced expression of GLUT3 in the hippocampus and parietal cortex. Reduced expression of GLUT3 can lead to decreased glucose uptake in neurons and contribute to energy deficits in the affected brain regions. However, GLUT1 has remained largely unexplored as a possible therapeutic target to alleviate cerebrovascular dysfunction and subsequent AD-associated neurodegeneration. Upregulating the expression of GLUTs at the BBB level (maintaining the apical versus basolateral distribution ratio) or brain cells can increase glucose flux into the brain [159]. This can be achieved through drugs or gene therapy [160].
In terms of drug-based approaches, several drugs have been investigated for their potential to upregulate GLUT expression and enhance glucose uptake in the brain. These drugs typically act through various mechanisms to increase the expression, trafficking, or activity of glucose transporters. Examples include:
- Insulin sensitizer drugs: insulin resistance has been associated with reduced GLUT expression and impaired glucose uptake in the brain. Insulin sensitizer drugs such as thiazolidinediones (TZDs) or peroxisome proliferator-activated receptor gamma (PPARγ) agonists have been shown to enhance GLUT expression and glucose transport in the brain [161].
- AMP-activated protein kinase (AMPK) activators: AMPK is an enzyme in cellular energy regulation. Activating AMPK can increase GLUT expression and glucose uptake. Compounds that activate AMPK, such as metformin, have been investigated for their potential to enhance brain glucose metabolism and improve cognitive function [134].
Gene therapy approaches are another option; these involve introducing specific genes into brain cells to enhance GLUT expression [167,168]. This can be achieved through viral vectors or other delivery systems. The introduced genes can encode specific GLUT isoforms or other factors that regulate GLUT expression. Gene therapy strategies can be targeted to brain endothelial cells at the BBB or directly to brain cells [169]. However, gene therapy for brain disorders, including AD, currently remains experimental and requires further research and development [170].
5. The Current Stage of AD Treatments: Targeting the Usual Culprits
The neuropathological characteristics of AD include widespread deposition of Aβ plaques in the neocortex and a hierarchically ordered network of neurofibrillary tangles primarily made up of tau aggregates in the limbic and cortical association regions. According to the amyloid hypothesis, the buildup of beta-amyloid protein in the brain is a major contributor to the onset of AD [62,171]. Amyloid-beta precursor protein (APP) is a protein normally found in the membranes of neurons. Nonamyloidogenic processing of APP involves α-secretase followed by γ-secretase. In AD, APP is cleaved by two enzymes, beta-secretase (BACE1) and gamma-secretase, to form amyloid beta peptides. These peptides have the potential to accumulate into plaques in the spaces between neurons, which are observed in the brains of AD patients [172,173]. These plaques are thought to disrupt communication between neurons and contribute to the death of brain cells [174,175]. This process is thought to involve the activation of the immune system along with inflammation and oxidative stress [122,176,177].
On the other hand, the tau hypothesis focuses on the function of the tau protein in AD. Under normal conditions, this protein aids in preserving the organization of the brain’s nerve cells. Tau helps to stabilize the structure of neurons by supporting the microtubules that form the neuron’s internal transport system. Neurofibrillary tangles are aberrant tau protein clusters present in AD [123]. These tangles and amyloid plaques prevent nerve cells from operating normally, which causes brain damage and cognitive deterioration. This is fueling a growing interest in developing targeted therapies [124,125,128]. Although tangles are more closely related to neuronal loss and clinical symptoms, genetic studies implicate Aβ as a critical disease initiator for autosomal-dominantly inherited AD. These studies show that mutations in APP or enzymes that produce Aβ cause AD [63,126,127,178].
The amyloid beta peptides can bind to various receptors in the brain, including N-methyl-D-aspartate (NMDA), which plays a key role in learning and memory, Alpha7 nicotinic acetylcholine receptor (α7nAChR), which is involved in the regulation of neurotransmitter release, and receptor for advanced glycation end products (RAGE), which is involved in the immune response [179,180,181]. Tau proteins have been shown to bind to several receptors, including the low-density lipoprotein receptor-related protein 1 (LRP1) [182,183] and the sortilin-related receptor (SORL1) [184,185,186]. These receptors are involved in the transport of proteins within cells, and are thought to play a role in the clearance of tau from the brain [187]. The enzymes and genes involved in the production and clearance of amyloid beta and tau and their respective receptors have been extensively investigated as potential therapeutic targets for AD [188]. There have been several medications developed for Alzheimer’s disease that have passed through clinical trials; however, none have demonstrated significant clinical benefits. Bapineuzumab is a monoclonal antibody that targets amyloid beta plaques [189,190]. Developed by Pfizer and Johnson & Johnson, it went through several clinical trials, ultimately failing to show significant cognitive benefits in patients with Alzheimer’s disease. Solanezumab is another monoclonal antibody that targets amyloid beta plaques. Developed by Eli Lilly, it underwent several clinical trials without demonstrating significant cognitive benefits in patients with mild to moderate Alzheimer’s disease [191,192]. Crenezumab is a monoclonal antibody that targets amyloid beta plaques. It was developed by Genentech and went through several clinical trials, demonstrating no significant cognitive benefits in patients with mild to moderate Alzheimer’s disease [193,194,195]. Semagacestat is a gamma-secretase inhibitor developed by Eli Lilly. Designed to block the production of amyloid beta peptides, it ultimately failed in clinical trials due to side effects and lack of clinical benefit [196,197,198].
A few monoclonal antibodies targeting beta-amyloid or drugs aimed at blocking gamma-secretase (involved in beta-amyloid synthesis) have managed to advance through phase I and II clinical trials; however, none of these have been successful in phase III [199,200]. Furthermore, several tau protein-targeting medications, despite being expected to enhance cognitive or functional outcomes, have been unsuccessful:
- LMTX (LMTM) is a small molecule that targets tau protein aggregation. It was evaluated in clinical trials for both AD and frontotemporal dementia (FTD). However, the Phase III trials of LMTX in AD did not significantly improve cognitive or functional outcomes compared to the placebo [201]. The reasons for this lack of efficacy are currently being investigated.
- Tideglusib is a glycogen synthase kinase-3 beta (GSK-3β) inhibitor that can indirectly affect tau phosphorylation. GSK-3β is an enzyme involved in the abnormal phosphorylation of tau protein. Clinical trials of tideglusib in AD showed mixed results. In a Phase II trial it did not significantly affect cognitive decline [202], and a Phase III trial was discontinued due to lack of efficacy (NCT02579252).
Carriers of Aβ-enhancing genetic forms of AD (i.e., mutant APP and PS1/2, E4 variant of apolipoprotein E (APOE4) and Down’s syndrome), in which Aβ accumulates earlier in the disease, are associated with a dramatic hastening in the age of onset, with a comparable rate of progression of clinical symptoms relative to sporadic illness. This indicates that there are “Aβ-dependent” and “Aβ-independent” illness stages that respectively affect the age of initiation and the rate of progression. If this is the case, anti-Aβ treatments may not impact the outcome measure used in current AD clinical studies, which is the rate of progression after symptoms appear. The difficulties in creating effective AD treatments outline the complexity and heterogeneity of AD and the need for more focused research in order to better comprehend the disease’s pathophysiology [203,204,205].
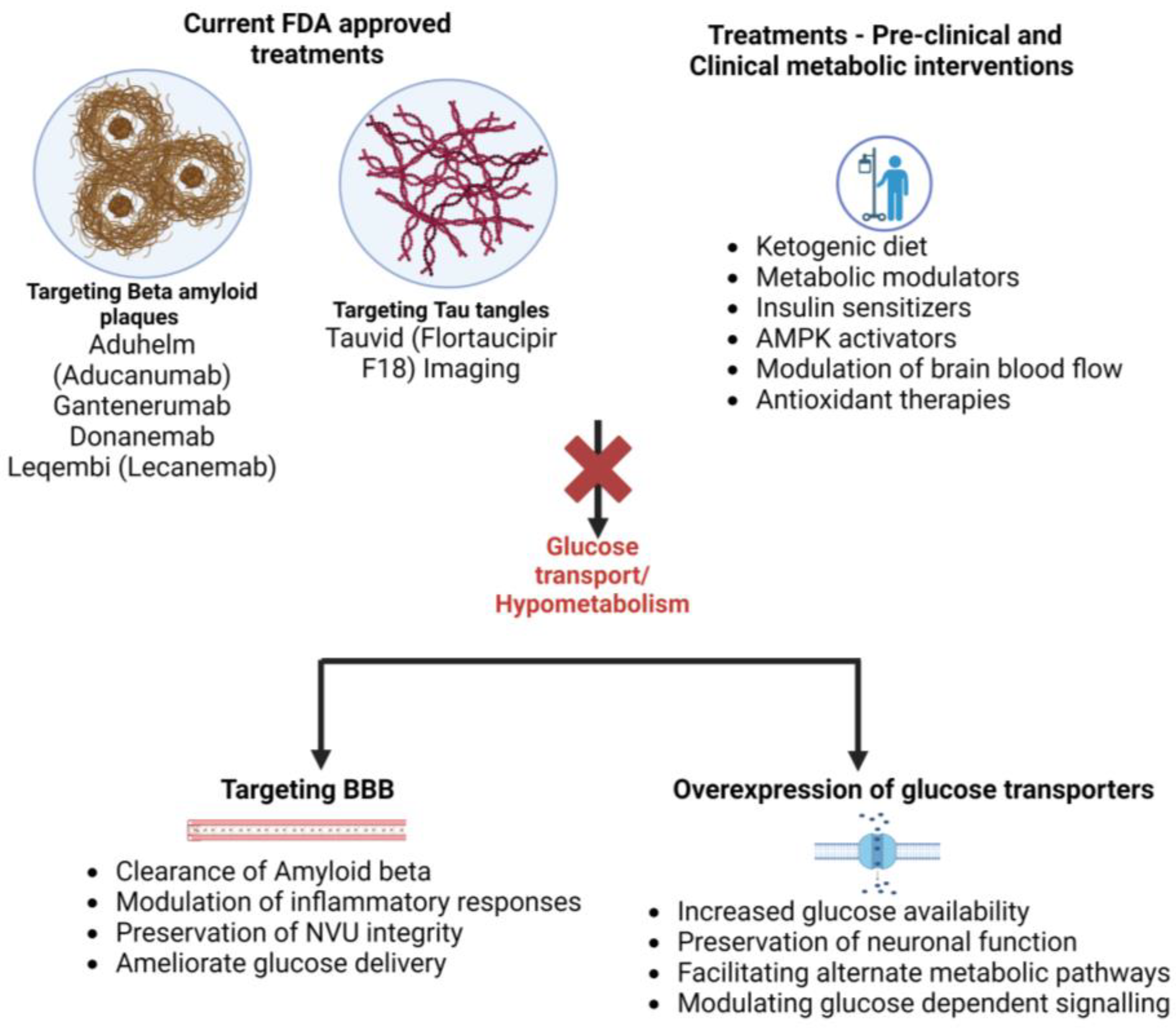
In the past few years the FDA has approved several drugs and treatments for Alzheimer’s disease. A number of the most recent are listed below.
| Drug/Treatment | MOA | Status | Conditions |
| Aduhelm (aducanumab) | A monoclonal antibody that targets beta-amyloid plaques in the brain | Approved in June 2021 | Mild cognitive impairment due to AD or mild AD |
| Gantenerumab | A monoclonal antibody that targets beta-amyloid plaques in the brain. | Phase III clinical trials | Mild AD |
| Donanemab | A monoclonal antibody that targets a modified form of beta-amyloid called N3pG | Phase III clinical trials | Early AD |
| Tauvid Imaging (flortaucipir F18) | A diagnostic imaging agent that targets tau protein | Approved on May 2020 | Detection of tau pathology in the brain |
| Leqembi (Lecanemab) | Remove sticky clumps of the toxic protein amyloid-β from the brain. | Approved on 6 January 2023 | Alzheimer’s disease |
It is important to note that while these drugs provide encouraging prospects for treating AD, much research must be undertaken in order to fully understand their effectiveness and potential risks. Additionally, many experts agree that a multifaceted approach that includes lifestyle modifications, social engagement, and cognitive stimulation is the most effective way to manage the symptoms of AD (Figure 3).
6. Discussion and Conclusions
While the exact cause of AD is unknown, researchers are actively exploring various avenues for treatment and prevention. These include developing new drugs to target specific pathways in the brain, using early diagnosis to start treatment earlier, and lifestyle changes such as regular exercise, a healthy diet, and mental stimulation to delay the onset of AD or slow its progression [206,207,208,209]. The amyloid and tau hypotheses provide a foundation for our understanding of the underlying causes of AD. It is a complex disorder that results from a combination of genetic, lifestyle, and medical factors. Further research is needed to develop and optimize strategies targeting glucose transport and uptake through the BBB. Nonetheless, the current evidence suggests that improving glucose delivery to the brain could provide a useful therapeutic approach to reducing the burden of AD and other neurological disorders [210]. Potential therapy approaches might try to restore the BBB’s functioning and integrity by focusing on BBB disturbance, which may indirectly affect AD glucose transport. It might be possible to improve energy metabolism and maintain neuronal function by restoring the selective permeability of the BBB, which would assist in controlling the passage of glucose and other nutrients into the brain.
Additionally, reducing the accumulation of toxic molecules through BBB repair could alleviate their negative effects on glucose transporters. It is important to note that while drug-based approaches or gene therapy strategies hold promise, several challenges are associated with upregulating GLUT expression for therapeutic purposes. The key considerations for developing such approaches include ensuring specific and targeted delivery to the brain, maintaining balanced glucose homeostasis, and avoiding potential side or off-target effects.
Hypometabolism is a decrease in the metabolic activity of brain cells and has been found to occur earlier in AD than other clinical symptoms, such as memory loss. However, hypometabolism is typically detected using advanced imaging techniques such as positron emission tomography (PET) scans, which are not routinely used in clinical practice for AD diagnosis [211,212]. Furthermore, hypometabolism alone is not specific to Alzheimer’s, and can be seen in other neurological conditions [213]. Therefore, a diagnosis of AD is typically based on a combination of clinical symptoms, imaging studies, and other tests such as neuropsychological evaluations [214].
In summary, although hypometabolism may be seen earlier in AD than other clinical symptoms, it is not considered a primary diagnostic criterion because it is not specific to this condition and requires advanced imaging techniques that are not routinely used in clinical practice. Overall, this review article highlights the growing interest in hypometabolism as a potential therapeutic target for AD and provides a comprehensive overview of the current knowledge regarding the underlying mechanisms and potential treatments. While much work remains to be done, the findings discussed in this review suggest that there are possibilities for developing new treatments for AD that target brain metabolism and related pathways.
Author Contributions
S.R. conceived the study and prepared the drafting of the manuscript. A.B., M.P., M.G. and S.M. edited and revised the manuscript. L.C. assisted with the drafting and editing of the manuscript, oversaw the entire project, and provided funding support. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health/National Institute on Drug Abuse 2R01DA029121-01A1, 1R01DA049737-01, and the National Institute of Neurological Disorders and Stroke 1R01NS117906-01 to Luca Cucullo.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data Availability Statements are available in the manuscript.
Acknowledgments
Figures were created with BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Oyarzabal, A.; Marin-Valencia, I. Synaptic energy metabolism and neuronal excitability, in sickness and health. J. Inherit. Metab. Dis. 2019, 42, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Engl, E.; Attwell, D. Non-signalling energy use in the brain. J. Physiol. 2015, 593, 3417–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain glucose metabolism: Integration of energetics with function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef]
- Cheng, J.; Korte, N.; Nortley, R.; Sethi, H.; Tang, Y.; Attwell, D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018, 136, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Saab, A.S.; Tzvetavona, I.D.; Trevisiol, A.; Baltan, S.; Dibaj, P.; Kusch, K.; Möbius, W.; Goetze, B.; Jahn, H.M.; Huang, W. Oligodendroglial NMDA receptors regulate glucose import and axonal energy metabolism. Neuron 2016, 91, 119–132. [Google Scholar] [CrossRef]
- Barros, L.F.; Brown, A.; Swanson, R.A. G lia in brain energy metabolism: A perspective. Glia 2018, 66, 1134–1137. [Google Scholar] [CrossRef]
- Lecrux, C.; Bourourou, M.; Hamel, E. How reliable is cerebral blood flow to map changes in neuronal activity? Auton. Neurosci. 2019, 217, 71–79. [Google Scholar] [CrossRef]
- Shah, K.; DeSilva, S.; Abbruscato, T. The role of glucose transporters in brain disease: Diabetes and Alzheimer’s disease. Int. J. Mol. Sci. 2012, 13, 12629–12655. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Cunnane, S.C.; Courchesne-Loyer, A.; Vandenberghe, C.; St-Pierre, V.; Fortier, M.; Hennebelle, M.; Croteau, E.; Bocti, C.; Fulop, T.; Castellano, C.-A. Can ketones help rescue brain fuel supply in later life? Implications for cognitive health during aging and the treatment of Alzheimer’s disease. Front. Mol. Neurosci. 2016, 53. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef]
- Gruetter, R.; Novotny, E.J.; Boulware, S.D.; Rothman, D.L.; Mason, G.F.; Shulman, G.I.; Shulman, R.G.; Tamborlane, W.V. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc. Natl. Acad. Sci. USA 1992, 89, 1109–1112. [Google Scholar] [CrossRef]
- Riedl, V.; Utz, L.; Castrillón, G.; Grimmer, T.; Rauschecker, J.P.; Ploner, M.; Friston, K.J.; Drzezga, A.; Sorg, C. Metabolic connectivity mapping reveals effective connectivity in the resting human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 428–433. [Google Scholar] [CrossRef]
- Shulman, R.G.; Hyder, F.; Rothman, D.L. Cerebral energetics and the glycogen shunt: Neurochemical basis of functional imaging. Proc. Natl. Acad. Sci. USA 2001, 98, 6417–6422. [Google Scholar] [CrossRef]
- Hyder, F.; Rothman, D.L.; Shulman, R.G. Total neuroenergetics support localized brain activity: Implications for the interpretation of fMRI. Proc. Natl. Acad. Sci. USA 2002, 99, 10771–10776. [Google Scholar] [CrossRef]
- Bentsen, M.A.; Mirzadeh, Z.; Schwartz, M.W. Revisiting how the brain senses glucose—And why. Cell Metab. 2019, 29, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzier-Sore, A.-K.; Merle, M.; Magistretti, P.J.; Pellerin, L. Feeding active neurons:(re) emergence of a nursing role for astrocytes. J. Physiol. 2002, 96, 273–282. [Google Scholar] [CrossRef]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Dick, A.P.; Harik, S.I. Distribution of the glucose transporter in the mammalian brain. J. Neurochem. 1986, 46, 1406–1411. [Google Scholar] [CrossRef]
- Vannucci, S.J. Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J. Neurochem. 1994, 62, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Boado, R.J.; Farrell, C.R. Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J. Biol. Chem. 1990, 265, 18035–18040. [Google Scholar] [CrossRef] [PubMed]
- Maher, F.; Vannucci, S.J.; Simpson, I.A. Glucose transporter proteins in brain. FASEB J. 1994, 8, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Morgello, S.; Uson, R.R.; Schwartz, E.J.; Haber, R.S. The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia 1995, 14, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Arluison, M.; Quignon, M.; Nguyen, P.; Thorens, B.; Leloup, C.; Penicaud, L. Distribution and anatomical localization of the glucose transporter 2 (GLUT2) in the adult rat brain—An immunohistochemical study. J. Chem. Neuroanat. 2004, 28, 117–136. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Maher, F.; Simpson, I.A. Glucose transporter proteins in brain: Delivery of glucose to neurons and glia. Glia 1997, 21, 2–21. [Google Scholar] [CrossRef]
- Yu, S.; Ding, W.-G. The 45 kDa form of glucose transporter 1 (GLUT1) is localized in oligodendrocyte and astrocyte but not in microglia in the rat brain. Brain Res. 1998, 797, 65–72. [Google Scholar] [CrossRef]
- Maher, F.; Davies-Hill, T.M.; Simpson, I.A. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem. J. 1996, 315, 827–831. [Google Scholar] [CrossRef]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch.-Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Tups, A.; Benzler, J.; Sergi, D.; Ladyman, S.R.; Williams, L.M. Central regulation of glucose homeostasis. Compr. Physiol. 2011, 7, 741–764. [Google Scholar]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 mobilization supports energetic demands of active synapses. Neuron 2017, 93, 606–615. e603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, I.A.; Carruthers, A.; Vannucci, S.J. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2007, 27, 1766–1791. [Google Scholar] [CrossRef]
- Kacem, K.; Lacombe, P.; Seylaz, J.; Bonvento, G. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: A confocal microscopy study. Glia 1998, 23, 1–10. [Google Scholar] [CrossRef]
- Chowdhury, E.A.; Noorani, B.; Alqahtani, F.; Bhalerao, A.; Raut, S.; Sivandzade, F.; Cucullo, L. Understanding the brain uptake and permeability of small molecules through the BBB: A technical overview. J. Cereb. Blood Flow. Metab. 2021, 41, 1797–1820. [Google Scholar] [CrossRef]
- Gandhi, G.K.; Cruz, N.F.; Ball, K.K.; Dienel, G.A. Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J. Neurochem. 2009, 111, 522–536. [Google Scholar] [CrossRef] [Green Version]
- Stobart, J.L.; Anderson, C.M. Multifunctional role of astrocytes as gatekeepers of neuronal energy supply. Front. Cell. Neurosci. 2013, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Castillo, E.; Frias, E.S.; Swanson, R.A. Bioenergetic regulation of microglia. Glia 2018, 66, 1200–1212. [Google Scholar] [CrossRef]
- Deczkowska, A.; Keren-Shaul, H.; Weiner, A.; Colonna, M.; Schwartz, M.; Amit, I. Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 2018, 173, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cryer, P.E. Hypoglycemia: Pathophysiology, Diagnosis, and Treatment; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Association, A.D. Standards of medical care in diabetes—2015 abridged for primary care providers. Clin. Diabetes Publ. Am. Diabetes Assoc. 2015, 33, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manninen, A.H. Metabolic effects of the very-low-carbohydrate diets: Misunderstood “villains” of human metabolism. J. Int. Soc. Sports Nutr. 2004, 1, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marik, P.E.; Bellomo, R. Stress hyperglycemia: An essential survival response! Crit. Care 2013, 17, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arky, R.A.; Veverbrants, E.; Abramson, E.A. Irreversible hypoglycemia: A complication of alcohol and insulin. JAMA 1968, 206, 575–578. [Google Scholar] [CrossRef]
- Takahashi, S. Lactate and ketone bodies act as energy substrates as well as signal molecules in the brain. Psychol. Pathophysiol. Outcomes Eat. 2021, 6, 21. [Google Scholar]
- Sokoloff, L. Metabolism of ketone bodies by the brain. Annu. Rev. Med. 1973, 24, 271–280. [Google Scholar] [CrossRef]
- Lin, A.-L.; Zhang, W.; Gao, X.; Watts, L. Caloric restriction increases ketone bodies metabolism and preserves blood flow in aging brain. Neurobiol. Aging 2015, 36, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Bergersen, L.H. Lactate transport and signaling in the brain: Potential therapeutic targets and roles in body—Brain interaction. J. Cereb. Blood Flow Metab. 2015, 35, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Blood-brain barrier transport of glucose, free fatty acids, and ketone bodies. Adv. Exp. Med. Biol. 1991, 291, 43–53. [Google Scholar]
- Henderson, S.T. High carbohydrate diets and Alzheimer’s disease. Med. Hypotheses 2004, 62, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr Metab. 2005, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Le Foll, C.; Levin, B.E. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2016, 310, R1186–R1192. [Google Scholar] [CrossRef] [PubMed]
- Scarfò, G.; Piccarducci, R.; Daniele, S.; Franzoni, F.; Martini, C. Exploring the Role of Lipid-Binding Proteins and Oxidative Stress in Neurodegenerative Disorders: A Focus on the Neuroprotective Effects of Nutraceutical Supplementation and Physical Exercise. Antioxidants 2022, 11, 2116. [Google Scholar] [CrossRef]
- Panov, A.V.; Gutekunst, C.-A.; Leavitt, B.R.; Hayden, M.R.; Burke, J.R.; Strittmatter, W.J.; Greenamyre, J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002, 5, 731–736. [Google Scholar] [CrossRef]
- Frere, S.; Slutsky, I. Alzheimer’s disease: From firing instability to homeostasis network collapse. Neuron 2018, 97, 32–58. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Parihar, M.; Hemnani, T. Alzheimer’s disease pathogenesis and therapeutic interventions. J. Clin. Neurosci. 2004, 11, 456–467. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mattson, M.P. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011, 10, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef] [Green Version]
- Adlimoghaddam, A.; Snow, W.M.; Stortz, G.; Perez, C.; Djordjevic, J.; Goertzen, A.L.; Ko, J.H.; Albensi, B.C. Regional hypometabolism in the 3xTg mouse model of Alzheimer’s disease. Neurobiol. Dis. 2019, 127, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Hirono, N.; Mori, E.; Ishii, K.; Ikejiri, Y.; Imamura, T.; Shimomura, T.; Hashimoto, M.; Yamashita, H.; Sasaki, M. Frontal lobe hypometabolism and depression in Alzheimer’s disease. Neurology 1998, 50, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J. Neurosci. Res. 2017, 95, 943–972. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Grimmer, T.; Miederer, I.; Henriksen, G.; Yousefi, B.H.; Graner, P.; Wester, H.-J.; Förstl, H.; Kurz, A.; Dickerson, B.C. Regional expansion of hypometabolism in Alzheimer’s disease follows amyloid deposition with temporal delay. Biol. Psychiatry 2012, 71, 792–797. [Google Scholar] [CrossRef]
- Drzezga, A.; Lautenschlager, N.; Siebner, H.; Riemenschneider, M.; Willoch, F.; Minoshima, S.; Schwaiger, M.; Kurz, A. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: A PET follow-up study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1104–1113. [Google Scholar]
- Small, G.W.; Ercoli, L.M.; Silverman, D.H.; Huang, S.-C.; Komo, S.; Bookheimer, S.Y.; Lavretsky, H.; Miller, K.; Siddarth, P.; Rasgon, N.L. Cerebral metabolic and cognitive decline in persons at genetic risk for Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 6037–6042. [Google Scholar] [CrossRef]
- Hampel, H.; Hu, Y.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M. The amyloid-β pathway in Alzheimer’s disease: A plain language summary. Neurodegener. Dis. Manag. 2023. [Google Scholar] [CrossRef]
- Joo, I.L.; Lam, W.W.; Oakden, W.; Hill, M.E.; Koletar, M.M.; Morrone, C.D.; Stanisz, G.J.; McLaurin, J.; Stefanovic, B. Early alterations in brain glucose metabolism and vascular function in a transgenic rat model of Alzheimer’s disease. Prog. Neurobiol. 2022, 217, 102327. [Google Scholar] [CrossRef]
- Colavitta, M.; Grasso, L.; Barrantes, F.J. Environmental Enrichment in Murine Models and Its Translation to Human Factors Improving Conditions in Alzheimer Disease. J. Prev. Alzheimer’s Dis. 2023, 10, 287–300. [Google Scholar] [CrossRef]
- Ashleigh, T.; Swerdlow, R.H.; Beal, M.F. The role of mitochondrial dysfunction in Alzheimer’s disease pathogenesis. Alzheimer’s Dement. 2023, 19, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Dodart, J.-C.; Mathis, C.; Bales, K.R.; Paul, S.M.; Ungerer, A. Early regional cerebral glucose hypometabolism in transgenic mice overexpressing the V717F β-amyloid precursor protein. Neurosci. Lett. 1999, 277, 49–52. [Google Scholar] [CrossRef]
- Chadwick, W.; Maudsley, S.; Hull, W.; Havolli, E.; Boshoff, E.; Hill, M.D.; Goetghebeur, P.J.; Harrison, D.C.; Nizami, S.; Bedford, D.C. The oDGal Mouse: A Novel, Physiologically Relevant Rodent Model of Sporadic Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 6953. [Google Scholar] [CrossRef]
- López Oliva, E.; Trujillo-Estrada, L.I.; Sanchez Mejias, E.; Mejias-Ortega, M.; Fernandez-Valenzuela, J.J.; Gomez-Arboledas, A.; Davila, J.C.; Vitorica, F.J.; Gutierrez-Perez, A. Astroglial reactivity in response to b-amyloidosis is associated with mithocondrial pathology in the hippocampus of Alzheimer’s transgenic mice. In Proceedings of the International Conference on Alzheimer’s and Parkinson’s Diseases and Related Neurological Disorders, Gothenburg, Sweden, 28 March–1 April 2023. [Google Scholar]
- Mosconi, L.; Sorbi, S.; de Leon, M.J.; Li, Y.; Nacmias, B.; Myoung, P.S.; Tsui, W.; Ginestroni, A.; Bessi, V.; Fayyazz, M. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. 2006, 47, 1778–1786. [Google Scholar] [PubMed]
- Nestor, P.J.; Fryer, T.D.; Smielewski, P.; Hodges, J.R. Limbic hypometabolism in Alzheimer’s disease and mild cognitive impairment. Ann. Neurol. 2003, 54, 343–351. [Google Scholar] [CrossRef]
- Bailly, M.; Destrieux, C.; Hommet, C.; Mondon, K.; Cottier, J.-P.; Beaufils, E.; Vierron, E.; Vercouillie, J.; Ibazizene, M.; Voisin, T. Precuneus and cingulate cortex atrophy and hypometabolism in patients with Alzheimer’s disease and mild cognitive impairment: MRI and 18F-FDG PET quantitative analysis using FreeSurfer. BioMed Res. Int. 2015, 2015, 583931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, A.M.; Frackowiak, R.S.; Newman, S.K.; Bloomfield, P.M.; Seaward, J.; Roques, P.; Lewington, G.; Cunningham, V.J.; Rossor, M.N. Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci. Lett. 1995, 186, 17–20. [Google Scholar] [CrossRef]
- Petersen, R.C.; Stevens, J.C.; Ganguli, M.; Tangalos, E.G.; Cummings, J.L.; DeKosky, S.T. Practice parameter: Early detection of dementia: Mild cognitive impairment (an evidence-based review) [RETIRED]: Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2001, 56, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.; Newman, S.; Frackowiak, R.; Cunningham, V.; Roques, P.; Stevens, J.; Neary, D.; Bruton, C.; Warrington, E.; Rossor, M. Chromosome 14 linked familial Alzheimer’s disease: A clinico-pathological study of a single pedigree. Brain 1995, 118, 185–205. [Google Scholar] [CrossRef]
- Small, G.W.; Mazziotta, J.C.; Collins, M.T.; Baxter, L.R.; Phelps, M.E.; Mandelkern, M.A.; Kaplan, A.; La Rue, A.; Adamson, C.F.; Chang, L. Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 1995, 273, 942–947. [Google Scholar] [CrossRef]
- Arnaiz, E.; Jelic, V.; Almkvist, O.; Wahlund, L.; Winblad, B.; Valind, S.; Nordberg, A. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport 2001, 12, 851–855. [Google Scholar] [CrossRef]
- Raut, S.; Patel, R.; Al-Ahmad, A.J. Presence of a mutation in PSEN1 or PSEN2 gene is associated with an impaired brain endothelial cell phenotype in vitro. Fluids Barriers CNS 2021, 18, 3. [Google Scholar] [CrossRef] [PubMed]
- Raut, S.; Patel, R.; Pervaiz, I.; Al-Ahmad, A.J. Abeta peptides disrupt the barrier integrity and glucose metabolism of human induced pluripotent stem cell-derived brain microvascular endothelial cells. Neurotoxicology 2022, 89, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The Role of Insulin in Human Brain Glucose Metabolism: An 18Fluoro-Deoxyglucose Positron Emission Tomography Study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef] [Green Version]
- Manco, M.; Guerrera, S.; Ravà, L.; Ciofi degli Atti, M.; Di Vara, S.; Valeri, G.; Vicari, S. Cross-sectional investigation of insulin resistance in youths with autism spectrum disorder. Any role for reduced brain glucose metabolism? Transl. Psychiatry 2021, 11, 229. [Google Scholar] [CrossRef]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimers Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the brain: Sources, localization and functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- González-García, I.; Gruber, T.; García-Cáceres, C. Insulin action on astrocytes: From energy homeostasis to behaviour. J. Neuroendocr. 2021, 33, e12953. [Google Scholar] [CrossRef] [PubMed]
- Grillo, C.A.; Woodruff, J.L.; Macht, V.A.; Reagan, L.P. Insulin resistance and hippocampal dysfunction: Disentangling peripheral and brain causes from consequences. Exp. Neurol. 2019, 318, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, M.; Agrawal, A.K. Pathophysiological Association Between Diabetes Mellitus and Alzheimer’s Disease. Cureus 2022, 14, e29120. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Reno, C.M.; Sharma, S.; Christensen, C.; Huang, Y.; Fisher, S.J. Insulin action in the brain regulates both central and peripheral functions. Am. J. Physiol. Endocrinol. Metab. 2021, 321, E156–E163. [Google Scholar] [CrossRef]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. S2), S16. [Google Scholar] [CrossRef] [Green Version]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.-X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [Green Version]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Kalaria, R.N.; Gravina, S.A.; Schmidley, J.W.; Perry, G.; Harik, S.I. The glucose transporter of the human brain and blood-brain barrier. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1988, 24, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.A.; Vannucci, S.J.; Maher, F. Glucose transporters in mammalian brain. Biochem. Soc. Trans. 1994, 22, 671–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, M.H.; Geomor, H.K.; Bürgers, H.F.; Schelshorn, D.W.; Kuschinsky, W. Adult neural stem cells express glucose transporters GLUT1 and GLUT3 and regulate GLUT3 expression. FEBS Lett. 2006, 580, 4430–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harr, S.D.; Simonian, N.A.; Hyman, B.T. Functional alterations in Alzheimer’s disease: Decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol. 1995, 54, 38–41. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- McCall, A.L.; Van Bueren, A.M.; Huang, L.; Stenbit, A.; Celnik, E.; Charron, M.J. Forebrain endothelium expresses GLUT4, the insulin-responsive glucose transporter. Brain Res. 1997, 744, 318–326. [Google Scholar] [CrossRef]
- Vannucci, S.J.; Koehler-Stec, E.M.; Li, K.; Reynolds, T.H.; Clark, R.; Simpson, I.A. GLUT4 glucose transporter expression in rodent brain: Effect of diabetes. Brain Res. 1998, 797, 1–11. [Google Scholar] [CrossRef]
- McNay, E.C.; Pearson-Leary, J. GluT4: A central player in hippocampal memory and brain insulin resistance. Exp. Neurol. 2020, 323, 113076. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J. Neurochem. 2009, 111, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Folch, J.; Ettcheto, M.; Busquets, O.; Sánchez-López, E.; Castro-Torres, R.D.; Verdaguer, E.; Manzine, P.R.; Poor, S.R.; García, M.L.; Olloquequi, J. The implication of the brain insulin receptor in late onset Alzheimer’s disease dementia. Pharmaceuticals 2018, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Lee, H.; Seo, J. Impact of genetic risk factors for Alzheimer’s disease on brain glucose metabolism. Mol. Neurobiol. 2021, 58, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.; Balazs, R.; Thornton, P.L.; Cotman, C.W. β-amyloid peptide at sublethal concentrations downregulates brain-derived neurotrophic factor functions in cultured cortical neurons. J. Neurosci. 2004, 24, 6799–6809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, S.M.; Cha, M.-Y.; Choi, H.; Kang, S.; Choi, H.; Lee, M.-S.; Park, S.A.; Mook-Jung, I. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 2016, 12, 784–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guglielmotto, M.; Aragno, M.; Autelli, R.; Giliberto, L.; Novo, E.; Colombatto, S.; Danni, O.; Parola, M.; Smith, M.A.; Perry, G. The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1α. J. Neurochem. 2009, 108, 1045–1056. [Google Scholar] [CrossRef]
- Versele, R.; Sevin, E.; Gosselet, F.; Fenart, L.; Candela, P. TNF-α and IL-1β modulate blood-brain barrier permeability and decrease amyloid-β peptide efflux in a human blood-brain barrier model. Int. J. Mol. Sci. 2022, 23, 10235. [Google Scholar] [CrossRef] [PubMed]
- Alexandraki, K.; Piperi, C.; Kalofoutis, C.; Singh, J.; Alaveras, A.; Kalofoutis, A. Inflammatory process in type 2 diabetes: The role of cytokines. Ann. N. Y. Acad. Sci. 2006, 1084, 89–117. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Calabrese, V.; Scapagnini, G.; Colombrita, C.; Ravagna, A.; Pennisi, G.; Giuffrida Stella, A.; Galli, F.; Butterfield, D. Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: A nutritional approach. Amino Acids 2003, 25, 437–444. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.S.; Oliver, P.L. ROS generation in microglia: Understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Gerson, J.; Kayed, R. Therapeutic Approaches Targeting Pathological Tau Aggregates. Curr. Pharm. Des. 2016, 22, 4028–4039. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, M.; Warnecke, T. Causal treatment of Alzheimer’s disease: Amyloid antibodies. Inn. Med. 2022, 63, 1000–1008. [Google Scholar] [CrossRef]
- Anand, K.; Sabbagh, M. Early investigational drugs targeting tau protein for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2015, 24, 1355–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, T.-P.V.; Holtzman, D.M. In search of an identity for amyloid plaques. Trends Neurosci. 2018, 41, 483–486. [Google Scholar] [CrossRef]
- Schindler, S.E.; Fagan, A.M. Autosomal dominant Alzheimer disease: A unique resource to study CSF biomarker changes in preclinical AD. Front. Neurol. 2015, 6, 142. [Google Scholar] [CrossRef] [Green Version]
- Vaz, M.; Silvestre, S. Alzheimer’s disease: Recent treatment strategies. Eur. J. Pharmacol. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Testai, L.; Martelli, A.; Flori, L.; Cicero, A.F.G.; Colletti, A. Coenzyme Q(10): Clinical Applications beyond Cardiovascular Diseases. Nutrients 2021, 13, 1697. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Y.; Xu, H.; Luo, X.; Yu, J.; Liu, J.; Chang, R.C. Neuroprotection of Coenzyme Q10 in Neurodegenerative Diseases. Curr. Top Med. Chem. 2016, 16, 858–866. [Google Scholar] [CrossRef]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Jiménez, F.J.; Alonso-Navarro, H.; García-Martín, E.; Agúndez, J.A. Coenzyme Q10 and Dementia: A Systematic Review. Antioxidants 2023, 12, 533. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, B.; Gupta, S. A review of antioxidants and Alzheimer’s disease. Ann. Clin. Psychiatry 2005, 17, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Adalier, N.; Parker, H. Vitamin E, Turmeric and Saffron in Treatment of Alzheimer’s Disease. Antioxidants 2016, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879. [Google Scholar] [CrossRef] [Green Version]
- Dysken, M.W.; Sano, M.; Asthana, S.; Vertrees, J.E.; Pallaki, M.; Llorente, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; Malphurs, J. Effect of vitamin E and memantine on functional decline in Alzheimer disease: The TEAM-AD VA cooperative randomized trial. JAMA 2014, 311, 33–44. [Google Scholar] [CrossRef]
- Petersen, R.C.; Thomas, R.G.; Grundman, M.; Bennett, D.; Doody, R.; Ferris, S.; Galasko, D.; Jin, S.; Kaye, J.; Levey, A. Vitamin E and donepezil for the treatment of mild cognitive impairment. N. Engl. J. Med. 2005, 352, 2379–2388. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [Green Version]
- Rainey-Smith, S.R.; Brown, B.M.; Sohrabi, H.R.; Shah, T.; Goozee, K.G.; Gupta, V.B.; Martins, R.N. Curcumin and cognition: A randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br. J. Nutr. 2016, 115, 2106–2113. [Google Scholar] [CrossRef] [Green Version]
- Frautschy, S.; Hu, W.; Kim, P.; Miller, S.; Chu, T.; Harris-White, M.; Cole, G. Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiol. Aging 2001, 22, 993–1005. [Google Scholar] [CrossRef]
- Ege, D. Action mechanisms of curcumin in Alzheimer’s disease and its brain targeted delivery. Materials 2021, 14, 3332. [Google Scholar] [CrossRef] [PubMed]
- Goozee, K.; Shah, T.; Sohrabi, H.R.; Rainey-Smith, S.; Brown, B.; Verdile, G.; Martins, R. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br. J. Nutr. 2016, 115, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef]
- Sharma, R.; Gescher, A.; Steward, W. Curcumin: The story so far. Eur. J. Cancer 2005, 41, 1955–1968. [Google Scholar] [CrossRef] [PubMed]
- Kuepeli Akkol, E.; Bardakcı, H.; Yücel, Ç.; Şeker Karatoprak, G.; Karpuz, B.; Khan, H. A New Perspective on the Treatment of Alzheimer’s Disease and Sleep Deprivation-Related Consequences: Can Curcumin Help? Oxidative Med. Cell. Longev. 2022, 2022. [Google Scholar] [CrossRef] [PubMed]
- Wollen, K.A. Alzheimer’s disease: The pros and cons of pharmaceutical, nutritional, botanical, and stimulatory therapies, with a discussion of treatment strategies from the perspective of patients and practitioners. Altern Med. Rev. 2010, 15, 223–244. [Google Scholar]
- Chin, D.; Huebbe, P.; Pallauf, K.; Rimbach, G. Neuroprotective properties of curcumin in Alzheimer’s disease-merits and limitations. Curr. Med. Chem. 2013, 20, 3955–3985. [Google Scholar] [CrossRef]
- Formicola, B.; Cox, A.; Dal Magro, R.; Masserini, M.; Re, F. Nanomedicine for the Treatment of Alzheimer’s Disease. J. Biomed. Nanotechnol. 2019, 15, 1997–2024. [Google Scholar] [CrossRef]
- Han, L.; Jiang, C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm. Sin. B 2021, 11, 2306–2325. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J. Vascular dysfunction—The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef] [Green Version]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in Mural Vascular Cells Coincides with Blood–Brain Barrier Disruption in A lzheimer’s Disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijmans, C.R.; Graven, C.; Dederen, P.J.; Tanila, H.; van Groen, T.; Kiliaan, A.J. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007, 1181, 93–103. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Hall, J.L.; Gonder-Frederick, L.A.; Chewning, W.W.; Silveira, J.; Gold, P.E. Glucose enhancement of performance on memory tests in young and aged humans. Neuropsychologia 1989, 27, 1129–1138. [Google Scholar] [CrossRef]
- Sun, J.; Roy, S. Gene-based therapies for neurodegenerative diseases. Nat. Neurosci. 2021, 24, 297–311. [Google Scholar] [CrossRef]
- Hacıhamdioğlu, B.; Kendirli, T.; Öçal, G.; Şıklar, Z.; Savaş Erdeve, Ş.; Ince, E.; Berberoğlu, M. Pathophysiology of critical illness hyperglycemia in children. J. Pediatr. Endocrinol. Metab. 2013, 26, 715–720. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Identification of novel class I and class IIb histone deacetylase inhibitor for Alzheimer’s disease therapeutics. Life Sci. 2020, 256, 117912. [Google Scholar] [CrossRef]
- Tseng, H.-J.; Lin, M.-H.; Shiao, Y.-J.; Yang, Y.-C.; Chu, J.-C.; Chen, C.-Y.; Chen, Y.-Y.; Lin, T.E.; Su, C.-J.; Pan, S.-L. Synthesis and biological evaluation of acridine-based histone deacetylase inhibitors as multitarget agents against Alzheimer’s disease. Eur. J. Med. Chem. 2020, 192, 112193. [Google Scholar] [CrossRef]
- Song, J.-H.; Yu, J.-T.; Tan, L. Brain-derived neurotrophic factor in Alzheimer’s disease: Risk, mechanisms, and therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef]
- Pezet, S.; Malcangio, M. Brain-derived neurotrophic factor as a drug target for CNS disorders. Expert Opin. Ther. Targets 2004, 8, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.V.; Dave, K.R.; Perez-Pinzon, M.A. Ischemic preconditioning and clinical scenarios. Curr. Opin. Neurol. 2013, 26, 1. [Google Scholar] [CrossRef]
- Glat, M.J.; Offen, D. Cell and gene therapy in Alzheimer’s disease. Stem Cells Dev. 2013, 22, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Osaka, H.; Muramatsu, S.-i.; Takino, N.; Ito, M.; Aoki, S.; Jimbo, E.F.; Shimazaki, K.; Onaka, T.; Ohtsuki, S. Gene therapy for a mouse model of glucose transporter-1 deficiency syndrome. Mol. Genet. Metab. Rep. 2017, 10, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, P.; Iwata, N.; Muramatsu, S.i.; Tjernberg, L.O.; Winblad, B.; Saido, T.C. Gene therapy in Alzheimer’s disease–potential for disease modification. J. Cell. Mol. Med. 2010, 14, 741–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinuevo, J.L.; Ayton, S.; Batrla, R.; Bednar, M.M.; Bittner, T.; Cummings, J.; Fagan, A.M.; Hampel, H.; Mielke, M.M.; Mikulskis, A. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018, 136, 821–853. [Google Scholar] [CrossRef] [Green Version]
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef]
- Reiss, A.B.; Arain, H.A.; Stecker, M.M.; Siegart, N.M.; Kasselman, L.J. Amyloid toxicity in Alzheimer’s disease. Rev. Neurosci. 2018, 29, 613–627. [Google Scholar] [CrossRef]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef]
- Xie, H.; Hou, S.; Jiang, J.; Sekutowicz, M.; Kelly, J.; Bacskai, B.J. Rapid cell death is preceded by amyloid plaque-mediated oxidative stress. Proc. Natl. Acad. Sci. USA 2013, 110, 7904–7909. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.-H.; Lam, K.S.-L.; Cheng, O.-Y.; Kwan, J.S.-C.; Ho, P.W.-L.; Cheng, K.K.-Y.; Chung, S.K.; Ho, J.W.-M.; Guo, V.Y.; Xu, A. Adiponectin is protective against oxidative stress induced cytotoxicity in amyloid-beta neurotoxicity. PLoS ONE 2012, 7, e52354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojas-Gutierrez, E.; Muñoz-Arenas, G.; Treviño, S.; Espinosa, B.; Chavez, R.; Rojas, K.; Flores, G.; Díaz, A.; Guevara, J. Alzheimer’s disease and metabolic syndrome: A link from oxidative stress and inflammation to neurodegeneration. Synapse 2017, 71, e21990. [Google Scholar] [CrossRef]
- Muthaiyah, B.; Essa, M.M.; Chauhan, V.; Chauhan, A. Protective effects of walnut extract against amyloid beta peptide-induced cell death and oxidative stress in PC12 cells. Neurochem. Res. 2011, 36, 2096–2103. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Fu, Y.; Yasvoina, M.; Shao, P.; Hitt, B.; O’Connor, T.; Logan, S.; Maus, E.; Citron, M.; Berry, R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. J. Neurosci. 2007, 27, 3639–3649. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.-f.; Xu, T.-h.; Yan, Y.; Zhou, Y.-r.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [Green Version]
- Verdier, Y.; Zarándi, M.; Penke, B. Amyloid β-peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer’s disease. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2004, 10, 229–248. [Google Scholar]
- Verdier, Y.; Penke, B. Binding sites of amyloid β-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr. Protein Pept. Sci. 2004, 5, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Lathuiliere, A.; Migliorini, M.; Arai, A.L.; Wani, M.M.; Dujardin, S.; Muratoglu, S.C.; Hyman, B.T.; Strickland, D.K. Regulation of tau internalization, degradation, and seeding by LRP1 reveals multiple pathways for tau catabolism. J. Biol. Chem. 2021, 296, 100715. [Google Scholar] [CrossRef]
- Fein, J.A.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Yang, F.; Cole, G.M.; Gylys, K.H. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. Am. J. Pathol. 2008, 172, 1683–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, J.M.; Lathuiliere, A.; Migliorini, M.; Arai, A.L.; Wani, M.M.; Dujardin, S.; Muratoglu, S.C.; Hyman, B.T.; Strickland, D.K. LRP1 and SORL1 regulate tau internalization and degradation and enhance tau seeding. BioRxiv 2020. [Google Scholar] [CrossRef]
- Yin, R.-H.; Yu, J.-T.; Tan, L. The role of SORL1 in Alzheimer’s disease. Mol. Neurobiol. 2015, 51, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.-H.; Westerteicher, C.; Wang, X.-H.; Kratzer, M.; Tsolakidou, A.; Jiang, M.; Grimmer, T.; Laws, S.M.; Alexopoulos, P.; Bujo, H. SORL1 genetic variants and cerebrospinal fluid biomarkers of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Wu, Z.; Zlokovic, B.V. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid β-peptide clearance through transport across the blood–brain barrier. Stroke 2004, 35, 2628–2631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deane, R.; Sagare, A.; Hamm, K.; Parisi, M.; LaRue, B.; Guo, H.; Wu, Z.; Holtzman, D.M.; Zlokovic, B.V. IgG-assisted age-dependent clearance of Alzheimer’s amyloid β peptide by the blood–brain barrier neonatal Fc receptor. J. Neurosci. 2005, 25, 11495–11503. [Google Scholar] [CrossRef] [Green Version]
- Miles, L.A.; Crespi, G.A.; Doughty, L.; Parker, M.W. Bapineuzumab captures the N-terminus of the Alzheimer’s disease amyloid-beta peptide in a helical conformation. Sci. Rep. 2013, 3, 1302. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Frisardi, V.; P Imbimbo, B.; Seripa, D.; Paris, F.; Santamato, A.; D’Onofrio, G.; Logroscino, G.; Pilotto, A.; Solfrizzi, V. Anti-β-amyloid immunotherapy for Alzheimer’s disease: Focus on bapineuzumab. Curr. Alzheimer Res. 2011, 8, 808–817. [Google Scholar] [CrossRef]
- Siemers, E.R.; Sundell, K.L.; Carlson, C.; Case, M.; Sethuraman, G.; Liu-Seifert, H.; Dowsett, S.A.; Pontecorvo, M.J.; Dean, R.A.; Demattos, R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimer’s Dement. 2016, 12, 110–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyck, C.H. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise. Biol. Psychiatry 2018, 83, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Bouter, Y.; Noguerola, J.S.L.; Tucholla, P.; Crespi, G.A.; Parker, M.W.; Wiltfang, J.; Miles, L.A.; Bayer, T.A. Abeta targets of the biosimilar antibodies of Bapineuzumab, Crenezumab, Solanezumab in comparison to an antibody against N-truncated Abeta in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol. 2015, 130, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Honigberg, L.A.; Cho, W.; Ward, M.; Friesenhahn, M.; Brunstein, F.; Quartino, A.; Clayton, D.; Mortensen, D.; Bittner, T. Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimer’s Res. Ther. 2018, 10, 96. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Dang, Y.; Ostaszewski, B.; Mengel, D.; Steffen, V.; Rabe, C.; Bittner, T.; Walsh, D.M.; Selkoe, D.J. Target engagement in an alzheimer trial: Crenezumab lowers amyloid β oligomers in cerebrospinal fluid. Ann. Neurol. 2019, 86, 215–224. [Google Scholar] [CrossRef]
- Henley, D.B.; May, P.C.; Dean, R.A.; Siemers, E.R. Development of semagacestat (LY450139), a functional γ-secretase inhibitor, for the treatment of Alzheimer’s disease. Expert Opin. Pharmacother. 2009, 10, 1657–1664. [Google Scholar] [CrossRef]
- Roher, A.E.; Maarouf, C.L.; Kokjohn, T.A.; Whiteside, C.M.; Kalback, W.M.; Serrano, G.; Belden, C.; Liebsack, C.; Jacobson, S.A.; Sabbagh, M.N. Neuropathological and biochemical assessments of an Alzheimer’s disease patient treated with the γ-secretase inhibitor semagacestat. Am. J. Neurodegener. Dis. 2014, 3, 115. [Google Scholar] [PubMed]
- Henley, D.B.; Sundell, K.L.; Sethuraman, G.; Dowsett, S.A.; May, P.C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med. Res. Opin. 2014, 30, 2021–2032. [Google Scholar] [CrossRef]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Sun, B.L.; Li, W.W.; Zhu, C.; Jin, W.S.; Zeng, F.; Liu, Y.H.; Bu, X.L.; Zhu, J.; Yao, X.Q.; Wang, Y.J. Clinical Research on Alzheimer’s Disease: Progress and Perspectives. Neurosci. Bull. 2018, 34, 1111–1118. [Google Scholar] [CrossRef]
- Shoeibi, A.; Olfati, N.; Litvan, I. Frontrunner in Translation: Progressive Supranuclear Palsy. Front. Neurol. 2019, 10, 1125. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andres, M.V.; Gómez-Carrillo, B.; Leon, T. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef]
- Tatulian, S.A. Challenges and hopes for Alzheimer’s disease. Drug Discov. Today 2022, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Chancellor, B.; Duncan, A.; Chatterjee, A. Art therapy for Alzheimer’s disease and other dementias. J. Alzheimer’s Dis. 2014, 39, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, A.; Olaso-Gonzalez, G.; Arc-Chagnaud, C.; Millan, F.; Salvador-Pascual, A.; García-Lucerga, C.; Blasco-Lafarga, C.; Garcia-Dominguez, E.; Carretero, A.; Correas, A.G.; et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J. Sport Health Sci. 2020, 9, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Cass, S.P. Alzheimer’s Disease and Exercise: A Literature Review. Curr. Sports Med. Rep. 2017, 16, 19–22. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 1106–1125. [Google Scholar] [CrossRef]
- Ouyang, Q.; Meng, Y.; Zhou, W.; Tong, J.; Cheng, Z.; Zhu, Q. New advances in brain-targeting nano-drug delivery systems for Alzheimer’s disease. J. Drug Target 2022, 30, 61–81. [Google Scholar] [CrossRef]
- Jagust, W.J.; Bandy, D.; Chen, K.; Foster, N.L.; Landau, S.M.; Mathis, C.A.; Price, J.C.; Reiman, E.M.; Skovronsky, D.; Koeppe, R.A. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimer’s Dement. 2010, 6, 221–229. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Tsui, W.H.; Herholz, K.; Pupi, A.; Drzezga, A.; Lucignani, G.; Reiman, E.M.; Holthoff, V.; Kalbe, E.; Sorbi, S. Multicenter standardized 18F-FDG PET diagnosis of mild cognitive impairment, Alzheimer’s disease, and other dementias. J. Nucl. Med. 2008, 49, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L. Glucose metabolism in normal aging and Alzheimer’s disease: Methodological and physiological considerations for PET studies. Clin. Transl. Imaging 2013, 1, 217–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer’s Association. 2018 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2018, 14, 367–429. [Google Scholar] [CrossRef]
Figure 1.
Healthy brain and brain with AD. The neurovascular unit comprises the brain’s microvascular endothelial cells, pericytes, astrocytes, oligodendrocytes, and neurons. Glucose transport is accomplished by the coordinated activation of glucose transporters on the capillary endothelium, astrocytes, oligodendrocytes, and neurons. After being transported, it is converted into ATP through biochemical reactions involving glycolysis, the TCA, and oxidative phosphorylation. It is utilized in many other energy pathways to support a variety of intricate cellular processes. Brain energetics has been implicated in various neurological disorders, including AD, as it leads to neurodegeneration and neuronal death.
Figure 1.
Healthy brain and brain with AD. The neurovascular unit comprises the brain’s microvascular endothelial cells, pericytes, astrocytes, oligodendrocytes, and neurons. Glucose transport is accomplished by the coordinated activation of glucose transporters on the capillary endothelium, astrocytes, oligodendrocytes, and neurons. After being transported, it is converted into ATP through biochemical reactions involving glycolysis, the TCA, and oxidative phosphorylation. It is utilized in many other energy pathways to support a variety of intricate cellular processes. Brain energetics has been implicated in various neurological disorders, including AD, as it leads to neurodegeneration and neuronal death.

Figure 2.
Neuropathological hallmarks of AD. The neuropathological hallmarks of AD include lesions such as amyloid plaques and neurofibrillary tangles, neuronal and synaptic loss, oxidative stress, hyperactivated glial responses leading to neuroinflammation, and BBB leakage.
Figure 2.
Neuropathological hallmarks of AD. The neuropathological hallmarks of AD include lesions such as amyloid plaques and neurofibrillary tangles, neuronal and synaptic loss, oxidative stress, hyperactivated glial responses leading to neuroinflammation, and BBB leakage.

Figure 3.
Current FDA-approved treatments and proposed targets for AD. In the past few years, the FDA has approved several drugs and treatments for AD, targeting the usual culprits, namely, beta-amyloid plaques and tau tangles. The potential therapeutic targets for hypometabolism in AD include targeting the BBB and glucose transporter overexpression.
Figure 3.
Current FDA-approved treatments and proposed targets for AD. In the past few years, the FDA has approved several drugs and treatments for AD, targeting the usual culprits, namely, beta-amyloid plaques and tau tangles. The potential therapeutic targets for hypometabolism in AD include targeting the BBB and glucose transporter overexpression.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Raut, S.; Bhalerao, A.; Powers, M.; Gonzalez, M.; Mancuso, S.; Cucullo, L. Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview. Cells 2023, 12, 2019. https://doi.org/10.3390/cells12162019
AMA Style
Raut S, Bhalerao A, Powers M, Gonzalez M, Mancuso S, Cucullo L. Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview. Cells. 2023; 12(16):2019. https://doi.org/10.3390/cells12162019
Chicago/Turabian StyleRaut, Snehal, Aditya Bhalerao, Michael Powers, Minelly Gonzalez, Salvatore Mancuso, and Luca Cucullo. 2023. "Hypometabolism, Alzheimer’s Disease, and Possible Therapeutic Targets: An Overview" Cells 12, no. 16: 2019. https://doi.org/10.3390/cells12162019
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.