
The Role of Mitophagy in Glaucomatous Neurodegeneration
by
 , , and
, , and
Dimitrios Stavropoulos
1,2,
Manjot K. Grewal
3,4,
Bledi Petriti
3,5,
Kai-Yin Chau
5,
Christopher J. Hammond
6,7,
David F. Garway-Heath
3 and
Gerassimos Lascaratos
1,6,* 1
Department of Ophthalmology, King’s College Hospital, London SE5 9RS, UK
2
Department of Ophthalmology, 417 Veterans Army Hospital (NIMTS), 11521 Athens, Greece
3
NIHR Biomedical Research Center, Moorfields Eye Hospital and UCL Institute of Ophthalmology, London EC1V 9EL, UK
4
Division of Optometry and Visual Science, School of Health Sciences, City, University of London, London EC1V 0HB, UK
5
Department of Clinical & Movement Neurosciences, UCL Queens Square Institute of Neurology, London NW3 2PF, UK
6
Section of Ophthalmology, School of Life Course Sciences, King’s College London, London SE1 7EH, UK
7
Department of Ophthalmology, St Thomas’ Hospital, London SE1 7EH, UK
*
Author to whom correspondence should be addressed.
Cells 2023, 12(15), 1969; https://doi.org/10.3390/cells12151969
Submission received: 3 June 2023
/
Revised: 15 July 2023
/
Accepted: 19 July 2023
/
Published: 30 July 2023
(This article belongs to the Special Issue Recent Research on the Role of Mitochondria in Neurodegeneration)
Abstract
:This review aims to provide a better understanding of the emerging role of mitophagy in glaucomatous neurodegeneration, which is the primary cause of irreversible blindness worldwide. Increasing evidence from genetic and other experimental studies suggests that mitophagy-related genes are implicated in the pathogenesis of glaucoma in various populations. The association between polymorphisms in these genes and increased risk of glaucoma is presented. Reduction in intraocular pressure (IOP) is currently the only modifiable risk factor for glaucoma, while clinical trials highlight the inadequacy of IOP-lowering therapeutic approaches to prevent sight loss in many glaucoma patients. Mitochondrial dysfunction is thought to increase the susceptibility of retinal ganglion cells (RGCs) to other risk factors and is implicated in glaucomatous degeneration. Mitophagy holds a vital role in mitochondrial quality control processes, and the current review explores the mitophagy-related pathways which may be linked to glaucoma and their therapeutic potential.
1. Introduction
Glaucoma is the primary cause of irreversible blindness worldwide [1], the second cause in the UK, and a leading cause of certifications for sight impairment [2]. The term glaucoma refers to a group of conditions characterized by progressive optic neuropathy, which occurs through slow and continuing apoptotic degeneration of retinal ganglion cells’ (RGCs) somata and axons, leading to a loss of visual function [3,4,5]. Our understanding of the pathophysiological mechanisms of glaucoma remains incomplete. Hypotheses, such as the vascular theory and the mechanical theory of glaucoma, were formed to explain how elevated intraocular pressure (IOP) harms the optic nerve (ON) [6,7]. Reduction in IOP is currently the only modifiable risk factor, established by several long-term clinical trials [8,9,10], to reduce the progression of glaucoma. However, patients deteriorate at all IOP levels and despite receiving IOP-lowering therapy [11], suggesting that other factors confer susceptibility. Furthermore, clinical studies emphasize the insufficient results of current IOP-lowering therapeutic approaches, with over 40% of glaucoma patients progressing after seven years in one study [12] and 60% showing statistically significant deterioration after an average of eight years of follow-up in another study [13]. As a result, it is widely accepted that other risk factors modulate the susceptibility of an eye to IOP. This has led to a considerable body of research on neurodegenerative mechanisms and the role of mitochondrial dysfunction in glaucoma [14]. Dysfunction of mitochondria has been strongly implicated in glaucomatous degeneration in multiple glaucoma models in recent years [15,16,17,18]. Mitochondrial dysfunction is capable of increasing the susceptibility of RGCs to stress from other risk factors, such as elevated intraocular pressure (IOP), light exposure and vascular insufficiency [19]. Mitophagy is a mitochondrial quality control process crucial in securing mitochondrial quantity and quality [20], and increasing evidence from various experimental studies indicates the importance of impaired mitophagy as a feature of glaucomatous degeneration [21,22,23]. This paper summarizes the evidence for the role of mitophagy-related genes in the pathogenesis of human glaucoma and in animal glaucoma models, and potential therapeutic avenues for glaucoma based on a modification of the mitophagy-related pathways.
2. Mitochondria in Glaucoma
The role of mitochondria in the pathogenesis of glaucoma has gained increasing interest as they are considered potential therapeutic targets [24,25]. Mitochondria are cytosolic organelles that hold a key role in generating energy in human cells in the form of adenosine triphosphate (ATP) through oxidative phosphorylation (OXPHOS) and other pathways [18]. Mitochondria also regulate calcium homeostasis in cells, intermediary metabolism, and the balance between reactive oxygen species (ROS) generation and scavenging [26]. The number of mitochondria per cell varies, depending on the energy demands of a cell. RGCs consume enormous amounts of energy to support axon potential generation in unmyelinated axons and to maintain axonal transport across the high-pressure gradient as axons leave the eye at the optic nerve head (ONH). The former is evidenced by the distribution and trafficking of mitochondria within RGC axons [27], and the latter is evidenced by the presence of giant mitochondria in ONH astrocytes, which provide support to RGC axonal metabolism at an anatomical site of particular vulnerability [28]. These huge energy demands make this part of the ON in the eye particularly susceptible to mitochondrial dysfunction [29]. ATP production and efficiency and number of mitochondria in RGCs decline with aging [30]. ROS are mainly produced through leakage of electrons from the respiratory chain and, under physiological conditions, ROS are detoxified by cellular antioxidants. When an impairment is present in this equilibrium, oxidative damage may induce apoptotic RGC death [31]. Studies have shown that although apoptosis is actively executed by caspases, RGC death may happen independently of caspase-mediated pathways [32]. These data suggest that both apoptosis and non-apoptotic cell death play a critical role in the death of RGCs. Wang et al. [33] suggest a non-apoptotic cell death mechanism of RGCs termed “paraptosis”, which may occur simultaneously with apoptosis and autophagy in glaucomatous RGCs and happens in two stages. Excessive production of ROS leads to mitochondrial impairment in glaucomatous RGCs, and mitochondrial damage may provoke RGC paraptosis.
Furthermore, nitric oxide, tumor necrosis factor alpha (TNFα), glutamate and other toxic substances released from glial cells in glaucoma are considered a possible path that leads to RGCs’ death due to a compromised energetic state when mitochondria are unable to maintain their expected function [34]. Glutamate excitotoxicity in glaucoma has been linked to increased calcium influx into mitochondria, causing the opening of the mitochondrial permeability transition pore (MPTP) of the inner mitochondrial membrane (IMM), increased ROS production, activation of apoptotic pathways and RGC degradation [35,36,37]. The human eye is also exposed every day to sunlight and artificial light, and it has been proposed that certain wavelengths of light can cause an enhanced generation of ROS in RGCs’ mitochondria and may lead to neuronal apoptosis [38,39].
In human glaucoma, mitochondrial dysfunction has been reported in peripheral blood mononuclear cells, tenon’s ocular fibroblasts, the trabecular meshwork and the lamina cribrosa. Vallabh et al. [40] demonstrated that basal respiration (oxygen consumption rate, OCR) was significantly reduced in glaucomatous tenon’s ocular fibroblasts compared to non-glaucomatous ones and demonstrated their impaired mitochondrial cellular bioenergetics. There is also evidence that susceptibility to IOP is related to mitochondrial function. Lascaratos et al. [41] demonstrated that patients with ocular hypertension who have not developed glaucoma despite experiencing raised IOP over several years have better lymphocyte mitochondrial function when compared to glaucoma patients with normal IOP (normal-tension glaucoma) and controls of a similar age. Additionally, Petriti et al. [42] showed that systemic mitochondrial function in the peripheral blood lymphocytes of patients with normal-tension glaucoma (NTG) was lower compared to patients with glaucoma and high IOP (high-tension glaucoma; HTG), suggesting that, in the absence of raised IOP as a clear risk factor for glaucoma, systemic mitochondrial dysfunction may play a critical role in the pathogenesis of glaucomatous neurodegeneration. Degeneration of RGCs’ mitochondria is an early feature of human glaucoma [43], and mitochondrial dysfunction has been identified in lamina cribrosa [44] and trabecular meshwork (TM) cells [45] of glaucomatous eyes.
Studies with animal glaucoma models, which investigated the effects of elevated IOP, indicate a reduction in the number of healthy mitochondria with normal membrane potential, decreased mitochondrial cristae volume, smaller mitochondrial size and decreased mitochondrial movement within ONH axons after chronic IOP elevation [46,47,48,49]. Differential expression of genes involved in mitochondrial dysfunction and oxidative stress pathways is an early feature in a DBA/2J mouse glaucoma model, and age-dependent declines of nicotinamide adenine dinucleotide (NAD+) and glutathione in the retina render RGCs susceptible to damage from raised IOP [49].
The role of mitochondria in glaucoma susceptibility is further supported by genetic studies [50]. The three Mendelian genes, myocilin, TANK-binding kinase 1 (TBK1) and optineurin (OPTN), have been linked to glaucoma, with the last two genes being mitophagy-related genes [51]. Khawaja et al. [52], using gene set analyses of a large cohort of patients, identified mitochondrial enzyme pathways critical to the pathogenesis of primary open-angle glaucoma (POAG). Moreover, polymorphisms in the OPTN gene (which mediates mitophagy) are associated with open-angle glaucoma (OAG) with normal lOP (NTG) and younger age of onset of HTG [53]. Inherited defects in mitochondria are associated with characteristic optic neuropathies, such as Leber’s hereditary optic neuropathy (mitochondrial genome mutations) and dominant optic atrophy (DOA) (mutations in a nuclear gene encoding inner mitochondrial membrane proteins) [54]. Interestingly, the clinical phenotype of these mitochondrial optic neuropathies may mimic NTG [55].
3. Mitophagy
Autophagy refers to a procedure of degradation and recycling of intracellular materials and inoperative organelles, which preserves cellular homeostasis [56]. It is subclassified into macroautophagy, microautophagy and chaperone-mediated autophagy. These three types of autophagy are completed through different mechanisms, but the final goal is the same: transportation of intracellular materials to lysosomes, where they can be degraded by hydrolytic enzymes [57]. In order to remain intact, mitochondria undergo several processes such as fusion (junction of two mitochondria into one), fission (separation of one mitochondrion into two), biogenesis (creation of new mitochondria) and mitophagy (degradation of mitochondria by autophagy) [58]. Mitophagy is a selective subtype of macroautophagy and a mitochondrial quality control process in which dysfunctional mitochondria are degraded in lysosomes [59] (Figure 1). Based on the targeting signals in impaired mitochondria that commence the process, it is divided into ubiquitin-dependent mitophagy, ubiquitin-independent or receptor-based mitophagy, lipid-based mitophagy and micromitophagy, with the first two categories being the most common [60]. An accumulation of mitochondrial DNA (mtDNA) mutations and diminished mitochondrial activity are thought to contribute to the normal aging process [61] and are associated with various systemic diseases, including heart failure, diabetes and cancer [62]. Mitophagy, as a quality control mechanism, is considered critical to the prevention of human disease by retaining normal cellular function, while aberrant mitophagy is implicated in the pathogenesis of multiple metabolic, cardiovascular, skeletal muscle and neurodegenerative diseases, including Alzheimer’s disease, amyotrophic lateral sclerosis, and Duchenne muscle dystrophy [63]. Several studies have demonstrated the importance of mitophagy in glaucomatous degeneration [21,22,23], as well as in other ocular diseases [64]. In the next section, we explore in more detail the different mitophagy-related genes (Table 1) and the growing scientific evidence that links mitophagy to glaucoma.
4. Mitophagy-Related Genes and Glaucoma Pathogenesis
4.1. TBK1
The TBK1 gene (TANK-binding kinase 1; MIM#604834), localized on 12q14.2, encodes the serine/threonine protein kinase TANK-binding kinase 1 [119], which controls the activation of genes in the nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) pathway [120]. TBK1 holds a major role in cell proliferation, innate immune response, autophagy and apoptosis [121]. TBK1 is activated upon mitochondrial depolarization and regulates mitophagy via phosphorylation of major autophagy receptors, such as OPTN, calcium-binding and coiled-coil domain 2 (NDP52/CALCOCO2), sequestosome-1 (SQSTM1/p62) and tax1-binding protein 1 (TAX1BP1), enhancing their ubiquitin- and microtubule-associated protein 1A/1B-light chain 3 (LC3)-binding domains and, therefore, facilitating their autophagosomal engulfment [122]. TBK1 gene duplications and triplications have been associated with approximately 1% of NTG cases, including Asian [65,66], Caucasian [67,68] and African American patients [69]. A single case of a patient with exfoliation glaucoma and TBK1 gene duplication was reported by Fingert et al. [70]. Moreover, it is proven that TBK1 is expressed in RGCs, which constitute a crucial site of NTG pathogenesis [69,123], while in induced pluripotent stem cell (iPSC)-derived retinal ganglion cells, an extra copy of TBK1 has been found to activate the crucial autophagy protein LC3-II [124]. Transgenic TBK1 mice show traits of NTG and significant progressive loss of RGCs [71].
4.2. OPA1
The OPA1 gene (optic atrophy-1; MIM#605290; chr3q28-q29), which encodes a dynamin-related protein located in the inter-membrane space and is implicated in mitochondrial morphology [125], was identified by Delettre et al. [126]. OPA1 orchestrates fusion of the IMM [127] and ensures the maintenance of cristae integrity [128]. Studies have shown that mutations of OPA1 disrupt IMM fusion, resulting in DOA [129,130]. In addition, Liao et al. [131] showed that apart from mitochondrial fragmentation, OPA1 loss leads to increased mitophagy and mitochondrial mislocalization. Moreover, Frezza et al. [132] proved, via electron microscopy, that OPA1 controls apoptosis by regulating cytochrome c redistribution and cristae remodeling. OPA1 is expressed in the inner plexiform layer, the outer plexiform layer, the inner nuclear layer and the myelinated region beyond the lamina cribrosa within the ON [133,134,135]. Wang et al. [136] used three donated human eyeballs with no history of ocular disease and proposed that OPA1 is expressed all the way from the prelaminar portion through the lamina cribrosa to the retrolaminar myelinated portion. Furthermore, Hu et al. [21] used purified RGCs from rats and chronic hypertensive glaucoma rats and proved that OPA1 overexpression protects RGCs by intensifying fusion and enhancing parkin RBR E3 ubiquitin protein ligase (PARKIN)-mediated mitophagy. A study that investigated peripheral blood leucocytes from 43 POAG patients and 27 controls conducted by Bosley et al. [72] evidenced that OPA1 expression was significantly lower in the POAG patients compared to the controls. Aung et al. [73] found that polymorphisms in the OPA1 gene were associated with NTG in two Caucasian patient cohorts. The single-nucleotide polymorphism (SNP) IVS8+4C/T was strongly associated with NTG in both cohorts, and the second SNP IVS8+32T/C was only associated with NTG occurrence in the first cohort. Another study by Aung et al. [137] could not demonstrate an association of these two SNPs with HTG. A study by Yu-Wai-Man et al. [74] in the North East England also demonstrated that the CT/TT compound genotype at VS8+4 and IVS8+32 is a strong genetic causal factor for NTG, but not HTG. Milanowski et al. [75] investigated OPA1 polymorphisms in the Polish population and showed that the genotype CC and allele C of rs9851685 OPA1 polymorphism are NTG risk factors, whereas the TT genotype and T allele of this polymorphism are protective factors against NTG. Mabuchi et al. [138] investigated 194 Japanese patients with NTG and 191 with HTG and identified an association of the SNP IVS8+32T/C with NTG, but their study also revealed that this polymorphism is associated with earlier age of disease onset in HTG patients and should be considered a genetic risk factor for both. This result was not validated by the study conducted by Liu et al. [139], which demonstrated no association between the OPA1 polymorphisms and a POAG phenotype that includes increased IOP in Caucasian, African American and Ghanaian populations. Several other studies also failed to demonstrate a correlation between OPA1 and NTG in Greek [140], Korean [141], African Caribbean [142], Chinese [143], Mexican [144], German [76], Singaporean and Indian patient populations [145].
4.3. MFN1 and MFN2
MFN1 (mitofusin 1; MIM#608506) and MFN2 (mitofusin 2; MIM#608507) are genes that encode the transmembrane guanosine triphosphatases (GTPases) MFN1 and MFN2, respectively, which regulate fusion of the outer mitochondrial membrane (OMM) [146] where they are situated [147]. Chen et al. [148] demonstrated via linkage analysis that MFN1 is localized on chromosome 3 at 3q25-26 and MFN2 is localized on chromosome 1 at 1p36. MFN2 has a direct role in mitochondrial transport [149] and is proposed to regulate tethering and distance between mitochondria and the endoplasmic reticulum (ER) [150], modulating Ca2+ exchange between them [151]. A loss of mitofusins is associated with reduced autophagosome formation and fusion deficiencies of autophagosomes with lysosomes [152,153], which are a vital part of mitophagy. Mitofusins are ubiquitination targets by E3 ligases at the OMM [154]. MFN1 and MFN2 are polyubiquitinated [155] and degraded [156] following PARKIN activation. Mitochondrial fragmentation is necessary for the efficiency of mitophagy [157], and oxidative stress has been shown to cause mitochondrial fragmentation by regulating MFN2 [158]. Additionally, NTG has been associated in a German patient population with one SNP (rs2111534) in MFN1 and three almost-consecutive SNPs (rs873458, rs2295281 and rs11588779) in MFN2 [76]. Furthermore, an animal study with DBA and D2G mice conducted by Nivison et al. [159] demonstrated that MFN2 accumulates selectively in RGCs during glaucomatous degeneration and identified a phosphorylated MFN2 form that selectively accumulates in RGCs, but is absent in the ON. Finally, Milanowski et al. [75] investigated polymorphisms of MFN1 and MFN2 in the Polish population and showed that genotype GA of rs2111534 MFN1 polymorphism is a risk factor for HTG, while genotype AA of this polymorphism is protective against HTG, and genotype combinations of OPA1 and MFN2 are significantly associated with a higher or a lower risk of glaucoma.
4.4. AMBRA1
AMBRA1 (autophagy and beclin 1 regulator 1; MIM#611359) is localized on chromosome 11 at 11p11.2, encoding a 1.300-amino-acid-long protein that acts as an initiator of phagophore formation in the nervous system, and its impairment leads to dysfunctional mitophagy and immoderate apoptosis [160]. Van Humbeeck et al. [161] showed that AMBRA1 interacts with PARKIN in adult mouse brain and under basal culture conditions, and PARKIN-AMBRA1 interaction increases during mitochondrial depolarization. In addition, Strappazzon et al. [162] demonstrated that AMBRA1 connects via its LC3-interacting region (LIR) motif to the autophagosome LC3 adapter after the induction of mitophagy, thereby enhancing PARKIN-mediated clearance and also regulating PARKIN-independent mitophagy. Furthermore, Di Rienzo et al. [163] proved, by using cell cultures, that AMBRA1 obstruction leads to increased apoptosis and decreased mitophagy induction, and upon mitochondrial depolarization, AMBRA1 is recruited to the OMM and connects to the ATPase family AAA domain-containing 3A (ATAD3A)-translocase of outer mitochondrial membrane 20 (TOMM20)-PTEN induced kinase 1 (PINK1) complex to enhance PINK1 aggregation. Old autophagy-impaired-AMBRA1 mice show higher RGC susceptibility [77], and heterozygous ablation of AMBRA1 leads to a remarkable reduction in RGC survivability after ischemia [78].
4.5. CAV1
CAV1 (caveolin-1; MIM#601047) is mapped to chromosome 7q31.1 [164]. Nah et al. [165] showed that phosphorylated CAV1 triggers autophagy via interaction with beclin-1 (BECN-1) under oxidative stress. On the other hand, a study reported that CAV1 negatively modulates autophagy under deprivation circumstances by adjusting lysosomal function or interacting with autophagy-related 12-autophagy-related 5 (ATG12-ATG5) complex [166], and it binds in a phosphorylation-dependent manner to dynamin-related protein 1 (DRP1) and MFN2, negatively regulating mitophagy, fission and fusion and preventing PARKIN-mediated mitophagy [167]. A large genome-wide association study (GWAS) on POAG involving 1,236 cases and 34,877 controls in Iceland identified a common sequence variant at 7q31 and then replicated the association in 2,175 POAG cases and 2,064 controls, identifying a risk variant close to CAV1 and caveolin-2 (CAV2), which are expressed in the TM and RGCs [79]. Ozel et al. [80] conducted a GWAS and meta-analysis that identified CAV1/CAV2 as a susceptibility locus for intraocular pressure. Moreover, 10 CAV1/CAV2 SNPs were significantly associated with POAG, particularly among women [81], which was supported by another study in a Caucasian US population [82]. The polymorphism rs4236601 at the CAV1-CAV2 locus was also associated with POAG and NTG in Chinese populations [83,84]. Association of rs4236601 and POAG was not detected in a Saudi cohort [168] and in a US cohort from Iowa [169].
4.6. OPTN
The OPTN gene (optineurin; MIM#602432) is localized on chromosome 10 at 10p13, encoding the protein optineurin which regulates vesicular transport [170] and NF-KB signaling [171]. Wong and Holzbaur [172] found, using live-cell microscopy, that optineurin is recruited to impaired mitochondria after PARKIN recruitment and stabilised via ubiquitin binding to ABIN and NEMO (UBAN) domain and eventually recruits LC3 via its LIR domain, leading to autophagosome formation. TBK1 phosphorylates OPTN’s UBAN domain, thereby enhancing its binding with the ubiquitin chains on mitochondria [122]. A study by Rezaie et al. [53] speculated that OPTN plays a neuroprotective role and identified OPTN mutations E50K and M98K as adult-onset glaucoma risk-associated factors. Studies using E50K transgenic animal models demonstrated that these animals experienced autophagosome–lysosome fusion impairment, age-dependent energy deficiencies, protein synthesis malfunction [85], visual impairment in contrast sensitivity [86] and retinal pathological phenotypes [87]. Furthermore, studies have shown an association of OPTN with glaucoma in Japanese [88], Indian [89,90], Chinese [91,92,93], Korean [94] and Finnish [95] patient populations. On the contrary, studies in Faroese [173], Spanish [174] and Taiwanese [175] glaucoma patients showed no association, and the last study also demonstrated a protective role of the variant c.-233+25C>G against glaucoma. Lastly, NTG patients with the OPTN E50K mutation were found to have a more severe glaucomatous phenotype with advanced optic disc cupping and smaller neuroretinal rim area at diagnosis, when compared to NTG patients without the mutation [96].
4.7. HK2
The HK2 gene (hexokinase 2; MIM#601125) is localized on chromosome 2 at 2p12, encoding HK2, a glycolytic enzyme that boosts tumor glycolysis, progression and metastasis [176]. A study by McCoy et al. [177] identified HK2 as a modifier of PARKIN recruitment to mitochondria and demonstrated the crucial role of hexokinase activity for PARKIN recruitment and subsequent mitophagy. HK2 assists in the formation of a PINK1 complex that activates the PARKIN ubiquitin ligase to promote mitochondrial ubiquitylation and recruitment of ubiquitin-binding mitophagy receptors, such as OPTN [178]. A two-stage case-control study by Shi et al. [97] that tested 669 SNPs from the region of chromosome 2 in Japanese patients showed that HK2 had significant association with POAG and NTG. Lastly, a study that investigated the relationship of the HK2 gene and NTG in a Korean cohort showed that the HK2 gene polymorphism rs678350 may contribute to NTG genetic susceptibility [98].
4.8. PARL
PARL gene (presenilin-associated rhomboid-like gene; MIM#607858), located on chromosome 3 at 3q27.1, encodes a mitochondrial protease that regulates the function of OPA1 [179] and influences apoptosis by controlling cytochrome c release via OPA1-dependent cristae remodeling [180]. PARL mediates cleavage of PINK1, and PARL knockdown is thought to lead to an amassing of ectopic PINK1 expression at the mitochondrial surface [181], PARKIN recruitment and, subsequently, mitophagy activation [182]. Two neighboring SNPs in PARL (rs1000002 and rs1402003) were found to be associated with NTG in a German population, which was confirmed by using multimarker haplotype-based association testing [76]. In a Chinese cohort of 422 primary angle-closure glaucoma (PACG) patients and 400 control subjects, one SNP (rs3749446) in PARL was found to be associated with PACG, but following a conditional analysis, it did not remain significantly associated, suggesting that the association of this SNP was not independent [99].
4.9. TP53
The TP53 gene (tumor protein p53; MIM#191170), mapped to chromosome 17 at 17p13.1 [183], encodes the transcription factor p53, whose activation promotes cell cycle arrest to permit DNA repair and/or apoptosis in order to prevent the propagation of cells with significant DNA damage [184]. P53 also regulates mitophagy through the PINK1-PARKIN pathway by inhibiting the mitochondrial translocation and activation of PARKIN, which diminishes mitophagy and leads to failure of cells to effectively remove damaged mitochondria [185]. Two studies involving Caucasian patient populations showed significant association between POAG and two SNPs (rs1042522 and rs17878362) in TP53 [100,101]. The polymorphism rs1042522 was found to be associated with POAG in two Chinese cohorts, with the C allele frequency being higher in the POAG patients than in the controls in one study [102] and the G allele being more frequent in the other [143]. Blanco-Marchite et al. [103] showed that the TP53 p.R72P polymorphism acts as a glaucoma risk factor in Spanish patients and suggested a genetic interplay between TP53 and WD repeat domain 36 (WDR36) variants in POAG susceptibility. Furthermore, TP53 was found to be associated with POAG in a cohort of 65 unrelated POAG patients in Iran [104] and with PACG in a North Indian patient cohort [105]. In a meta-analysis with subgroup analyses controlling for ethnicity, the association between the TP53 codon 72 polymorphism and POAG risk was detected in Asian populations, but not in Caucasian populations [106]. Another study involving Chinese POAG patients suggested the rs4938723 SNP in TP53 may act as a protective factor against POAG [107], while Wiggs et al. [186] proposed that the P53 codon 72 PRO/PRO genotype is associated with early paracentral visual field defects in POAG patients. Nevertheless, various studies did not demonstrate significant association between rs1042522 and POAG in Polish [187], Turkish [188], Japanese [189], Australian [190], Brazilian [191] and Indian patient populations [192].
4.10. ROCK1
ROCK1 gene (Rho-associated coiled-coil containing protein kinase 1; MIM#601702) is localized on chromosome 18 at 18q11.1 and encodes a homonymous protein that regulates myosin light chain 2 (MLC2) phosphorylation and peripheral actomyosin contraction and is consequently involved in actin cytoskeleton remodeling [193]. A recent study showed that ROCK activates several substrates including phosphatase and tensin homolog (PTEN), which is a negative regulator of PARKIN [194], and identified ROCK as a negative PARKIN-mediated mitophagy regulator [195]. ROCK1 is expressed in the TM as demonstrated by a study using TM tissue derived from monkeys [196]. A study with 363 POAG patients and 213 healthy controls in Korea showed that the SNPs rs288979, rs1006881, rs35996865, rs10083915 and rs11873284 in ROCK1 are associated with an increased risk of HTG but not with NTG [108]. The ROCK1 polymorphism rs35996865 was found to have no association with POAG in a Turkish patient population [197].
4.11. TNFα
TNFα gene (tumor necrosis factor alpha; MIM#191160) is localized in chromosome 6 at 6p21.3, encoding a multifunctional proinflammatory cytokine that belongs to the tumor necrosis factor (TNF) superfamily [198]. TNFα overexpression increases autophagy and premature senescence in malignant melanoma cells due to mitochondrial dysfunction [199]. Moreover, quantitative proteomics demonstrates that TNFα activation of macrophages induces down-regulation of mitochondrial proteins through activation of mitophagy [200]. Interestingly, TNFα upregulation has been implicated in RGC apoptosis in human glaucomatous ONH [109] and in animal glaucoma models [110,111]. TNFα was also found to be associated with POAG in a study with 60 Chinese POAG patients and 103 controls, which demonstrated that the A allele (−308G>A) frequency of the promoter SNP rs1800629 was higher in the POAG patients compared to the controls [201]. Another study with a Chinese patient population showed that the G allele frequency of the promoter SNP rs1800629 was higher in HTG patients than in control subjects, while one TNF haplotype consisting of rs1799724 and rs1800629 was also significantly associated with HTG [143]. A study with a Caucasian patient population failed to prove the association between the two promoter SNPs (−308G>A and −238G>A) of TNFα and POAG [202], and a Japanese study revealed no significant association between three promoter SNPs (−308G>A, −857C>T and −863C>A) of TNFα and POAG, despite a potential interaction between OPTN and TNFα [203].
4.12. PARKIN
The PARKIN gene (Parkin RBR E3 ubiquitin protein ligase; MIM#602544) is localized on chromosome 6 at 6q26, encoding PARKIN, a member of RBR E3 ubiquitin ligases, which ubiquitinates outer mitochondrial membrane proteins upon the depolarization of mitochondria [204]. PARKIN orchestrates the autophagic clearance of impaired mitochondria [205]. PINK1 aggregation in depolarized mitochondria leads to PINK1-mediated phosphorylation of PARKIN and ubiquitin (UB) conjugates on mitochondrial substrates in close proximity, which ultimately triggers and recruits PARKIN to these mitochondria [206]. Overexpression of PARKIN exerts protective effects against age-related function loss in tissues and confers protection against senescence in various in vitro and in vivo models [112]. Additionally, PARKIN overexpression in a human neuroblastoma SH-SY5Y cell line was found to preserve mtDNA against oxidative damage and promote mtDNA repair [207]. PARKIN overexpression also repairs the mitophagy process and reduces mitochondrial abnormalities in Alzheimer’s disease [208], and decreases cell death in human mesenchymal stromal cells exposed to neurotoxicant 6-hydroxydopamine (6-OHDA) [209]. Overexpression of PARKIN has also been shown to protect retinal ganglion cells in experimental glaucoma [19]. Aging is a major risk factor for most neurodegenerative diseases, including glaucoma, and PARKIN is thought to hold a neuroprotective role in various models of neurodegeneration. Therefore, if we thoroughly investigate the mechanisms through which PARKIN exerts positive action on age-related neurodegenerative diseases, new therapeutic avenues may be revealed.
4.13. DRP1
The DRP1 gene (dynamin-related protein 1; MIM#603850), also known as DNM1L (dynamin 1-like), is localized on chromosome 12 at 12p11.21, encoding DRP1 GTPase which facilitates mitochondrial fission [210]. Phosphorylation or dephosphorylation of serine sites S616 (phosphorylation) and S637 (dephosphorylation) regulates DRP1 activity [211]. When recruited, DRP1 creates a ring-like formation around the OMM, which undergoes GTP hydrolysis, leading to membrane compression and consequently scission [212]. DRP1 recruitment promotes the entry of Zn2+ into the mitochondrial matrix, the mitochondrial membrane potential is decreased, and, upon fission, the fragmented healthy mitochondria manage to restore their membrane potential and join the fission–fusion cycle again, whereas mitochondria that cannot accomplish this are eliminated via mitophagy [213]. Obstruction of DRP1-mediated mitochondrial fission prevents PARKIN-mediated mitophagy in HeLa cells and mouse embryonic fibroblasts (MEFs) [156]. DRP1 also facilitates mitophagy activation during hypoxia in HeLa cells [214]. Downregulation of DRP1 inhibits mitophagy, suppresses autophagosome formation, and leads to cell death at baseline and under stress conditions, such as ischemia/reperfusion in cardiomyocytes [215]. A study by Kim et al. [113] showed that oxidative stress activates mitochondrial fission and loss of RGCs in a mouse glaucoma model through an increase in DRP1 activity, and DRP1 inhibition may protect RGCs by maintaining mitochondrial integrity. Glaucomatous human ONH astrocytes with healthy N-Methyl-D-aspartic acid (NMDA) receptors show upregulation of DRP1 and its phosphorylation at serine 616 [114]. Lastly, a study by Edwards et al. [115] showed an increase in total DRP1 levels and a reduction in DRP1 phosphorylation at serine 637 (Ser637) in the retina in a mouse glaucoma model, leading to mitochondrial fragmentation and loss, as well as mitophagosome formation in RGCs.
4.14. UCP2
The UCP2 gene (uncoupling protein 2; MIM#601693), is localized in chromosome 11 at 11q13.4, encoding the mitochondrial uncoupling protein 2, which belongs to the family of mitochondrial anion carrier proteins and uncouples oxygen consumption from ATP synthesis, while also minimizing ROS emission from the electron transport chain [216]. Forte et al. [217] showed that PARKIN is upregulated in cells overexpressing UCP2 and suggested that UCP2 modulates mitophagy, since UCP2 knockdown leads to mitophagy reduction and, consequently, to the accumulation of defective mitochondria. UCP2 expression is altered during different stages of glaucoma and increases with elevated IOP, and overexpression of UCP2 in RGCs significantly reduces cell death in mice with high IOP [116]. Moreover, a study that investigated the TM of patients with neovascular glaucoma who underwent trabeculectomy demonstrated that UCP2 was significantly decreased in the TM cells of neovascular glaucoma patients compared to the control group [117].
4.15. KEAP1 and NRF2
The KEAP1 gene (Kelch-like ECH-associated protein 1; MIM#606016) is mapped to chromosome 19 at 19p13.2, encoding the homonymous protein, and the NRF2 gene (NF-E2-related factor 2; #MIM600492) is localized in chromosome 2 at 2q31.2, encoding the transcription factor NRF2. Both KEAP1 and NRF2 are major regulators of redox homeostasis [218]. KEAP1 binds NRF2 through its C-terminal Kelch domain [219]. Two KEAP1 molecules and one NRF2 molecule form a trimer, and KEAP1 regulates the activity, stability and accumulation of NRF2 [220]. The KEAP-NRF2 stress response pathway is the main inducible defense against oxidative stress insults [221]. Zeb et al. [222] showed that KEAP1 is involved in the regulation of the PINK1-PARKIN pathway by ROS and ROS-induced mitophagy, since overexpression of KEAP1 blocks PARKIN translocation from the cytosol to mitochondria and KEAP1 silencing enhances mitophagy. Murata et al. [223] showed that NRF2 upregulates the PINK1 gene (Figure 2) as a response to oxidative stress conditions. Lastly, a study by Naguib et al. [118] suggested that oxidative stress is an early event in the pathogenesis of glaucoma and revealed an antioxidant response mediated by the NRF2-KEAP1 pathway, which is activated by NRF2 phosphorylation.
5. Mitophagy as a Therapeutic Target
Since impaired mitophagy is implicated in glaucomatous degeneration, as described previously, modulating mitophagy may prove to be an important strategy to treat glaucoma. Reduction in IOP and higher aqueous humor outflow through the TM are achieved by ROCK inhibitors, as demonstrated in animal models [224]. In a discovery pipeline study using thousands of compounds to identify small molecules that increase PARKIN recruitment to damaged mitochondria and ensuing mitophagy, ROCK inhibitors (such as SR3677) were found to trigger mitophagy through increased HK2 activation in flies exposed to a parkinsonian toxin that induces mitochondrial damage [195]. Apart from increasing HK2 activity to promote mitophagy, ROCK inhibitors may hold a neuroprotective role by inhibiting the release of inflammatory cytokines in microglial cells [225]. In addition, ROCK inhibitors promote RGC regeneration and survival as demonstrated in various optic nerve injury models when delivered intravitreally [226,227], but they also reduce RGC loss and promote axonal regeneration when administered topically [228]. In a randomized phase II clinical trial against latanoprost, the ROCK inhibitor Netarsudil 0.02% demonstrated significant reductions of 5.7 mmHg in IOP after four weeks of dosing [229]. The FDA approved Netarsudil for clinical use in 2017 [230].
In a rat glaucoma model, adeno-associated virus 2-PARKIN (AAV2-PARKIN), which was used to overexpress PARKIN, led to a significant decrease in RGC loss and partial restoration of mitophagy levels under elevated IOP conditions, suggesting that clearance of impaired mitochondria through mitophagy is directly linked to better RGC survival [22]. Fucoxanthin, a carotenoid of the xanthophyll family with antioxidant and anti-inflammatory properties, also exerts protective effects on RGCs by enhancing PARKIN-mediated mitophagy under glutamate excitotoxicity [231]. Interestingly, a chronic ocular hypertension rat glaucoma model showed that during short-term IOP elevations, fucoxanthin decreases PARKIN expression and reduces the number of mitophagosomes and autophagosomes to prevent excessive damage caused by unrestrained mitophagy, while during long-term IOP elevations, fucoxanthin protects RGCs, improves mitochondrial health, increases PARKIN expression and enhances the PARKIN-mediated mitophagy pathway [232].A recent study by Zhuang et al. [233] showed that the small natural molecule S3 derived from diterpenoids could protect RGCs by enhancing PARKIN-mediated mitophagy under NMDA-induced excitotoxicity. Metformin, an FDA-approved mammalian target of rapamycin (mTOR) inhibitor, stimulates mitophagy by restoring PARKIN-mediated mechanisms and mTOR-dependent autophagy initiation [234], is associated with a reduced risk of OAG development in people with diabetes [235], and has been correlated with a reduced risk of OAG that could not be explained by IOP [236].
Hu et al. [21] injected hypertensive glaucoma model rats with the adeno-associated virus 2-OPA1 (AAV2-OPA1) and reported healthier mitochondria in the optic nerve RGCs, bigger mitochondrial surface area and enhanced mitochondrial fusion. Overexpression of OPA1 increased PARKIN expression, induced PARKIN-mediated mitophagy and protected RGCs. Treatment with memantine, an NMDA glutamate receptor antagonist, promoted RGC survival in pre-glaucomatous mice by enhancing OPA1 expression, reducing DNM1 (a mouse homologue of DRP1) expression, and inhibiting OPA and cytochrome c release from mitochondria [237]. Two randomized double-masked, placebo-controlled, multicenter phase 3 clinical studies that lasted 48 months showed no benefit of orally administered memantine [238].
Moreover, an animal study by Hass and Barnstable [23] showed that deletion of mitochondrial uncoupling protein 2 (UCP2), despite uncoupling the electron transport chain from ATP synthase activity and increasing ROS production, promotes mitophagy in cortical astrocyte cultures and retinal tissue, and ultimately decreases RGC loss, in male and female mice. This demonstrates a potential neuroprotective role and pharmacological compounds that inhibit UCP2 may provide a novel therapeutic approach.
P62-mediated mitophagy inducer (PMI) is another promising neuroprotective agent that inhibits KEAP1 and activates NF-E2-related factor 2 (NRF2), leading to the expression of antioxidant genes, including sequestosome 1 (p62/SQSTM1), and enhanced mitophagy [239]. DRP1 may also serve as an auspicious target for a potential antibody-based therapy for glaucoma, since intravitreal injection of an anti-DRP1 antibody in an animal glaucoma model protected RGCs, decreased apoptosis as shown using western blot analysis, and improved retinal functionality as observed using electroretinography [240].
Coenzyme Q10 (CoQ10) is an essential component for the transport of electrons in mitochondria, and electron microscopy studies on fibroblasts from patients with CoQ10 deficiency showed increased lysosomal markers and mitochondrial degradation via mitophagy [241]. Using a retinal ischemia/reperfusion animal model, Nucci et al. [242] demonstrated that intraocular CoQ10 administration reduces glutamate increase and affords neuroprotection. A retrospective study by Alpogan et al. [243] evaluated the impact of CoQ10 eye drops combined with vitamin E on the retinal nerve fiber layer (RNFL) and ganglion cell complex (GCC) thickness in patients with OAG and showed a statistically significant beneficial effect of CoQ10 on the RNFL. Moreover, Parisi et al. [244] showed an improvement in inner retinal function using pattern electroretinogram and in visual evoked potential amplitudes in patients with OAG who were treated with CoQ10 and vitamin E eye drops over 12 months.
Pharmacological regulation of mitophagy beyond ophthalmology attracts great interest, as mitophagy holds a significant role in highly prevalent chronic diseases [63]. Bhansali et al. [245,246] showed that metformin promotes mitophagy by enhancing important mitophagy genes, such as PINK1, and preserves mitochondrial health in mononuclear cells of type 2 diabetes patients. Ursolic and oleanolic acids have also been suggested to have anti-tumorigenic effects that are dependent on their mitophagy-enhancing properties [247]. Finally, gerontoxanthone I and macluraxanthone have been shown to regulate mitophagy through the PINK1-PARKIN pathway and are thought to exert protective effects against myocardial ischemia-reperfusion injury [248].
6. Discussion/Conclusions
It is widely accepted that glaucoma is likely caused by the interactions between multiple genes and environmental factors [249]. Current IOP-lowering therapeutic approaches, as indicated by clinical studies, fail to adequately control glaucoma progression in the long term in a significant number of patients. Emerging evidence from genetic and experimental studies reveals an increasing number of genes involved in mitophagy that may contribute to glaucoma susceptibility. Mitophagy-related processes and pathways may act as targets for novel gene-based glaucoma therapies and pharmacological interventions. Understanding the mechanisms that confer resistance to glaucoma progression would be critical to enable the development of a genetic or biochemical test that may identify glaucoma patients with mitophagy defects who are at risk of progression and/or more likely to respond to treatment, thus promoting the field of personalized medicine in glaucoma. It is important to note that apart from single genes and single-gene variants, gene–gene and gene–environment interactions should be further investigated in pursuance of more thorough knowledge on the subject. This review presents the increasing body of evidence that mitochondrial dysfunction and mitophagy-related genetic pathways may contribute to glaucoma pathogenesis and therapeutics.
Author Contributions
Conceptualization and writing of the original draft, D.S. and G.L.; reference collection, table configuration and editing, D.S.; figure illustration, D.S.; reviewing, writing and editing manuscript, G.L., M.K.G., B.P., K.-Y.C., C.J.H. and D.F.G.-H.; supervision, G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAV2 | adeno-associated virus 2 |
| AMBRA1 | autophagy and beclin 1 regulator 1 |
| ATAD3A | ATPase family AAA domain-containing 3A |
| ATG5 | autophagy-related 5 |
| ATG12 | autophagy-related 12 |
| ATP | adenosine triphosphate |
| BECN-1 | beclin-1 |
| BNIP3 | BCL2-interacting protein 3 |
| CAV1 | caveolin-1 |
| CAV2 | caveolin-2 |
| CoQ10 | coenzyme Q10 |
| DNM1L | dynamin 1-like |
| DOA | dominant optic atrophy |
| DRP1 | dynamin-related protein 1 |
| ER | endoplasmic reticulum |
| GCC | ganglion cell complex |
| GTP | guanosine triphosphate |
| GWAS | genome-wide association study |
| HK2 | hexokinase 2 |
| HTG | high-tension glaucoma |
| IMM | inner mitochondrial membrane |
| IOP | intraocular pressure |
| iPSC | induced pluripotent stem cells |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| LC3 | microtubule-associated protein 1A/1B-light chain 3 |
| LIR | LC3-interacting region |
| MEFs | mouse embryonic fibroblasts |
| MFN1 | mitofusin 1 |
| MFN2 | mitofusin 2 |
| MLC2 | myosin light chain 2 |
| MPTP | mitochondrial permeability transition pore |
| mtDNA | mitochondrial DNA |
| mTOR | mammalian target of rapamycin |
| NAD+ | nicotinamide adenine dinucleotide |
| NDP52/CALCOCO2 | calcium-binding and coiled-coil domain 2 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NMDA | N-Methyl-D-aspartic acid |
| NRF2 | NF-E2-related factor 2 |
| NTG | normal-tension glaucoma |
| OAG | open-angle glaucoma |
| OCR | oxygen consumption rate |
| OMM | outer mitochondrial membrane |
| ON | optic nerve |
| ONH | optic nerve head |
| OPA1 | optic atrophy-1 |
| OPTN | optineurin |
| OXPHOS | oxidative phosphorylation |
| PACG | primary angle-closure glaucoma |
| PARKIN | Parkin RBR E3 ubiquitin protein ligase |
| PARL | presenilin-associated rhomboid-like |
| PINK1 | PTEN-induced kinase 1 |
| PMI | P62-mediated mitophagy inducer |
| POAG | primary open-angle glaucoma |
| RNFL | retinal nerve fiber layer |
| RGCs | retinal ganglion cells |
| ROCK1 | Rho-associated coiled-coil containing protein kinase 1 |
| ROS | reactive oxygen species |
| 6-OHDA | 6-hydroxydopamine |
| SNP | single-nucleotide polymorphism |
| SQSTM1/p62 | sequestosome-1 |
| TAX1BP1 | tax1-binding protein 1 |
| TBK1 | TANK-binding kinase 1 |
| TM | trabecular meshwork |
| TNFα | tumor necrosis factor alpha |
| TOMM20 | translocase of outer mitochondrial membrane 20 |
| TP53 | tumor protein p53 |
| UB | ubiquitin |
| UBAN | ubiquitin binding in ABIN and NEMO |
| UCP2 | uncoupling protein 2 |
| WDR36 | WD repeat domain 36 |
References
- GBD 2019 Blindness and Vision Impairment Collaborators; Vision Loss Expert Group of the Global Burden of Disease Study. Causes of Blindness and Vision Impairment in 2020 and Trends over 30 Years, and Prevalence of Avoidable Blindness in Relation to VISION 2020: The Right to Sight: An Analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Quartilho, A.; Simkiss, P.; Zekite, A.; Xing, W.; Wormald, R.; Bunce, C. Leading Causes of Certifiable Visual Loss in England and Wales during the Year Ending 31 March 2013. Eye 2016, 30, 602–607. [Google Scholar] [CrossRef]
- Kang, J.M.; Tanna, A.P. Glaucoma. Med. Clin. N. Am. 2021, 105, 493–510. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Dunkelberger, G.R.; Green, W.R. Chronic Human Glaucoma Causing Selectively Greater Loss of Large Optic Nerve Fibers. Ophthalmology 1988, 95, 357–363. [Google Scholar] [CrossRef]
- Ju, W.-K.; Perkins, G.A.; Kim, K.-Y.; Bastola, T.; Choi, W.-Y.; Choi, S.-H. Glaucomatous Optic Neuropathy: Mitochondrial Dynamics, Dysfunction and Protection in Retinal Ganglion Cells. Prog. Retin. Eye Res. 2022, 95, 101136. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Ganglion Cell Death in Glaucoma: Pathology Recapitulates Ontogeny. Aust. N. Z. J. Ophthalmol. 1995, 23, 85–91. [Google Scholar] [CrossRef]
- Begg, I.S.; Drance, S.M. Progress of the Glaucomatous Process Related to Recurrent Ischaemic Changes at the Optic Disc. Exp. Eye Res. 1971, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Heijl, A.; Leske, M.C.; Bengtsson, B.; Hyman, L.; Bengtsson, B.; Hussein, M.; Early Manifest Glaucoma Trial Group. Reduction of Intraocular Pressure and Glaucoma Progression: Results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002, 120, 1268–1279. [Google Scholar] [CrossRef]
- Collaborative Normal-Tension Glaucoma Study Group. The Effectiveness of Intraocular Pressure Reduction in the Treatment of Normal-Tension Glaucoma. Am. J. Ophthalmol. 1998, 126, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Garway-Heath, D.F.; Crabb, D.P.; Bunce, C.; Lascaratos, G.; Amalfitano, F.; Anand, N.; Azuara-Blanco, A.; Bourne, R.R.; Broadway, D.C.; Cunliffe, I.A.; et al. Latanoprost for Open-Angle Glaucoma (UKGTS): A Randomised, Multicentre, Placebo-Controlled Trial. Lancet Lond. Engl. 2015, 385, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Coleman, A.L. Glaucoma. Lancet Lond. Engl. 1999, 354, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Canadian Glaucoma Study Group Canadian Glaucoma Study: 1. Study Design, Baseline Characteristics, and Preliminary Analyses. Can. J. Ophthalmol. J. Can. Ophtalmol. 2006, 41, 566–575. [Google Scholar] [CrossRef]
- Heijl, A.; Buchholz, P.; Norrgren, G.; Bengtsson, B. Rates of Visual Field Progression in Clinical Glaucoma Care. Acta Ophthalmol. 2013, 91, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Q.; Xie, Z.; Chen, S.-Y.; Zhang, X. Mitochondrial Dysfunction in Glaucomatous Degeneration. Int. J. Ophthalmol. 2023, 16, 811–823. [Google Scholar] [CrossRef]
- Mittag, T.W.; Danias, J.; Pohorenec, G.; Yuan, H.M.; Burakgazi, E.; Chalmers-Redman, R.; Podos, S.M.; Tatton, W.G. Retinal Damage after 3 to 4 Months of Elevated Intraocular Pressure in a Rat Glaucoma Model. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3451–3459. [Google Scholar]
- Murphy, M.P. Mitochondrial Dysfunction Indirectly Elevates ROS Production by the Endoplasmic Reticulum. Cell Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial Dysfunction in Glaucoma and Emerging Bioenergetic Therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial Dysfunction in Ocular Disease: Focus on Glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar] [CrossRef]
- Kong, G.Y.X.; Van Bergen, N.J.; Trounce, I.A.; Crowston, J.G. Mitochondrial Dysfunction and Glaucoma. J. Glaucoma 2009, 18, 93–100. [Google Scholar] [CrossRef]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dai, Y.; Zhang, R.; Shang, K.; Sun, X. Overexpression of Optic Atrophy Type 1 Protects Retinal Ganglion Cells and Upregulates Parkin Expression in Experimental Glaucoma. Front. Mol. Neurosci. 2018, 11, 350. [Google Scholar] [CrossRef]
- Dai, Y.; Hu, X.; Sun, X. Overexpression of Parkin Protects Retinal Ganglion Cells in Experimental Glaucoma. Cell Death Dis. 2018, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Hass, D.T.; Barnstable, C.J. Mitochondrial Uncoupling Protein 2 Knock-out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 3582–3596. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucci, A.; Nucci, C. Evidence on Neuroprotective Properties of Coenzyme Q10 in the Treatment of Glaucoma. Neural Regen. Res. 2019, 14, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C. Mitochondria: Dynamic Organelles in Disease, Aging, and Development. Cell 2006, 125, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Casson, R.J.; Chidlow, G.; Crowston, J.G.; Williams, P.A.; Wood, J.P.M. Retinal Energy Metabolism in Health and Glaucoma. Prog. Retin. Eye Res. 2021, 81, 100881. [Google Scholar] [CrossRef]
- Li, Y.; Li, D.; Ying, X.; Khaw, P.T.; Raisman, G. An Energy Theory of Glaucoma. Glia 2015, 63, 1537–1552. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Trounce, I.A.; Crowston, J.G. Mechanisms of Retinal Ganglion Cell Injury in Aging and Glaucoma. Ophthalmic Res. 2010, 44, 173–178. [Google Scholar] [CrossRef]
- Pinazo-Durán, M.D.; Zanón-Moreno, V.; Gallego-Pinazo, R.; García-Medina, J.J. Oxidative Stress and Mitochondrial Failure in the Pathogenesis of Glaucoma Neurodegeneration. Prog. Brain Res. 2015, 220, 127–153. [Google Scholar] [CrossRef]
- Yang, X.-J.; Ge, J.; Zhuo, Y.-H. Role of Mitochondria in the Pathogenesis and Treatment of Glaucoma. Chin. Med. J. 2013, 126, 4358–4365. [Google Scholar] [PubMed]
- Tezel, G.; Yang, X. Caspase-Independent Component of Retinal Ganglion Cell Death, in Vitro. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4049–4059. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, K.; Zhang, H.; Zhao, J.; Zhu, X.; Wang, Y.; Wu, R. Retinal Ganglion Cell Death Is Triggered by Paraptosis via Reactive Oxygen Species Production: A Brief Literature Review Presenting a Novel Hypothesis in Glaucoma Pathology. Mol. Med. Rep. 2014, 10, 1179–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, N.N. Pathogenesis of Ganglion “Cell Death” in Glaucoma and Neuroprotection: Focus on Ganglion Cell Axonal Mitochondria. Prog. Brain Res. 2008, 173, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-C.; Huang, B.-Q.; Yang, J. Regulatory Mechanisms of Retinal Ganglion Cell Death in Normal Tension Glaucoma and Potential Therapies. Neural Regen. Res. 2023, 18, 87–93. [Google Scholar] [CrossRef]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- He, Y.; Ge, J.; Tombran-Tink, J. Mitochondrial Defects and Dysfunction in Calcium Regulation in Glaucomatous Trabecular Meshwork Cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4912–4922. [Google Scholar] [CrossRef]
- Osborne, N.N.; Lascaratos, G.; Bron, A.J.; Chidlow, G.; Wood, J.P.M. A Hypothesis to Suggest That Light Is a Risk Factor in Glaucoma and the Mitochondrial Optic Neuropathies. Br. J. Ophthalmol. 2006, 90, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Osborne, N.N.; Li, G.-Y.; Ji, D.; Mortiboys, H.J.; Jackson, S. Light Affects Mitochondria to Cause Apoptosis to Cultured Cells: Possible Relevance to Ganglion Cell Death in Certain Optic Neuropathies. J. Neurochem. 2008, 105, 2013–2028. [Google Scholar] [CrossRef]
- Vallabh, N.A.; Armstrong, J.; Czanner, G.; McDonagh, B.; Choudhary, A.; Criddle, D.N.; Willoughby, C.E. Evidence of Impaired Mitochondrial Cellular Bioenergetics in Ocular Fibroblasts Derived from Glaucoma Patients. Free Radic. Biol. Med. 2022, 189, 102–110. [Google Scholar] [CrossRef]
- Lascaratos, G.; Chau, D.; Zhu, H.; Schapira, A.; Garway-Heath, D. Mitochondrial Membrane Potential, as a Measure of Mitochondrial Function, in Patients with Normal Tension Glaucoma vs Ocular Hypertension. Investig. Ophthalmol. Vis. Sci. 2013, 54, 1604. [Google Scholar]
- Petriti, B.; Chau, D.; Lascaratos, G.; Campbell, P.; Lazaridis, G.; Garway-Heath, D.F. Normal Tension Glaucoma Patients Have Reduced Systemic Mitochondrial Function Compared to High Tension Glaucoma Patients. Investig. Ophthalmol. Vis. Sci. 2020, 61, 1009. [Google Scholar]
- Tribble, J.R.; Vasalauskaite, A.; Redmond, T.; Young, R.D.; Hassan, S.; Fautsch, M.P.; Sengpiel, F.; Williams, P.A.; Morgan, J.E. Midget Retinal Ganglion Cell Dendritic and Mitochondrial Degeneration Is an Early Feature of Human Glaucoma. Brain Commun. 2019, 1, fcz035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative Stress, Mitochondrial Dysfunction and Calcium Overload in Human Lamina Cribrosa Cells from Glaucoma Donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar]
- Izzotti, A.; Longobardi, M.; Cartiglia, C.; Saccà, S.C. Mitochondrial Damage in the Trabecular Meshwork Occurs Only in Primary Open-Angle Glaucoma and in Pseudoexfoliative Glaucoma. PLoS ONE 2011, 6, e14567. [Google Scholar] [CrossRef] [Green Version]
- Kimball, E.C.; Jefferys, J.L.; Pease, M.E.; Oglesby, E.N.; Nguyen, C.; Schaub, J.; Pitha, I.; Quigley, H.A. The Effects of Age on Mitochondria, Axonal Transport, and Axonal Degeneration after Chronic IOP Elevation Using a Murine Ocular Explant Model. Exp. Eye Res. 2018, 172, 78–85. [Google Scholar] [CrossRef]
- Munemasa, Y.; Kitaoka, Y.; Kuribayashi, J.; Ueno, S. Modulation of Mitochondria in the Axon and Soma of Retinal Ganglion Cells in a Rat Glaucoma Model. J. Neurochem. 2010, 115, 1508–1519. [Google Scholar] [CrossRef]
- Kimball, E.C.; Pease, M.E.; Steinhart, M.R.; Oglesby, E.N.; Pitha, I.; Nguyen, C.; Quigley, H.A. A Mouse Ocular Explant Model That Enables the Study of Living Optic Nerve Head Events after Acute and Chronic Intraocular Pressure Elevation: Focusing on Retinal Ganglion Cell Axons and Mitochondria. Exp. Eye Res. 2017, 160, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Harder, J.M.; Foxworth, N.E.; Cochran, K.E.; Philip, V.M.; Porciatti, V.; Smithies, O.; John, S.W.M. Vitamin B3 Modulates Mitochondrial Vulnerability and Prevents Glaucoma in Aged Mice. Science 2017, 355, 756–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lascaratos, G.; Garway-Heath, D.F.; Willoughby, C.E.; Chau, K.-Y.; Schapira, A.H.V. Mitochondrial Dysfunction in Glaucoma: Understanding Genetic Influences. Mitochondrion 2012, 12, 202–212. [Google Scholar] [CrossRef]
- Sears, N.C.; Boese, E.A.; Miller, M.A.; Fingert, J.H. Mendelian Genes in Primary Open Angle Glaucoma. Exp. Eye Res. 2019, 186, 107702. [Google Scholar] [CrossRef] [PubMed]
- Khawaja, A.P.; Cooke Bailey, J.N.; Kang, J.H.; Allingham, R.R.; Hauser, M.A.; Brilliant, M.; Budenz, D.L.; Christen, W.G.; Fingert, J.; Gaasterland, D.; et al. Assessing the Association of Mitochondrial Genetic Variation with Primary Open-Angle Glaucoma Using Gene-Set Analyses. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5046–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezaie, T.; Child, A.; Hitchings, R.; Brice, G.; Miller, L.; Coca-Prados, M.; Héon, E.; Krupin, T.; Ritch, R.; Kreutzer, D.; et al. Adult-Onset Primary Open-Angle Glaucoma Caused by Mutations in Optineurin. Science 2002, 295, 1077–1079. [Google Scholar] [CrossRef]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited Mitochondrial Optic Neuropathies. J. Med. Genet. 2009, 46, 145–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, E.C.; Danesh-Meyer, H.V.; Kong, G.X.Y.; Hewitt, A.W.; Coote, M.A.; Mackey, D.A.; Crowston, J.G.; Optic Nerve Study Group. Optic Disc Evaluation in Optic Neuropathies: The Optic Disc Assessment Project. Ophthalmology 2011, 118, 964–970. [Google Scholar] [CrossRef]
- Boya, P.; Esteban-Martínez, L.; Serrano-Puebla, A.; Gómez-Sintes, R.; Villarejo-Zori, B. Autophagy in the Eye: Development, Degeneration, and Aging. Prog. Retin. Eye Res. 2016, 55, 206–245. [Google Scholar] [CrossRef]
- Wang, X.-L.; Feng, S.-T.; Wang, Z.-Z.; Chen, N.-H.; Zhang, Y. Role of Mitophagy in Mitochondrial Quality Control: Mechanisms and Potential Implications for Neurodegenerative Diseases. Pharmacol. Res. 2021, 165, 105433. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic Regulation of Mitochondrial Dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef]
- Um, J.-H.; Yun, J. Emerging Role of Mitophagy in Human Diseases and Physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Zeb, A.; Kaasik, A. Molecular Mechanisms and Regulation of Mammalian Mitophagy. Cells 2021, 11, 38. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Hamasaki, N. Alterations of Mitochondrial DNA in Common Diseases and Disease States: Aging, Neurodegeneration, Heart Failure, Diabetes, and Cancer. Curr. Med. Chem. 2005, 12, 429–441. [Google Scholar] [CrossRef]
- Doblado, L.; Lueck, C.; Rey, C.; Samhan-Arias, A.K.; Prieto, I.; Stacchiotti, A.; Monsalve, M. Mitophagy in Human Diseases. Int. J. Mol. Sci. 2021, 22, 3903. [Google Scholar] [CrossRef] [PubMed]
- Skeie, J.M.; Nishimura, D.Y.; Wang, C.L.; Schmidt, G.A.; Aldrich, B.T.; Greiner, M.A. Mitophagy: An Emerging Target in Ocular Pathology. Investig. Ophthalmol. Vis. Sci. 2021, 62, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, Y.W.; Jeoung, J.W.; Seong, M.-W.; Lee, J.-S.; Kim, D.M.; Ahn, J.H.; Song, J.Y.; Cho, S.I.; Park, S.S.; et al. Genomic Characterization of TBK1 Duplication in Korean Normal-Tension Glaucoma Patients. J. Glaucoma 2020, 29, 331–336. [Google Scholar] [CrossRef]
- Kawase, K.; Allingham, R.R.; Meguro, A.; Mizuki, N.; Roos, B.; Solivan-Timpe, F.M.; Robin, A.L.; Ritch, R.; Fingert, J.H. Confirmation of TBK1 Duplication in Normal Tension Glaucoma. Exp. Eye Res. 2012, 96, 178–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritch, R.; Darbro, B.; Menon, G.; Khanna, C.L.; Solivan-Timpe, F.; Roos, B.R.; Sarfarzi, M.; Kawase, K.; Yamamoto, T.; Robin, A.L.; et al. TBK1 Gene Duplication and Normal-Tension Glaucoma. JAMA Ophthalmol. 2014, 132, 544–548. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Garrett, M.E.; Yaspan, B.L.; Bailey, J.C.; Loomis, S.J.; Brilliant, M.; Budenz, D.L.; Christen, W.G.; Fingert, J.H.; Gaasterland, D.; et al. DNA Copy Number Variants of Known Glaucoma Genes in Relation to Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 8251–8258. [Google Scholar] [CrossRef] [Green Version]
- Fingert, J.H.; Robin, A.L.; Stone, J.L.; Roos, B.R.; Davis, L.K.; Scheetz, T.E.; Bennett, S.R.; Wassink, T.H.; Kwon, Y.H.; Alward, W.L.M.; et al. Copy Number Variations on Chromosome 12q14 in Patients with Normal Tension Glaucoma. Hum. Mol. Genet. 2011, 20, 2482–2494. [Google Scholar] [CrossRef] [Green Version]
- Fingert, J.H.; Robin, A.L.; Scheetz, T.E.; Kwon, Y.H.; Liebmann, J.M.; Ritch, R.; Alward, W.L.M. Tank-Binding Kinase 1 (TBK1) Gene and Open-Angle Glaucomas (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2016, 114, T6. [Google Scholar]
- Fingert, J.H.; Miller, K.; Hedberg-Buenz, A.; Roos, B.R.; Lewis, C.J.; Mullins, R.F.; Anderson, M.G. Transgenic TBK1 Mice Have Features of Normal Tension Glaucoma. Hum. Mol. Genet. 2017, 26, 124–132. [Google Scholar] [CrossRef]
- Bosley, T.M.; Hellani, A.; Spaeth, G.L.; Myers, J.; Katz, L.J.; Moster, M.R.; Milcarek, B.; Abu-Amero, K.K. Down-Regulation of OPA1 in Patients with Primary Open Angle Glaucoma. Mol. Vis. 2011, 17, 1074–1079. [Google Scholar]
- Aung, T.; Ocaka, L.; Ebenezer, N.D.; Morris, A.G.; Krawczak, M.; Thiselton, D.L.; Alexander, C.; Votruba, M.; Brice, G.; Child, A.H.; et al. A Major Marker for Normal Tension Glaucoma: Association with Polymorphisms in the OPA1 Gene. Hum. Genet. 2002, 110, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Stewart, J.D.; Hudson, G.; Andrews, R.M.; Griffiths, P.G.; Birch, M.K.; Chinnery, P.F. OPA1 Increases the Risk of Normal but Not High Tension Glaucoma. J. Med. Genet. 2010, 47, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanowski, P.; Kosior-Jarecka, E.; Łukasik, U.; Wróbel-Dudzińska, D.; Milanowska, J.; Khor, C.C.; Aung, T.; Kocki, J.; Żarnowski, T. Associations between OPA1, MFN1, and MFN2 Polymorphisms and Primary Open Angle Glaucoma in Polish Participants of European Ancestry. Ophthalmic Genet. 2022, 43, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Gramer, E.; Müller-Myhsok, B.; Pasutto, F.; Reinthal, E.; Wissinger, B.; Weisschuh, N. Evaluation of Nine Candidate Genes in Patients with Normal Tension Glaucoma: A Case Control Study. BMC Med. Genet. 2009, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Bell, K.; Rosignol, I.; Sierra-Filardi, E.; Rodriguez-Muela, N.; Schmelter, C.; Cecconi, F.; Grus, F.; Boya, P. Age Related Retinal Ganglion Cell Susceptibility in Context of Autophagy Deficiency. Cell Death Discov. 2020, 6, 21. [Google Scholar] [CrossRef]
- Russo, R.; Varano, G.P.; Adornetto, A.; Nazio, F.; Tettamanti, G.; Girardello, R.; Cianfanelli, V.; Cavaliere, F.; Morrone, L.A.; Corasaniti, M.T.; et al. Rapamycin and Fasting Sustain Autophagy Response Activated by Ischemia/Reperfusion Injury and Promote Retinal Ganglion Cell Survival. Cell Death Dis. 2018, 9, 981. [Google Scholar] [CrossRef] [Green Version]
- Thorleifsson, G.; Walters, G.B.; Hewitt, A.W.; Masson, G.; Helgason, A.; DeWan, A.; Sigurdsson, A.; Jonasdottir, A.; Gudjonsson, S.A.; Magnusson, K.P.; et al. Common Variants near CAV1 and CAV2 Are Associated with Primary Open-Angle Glaucoma. Nat. Genet. 2010, 42, 906–909. [Google Scholar] [CrossRef] [Green Version]
- Ozel, A.B.; Moroi, S.E.; Reed, D.M.; Nika, M.; Schmidt, C.M.; Akbari, S.; Scott, K.; Rozsa, F.; Pawar, H.; Musch, D.C.; et al. Genome-Wide Association Study and Meta-Analysis of Intraocular Pressure. Hum. Genet. 2014, 133, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Loomis, S.J.; Kang, J.H.; Weinreb, R.N.; Yaspan, B.L.; Cooke Bailey, J.N.; Gaasterland, D.; Gaasterland, T.; Lee, R.K.; Lichter, P.R.; Budenz, D.L.; et al. Association of CAV1/CAV2 Genomic Variants with Primary Open-Angle Glaucoma Overall and by Gender and Pattern of Visual Field Loss. Ophthalmology 2014, 121, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L.; Kang, J.H.; Yaspan, B.L.; Mirel, D.B.; Laurie, C.; Crenshaw, A.; Brodeur, W.; Gogarten, S.; Olson, L.M.; Abdrabou, W.; et al. Common Variants near CAV1 and CAV2 Are Associated with Primary Open-Angle Glaucoma in Caucasians from the USA. Hum. Mol. Genet. 2011, 20, 4707–4713. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.Y.; Rong, S.S.; Wu, Z.; Huang, C.; Matsushita, K.; Ng, T.K.; Leung, C.K.S.; Kawashima, R.; Usui, S.; Tam, P.O.S.; et al. Association of the CAV1-CAV2 Locus with Normal-Tension Glaucoma in Chinese and Japanese. Clin. Experiment. Ophthalmol. 2020, 48, 658–665. [Google Scholar] [CrossRef]
- Rong, S.S.; Chen, L.J.; Leung, C.K.S.; Matsushita, K.; Jia, L.; Miki, A.; Chiang, S.W.Y.; Tam, P.O.S.; Hashida, N.; Young, A.L.; et al. Ethnic Specific Association of the CAV1/CAV2 Locus with Primary Open-Angle Glaucoma. Sci. Rep. 2016, 6, 27837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, Q.; Shao, Z.; Zhang, S.; Hou, M.; Jiang, M.; Du, M.; Li, J.; Yuan, H. Proteomic Analysis of Aged and OPTN E50K Retina in the Development of Normal Tension Glaucoma. Hum. Mol. Genet. 2021, 30, 1030–1044. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.C.; Riday, T.T.; McKee, C.; Braine, C.E.; Bomze, H.; Barak, I.; Marean-Reardon, C.; John, S.W.M.; Philpot, B.D.; Ehlers, M.D. Visual Impairment in an Optineurin Mouse Model of Primary Open-Angle Glaucoma. Neurobiol. Aging 2015, 36, 2201–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, Z.-L.; Akahori, M.; Obazawa, M.; Minami, M.; Noda, T.; Nakaya, N.; Tomarev, S.; Kawase, K.; Yamamoto, T.; Noda, S.; et al. Overexpression of Optineurin E50K Disrupts Rab8 Interaction and Leads to a Progressive Retinal Degeneration in Mice. Hum. Mol. Genet. 2010, 19, 2606–2615. [Google Scholar] [CrossRef] [Green Version]
- Funayama, T.; Mashima, Y.; Ohtake, Y.; Ishikawa, K.; Fuse, N.; Yasuda, N.; Fukuchi, T.; Murakami, A.; Hotta, Y.; Shimada, N.; et al. SNPs and Interaction Analyses of Noelin 2, Myocilin, and Optineurin Genes in Japanese Patients with Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 5368–5375. [Google Scholar] [CrossRef]
- Sripriya, S.; Nirmaladevi, J.; George, R.; Hemamalini, A.; Baskaran, M.; Prema, R.; Ve Ramesh, S.; Karthiyayini, T.; Amali, J.; Job, S.; et al. OPTN Gene: Profile of Patients with Glaucoma from India. Mol. Vis. 2006, 12, 816–820. [Google Scholar]
- Kumar, A.; Basavaraj, M.G.; Gupta, S.K.; Qamar, I.; Ali, A.M.; Bajaj, V.; Ramesh, T.K.; Prakash, D.R.; Shetty, J.S.; Dorairaj, S.K. Role of CYP1B1, MYOC, OPTN, and OPTC Genes in Adult-Onset Primary Open-Angle Glaucoma: Predominance of CYP1B1 Mutations in Indian Patients. Mol. Vis. 2007, 13, 667–676. [Google Scholar]
- Yao, H.; Cheng, C.; Fan, B.; Tam, O.; Tham, C.; Wang, D.; Lam, S.; Pang, C. Polymorphisms of myocilin and optineurin in primary open angle glaucoma patients. Zhonghua Yi Xue Za Zhi 2006, 86, 554–559. [Google Scholar] [PubMed]
- Fan, B.J.; Wang, D.Y.; Fan, D.S.P.; Tam, P.O.S.; Lam, D.S.C.; Tham, C.C.Y.; Lam, C.Y.; Lau, T.C.; Pang, C.P. SNPs and Interaction Analyses of Myocilin, Optineurin, and Apolipoprotein E in Primary Open Angle Glaucoma Patients. Mol. Vis. 2005, 11, 625–631. [Google Scholar] [PubMed]
- Huang, C.; Xie, L.; Wu, Z.; Cao, Y.; Zheng, Y.; Pang, C.-P.; Zhang, M. Detection of Mutations in MYOC, OPTN, NTF4, WDR36 and CYP1B1 in Chinese Juvenile Onset Open-Angle Glaucoma Using Exome Sequencing. Sci. Rep. 2018, 8, 4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kim, M.; Park, C.K.; Chae, H.; Lee, S.; Kim, Y.; Jang, W.; Chi, H.Y.; Park, H.-Y.L.; Park, S.H. Molecular Analysis of Myocilin and Optineurin Genes in Korean Primary Glaucoma Patients. Mol. Med. Rep. 2016, 14, 2439–2448. [Google Scholar] [CrossRef] [Green Version]
- Forsman, E.; Lemmelä, S.; Varilo, T.; Kristo, P.; Forsius, H.; Sankila, E.-M.; Järvelä, I. The Role of TIGR and OPTN in Finnish Glaucoma Families: A Clinical and Molecular Genetic Study. Mol. Vis. 2003, 9, 217–222. [Google Scholar] [PubMed]
- Aung, T.; Rezaie, T.; Okada, K.; Viswanathan, A.C.; Child, A.H.; Brice, G.; Bhattacharya, S.S.; Lehmann, O.J.; Sarfarazi, M.; Hitchings, R.A. Clinical Features and Course of Patients with Glaucoma with the E50K Mutation in the Optineurin Gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2816–2822. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Funayama, T.; Mashima, Y.; Takano, Y.; Shimizu, A.; Yamamoto, K.; Mengkegale, M.; Miyazawa, A.; Yasuda, N.; Fukuchi, T.; et al. Association of HK2 and NCK2 with Normal Tension Glaucoma in the Japanese Population. PLoS ONE 2013, 8, e54115. [Google Scholar] [CrossRef]
- Jung, S.-H.; Lee, Y.C.; Lee, M.Y.; Shin, H.-Y. Association of HK2 and NCK2 with Normal-Tension Glaucoma in a Population from the Republic of Korea. Graefes Arch. Clin. Exp. Ophthalmol. 2019, 257, 2717–2721. [Google Scholar] [CrossRef]
- Tang, F.Y.; Ma, L.; Tam, P.O.S.; Pang, C.P.; Tham, C.C.; Chen, L.J. Genetic Association of the PARL-ABCC5-HTR3D-HTR3C Locus With Primary Angle-Closure Glaucoma in Chinese. Investig. Ophthalmol. Vis. Sci. 2017, 58, 4384–4389. [Google Scholar] [CrossRef] [Green Version]
- Ressiniotis, T.; Griffiths, P.G.; Birch, M.; Keers, S.; Chinnery, P.F. Primary Open Angle Glaucoma Is Associated with a Specific P53 Gene Haplotype. J. Med. Genet. 2004, 41, 296–298. [Google Scholar] [CrossRef]
- Daugherty, C.L.; Curtis, H.; Realini, T.; Charlton, J.F.; Zareparsi, S. Primary Open Angle Glaucoma in a Caucasian Population Is Associated with the P53 Codon 72 Polymorphism. Mol. Vis. 2009, 15, 1939–1944. [Google Scholar]
- Lin, H.-J.; Chen, W.-C.; Tsai, F.-J.; Tsai, S.-W. Distributions of P53 Codon 72 Polymorphism in Primary Open Angle Glaucoma. Br. J. Ophthalmol. 2002, 86, 767–770. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Marchite, C.; Sánchez-Sánchez, F.; López-Garrido, M.-P.; Iñigez-de-Onzoño, M.; López-Martínez, F.; López-Sánchez, E.; Alvarez, L.; Rodríguez-Calvo, P.-P.; Méndez-Hernández, C.; Fernández-Vega, L.; et al. WDR36 and P53 Gene Variants and Susceptibility to Primary Open-Angle Glaucoma: Analysis of Gene-Gene Interactions. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8467–8478. [Google Scholar] [CrossRef] [PubMed]
- Neamatzadeh, H.; Soleimanizad, R.; Atefi, A.; Zare-Shehneh, M.; Gharibi, S.; Shekari, A.; Rahimzadeh, A.B. Association between P53 Codon 72 (Arg72Pro) Polymorphism and Primary Open-Angle Glaucoma in Iranian Patients. Iran. Biomed. J. 2015, 19, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Chatterjee, S.; Chandra, A.; Maurya, O.P.S.; Mishra, R.N.; Mukherjee, A.; Mutsuddi, M. TP53 Codon 72 Polymorphism and the Risk of Glaucoma in a North Indian Cohort: A Genetic Association Study. Ophthalmic Genet. 2018, 39, 228–235. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, H.; Chen, X.; Yang, X.; Cheng, W.; Zhao, K. Association of TP53 Polymorphisms with Primary Open-Angle Glaucoma: A Meta-Analysis. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3756–3763. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wang, L. Association between Rs4938723 Polymorphism and the Risk of Primary Open-Angle Glaucoma (POAG) in a Chinese Population. J. Cell. Biochem. 2019, 120, 12875–12886. [Google Scholar] [CrossRef]
- Kim, Y.-W.; Bak, E.; Wy, S.; Lee, S.-C.; Kim, Y.-J.; Kim, Y.-K.; Park, K.-H.; Jeoung, J.-W. Genetic Risk and Phenotype Correlation of Primary Open-Angle Glaucoma Based on Rho-Kinase Gene Polymorphisms. J. Clin. Med. 2021, 10, 1953. [Google Scholar] [CrossRef]
- Tezel, G. TNF-Alpha Signaling in Glaucomatous Neurodegeneration. Prog. Brain Res. 2008, 173, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, T.; Nakazawa, C.; Matsubara, A.; Noda, K.; Hisatomi, T.; She, H.; Michaud, N.; Hafezi-Moghadam, A.; Miller, J.W.; Benowitz, L.I. Tumor Necrosis Factor-Alpha Mediates Oligodendrocyte Death and Delayed Retinal Ganglion Cell Loss in a Mouse Model of Glaucoma. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 12633–12641. [Google Scholar] [CrossRef] [Green Version]
- Tezel, G.; Li, L.Y.; Patil, R.V.; Wax, M.B. TNF-Alpha and TNF-Alpha Receptor-1 in the Retina of Normal and Glaucomatous Eyes. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1787–1794. [Google Scholar]
- Leduc-Gaudet, J.; Hussain, S.N.; Gouspillou, G. Parkin: A Potential Target to Promote Healthy Ageing. J. Physiol. 2022, 600, 3405–3421. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-Y.; Perkins, G.A.; Shim, M.S.; Bushong, E.; Alcasid, N.; Ju, S.; Ellisman, M.H.; Weinreb, R.N.; Ju, W.-K. DRP1 Inhibition Rescues Retinal Ganglion Cells and Their Axons by Preserving Mitochondrial Integrity in a Mouse Model of Glaucoma. Cell Death Dis. 2015, 6, e1839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, W.-K.; Kim, K.-Y.; Noh, Y.H.; Hoshijima, M.; Lukas, T.J.; Ellisman, M.H.; Weinreb, R.N.; Perkins, G.A. Increased Mitochondrial Fission and Volume Density by Blocking Glutamate Excitotoxicity Protect Glaucomatous Optic Nerve Head Astrocytes. Glia 2015, 63, 736–753. [Google Scholar] [CrossRef]
- Edwards, G.; Perkins, G.A.; Kim, K.-Y.; Kong, Y.; Lee, Y.; Choi, S.-H.; Liu, Y.; Skowronska-Krawczyk, D.; Weinreb, R.N.; Zangwill, L.; et al. Loss of AKAP1 Triggers Drp1 Dephosphorylation-Mediated Mitochondrial Fission and Loss in Retinal Ganglion Cells. Cell Death Dis. 2020, 11, 254. [Google Scholar] [CrossRef] [Green Version]
- Hass, D.T.; Barnstable, C.J. Cell Autonomous Neuroprotection by the Mitochondrial Uncoupling Protein 2 in a Mouse Model of Glaucoma. Front. Neurosci. 2019, 13, 201. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-G.; Zhou, C.-J.; Li, X.-Y.; Sun, P.-R.; Li, S.-P.; Ren, B.-C. Alteration of UCP2 and ZO-1 Expression in Trabecular Meshwork of Neovascular Glaucoma Patients. J. Glaucoma 2015, 24, 291–296. [Google Scholar] [CrossRef]
- Naguib, S.; Backstrom, J.R.; Gil, M.; Calkins, D.J.; Rex, T.S. Retinal Oxidative Stress Activates the NRF2/ARE Pathway: An Early Endogenous Protective Response to Ocular Hypertension. Redox Biol. 2021, 42, 101883. [Google Scholar] [CrossRef]
- Goncalves, A.; Bürckstümmer, T.; Dixit, E.; Scheicher, R.; Górna, M.W.; Karayel, E.; Sugar, C.; Stukalov, A.; Berg, T.; Kralovics, R.; et al. Functional Dissection of the TBK1 Molecular Network. PLoS ONE 2011, 6, e23971. [Google Scholar] [CrossRef]
- Pomerantz, J.L.; Baltimore, D. NF-KappaB Activation by a Signaling Complex Containing TRAF2, TANK and TBK1, a Novel IKK-Related Kinase. EMBO J. 1999, 18, 6694–6704. [Google Scholar] [CrossRef] [Green Version]
- Helgason, E.; Phung, Q.T.; Dueber, E.C. Recent Insights into the Complexity of Tank-Binding Kinase 1 Signaling Networks: The Emerging Role of Cellular Localization in the Activation and Substrate Specificity of TBK1. FEBS Lett. 2013, 587, 1230–1237. [Google Scholar] [CrossRef] [Green Version]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 Enhances Its Binding to Ub Chains and Promotes Selective Autophagy of Damaged Mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Fingert, J.H.; Darbro, B.W.; Qian, Q.; Van Rheeden, R.; Miller, K.; Riker, M.; Solivan-Timpe, F.; Roos, B.R.; Robin, A.L.; Mullins, R.F. TBK1 and Flanking Genes in Human Retina. Ophthalmic Genet. 2014, 35, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, B.A.; Solivan-Timpe, F.; Roos, B.R.; Anfinson, K.R.; Robin, A.L.; Wiley, L.A.; Mullins, R.F.; Fingert, J.H. Duplication of TBK1 Stimulates Autophagy in IPSC-Derived Retinal Cells from a Patient with Normal Tension Glaucoma. J. Stem Cell Res. Ther. 2014, 3, 161. [Google Scholar] [CrossRef] [PubMed]
- Olichon, A.; Emorine, L.J.; Descoins, E.; Pelloquin, L.; Brichese, L.; Gas, N.; Guillou, E.; Delettre, C.; Valette, A.; Hamel, C.P.; et al. The Human Dynamin-Related Protein OPA1 Is Anchored to the Mitochondrial Inner Membrane Facing the Inter-Membrane Space. FEBS Lett. 2002, 523, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Delettre, C.; Griffoin, J.M.; Kaplan, J.; Dollfus, H.; Lorenz, B.; Faivre, L.; Lenaers, G.; Belenguer, P.; Hamel, C.P. Mutation Spectrum and Splicing Variants in the OPA1 Gene. Hum. Genet. 2001, 109, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Muench, N.A.; Patel, S.; Maes, M.E.; Donahue, R.J.; Ikeda, A.; Nickells, R.W. The Influence of Mitochondrial Dynamics and Function on Retinal Ganglion Cell Susceptibility in Optic Nerve Disease. Cells 2021, 10, 1593. [Google Scholar] [CrossRef]
- Olichon, A.; Baricault, L.; Gas, N.; Guillou, E.; Valette, A.; Belenguer, P.; Lenaers, G. Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome c Release and Apoptosis. J. Biol. Chem. 2003, 278, 7743–7746. [Google Scholar] [CrossRef] [Green Version]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 Deficiency in a Mouse Model of Autosomal Dominant Optic Atrophy Impairs Mitochondrial Morphology, Optic Nerve Structure and Visual Function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef] [Green Version]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 Mutations Induce Mitochondrial DNA Instability and Optic Atrophy “plus” Phenotypes. Brain J. Neurol. 2008, 131, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.; Ashley, N.; Diot, A.; Morten, K.; Phadwal, K.; Williams, A.; Fearnley, I.; Rosser, L.; Lowndes, J.; Fratter, C.; et al. Dysregulated Mitophagy and Mitochondrial Organization in Optic Atrophy Due to OPA1 Mutations. Neurology 2017, 88, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Aijaz, S.; Erskine, L.; Jeffery, G.; Bhattacharya, S.S.; Votruba, M. Developmental Expression Profile of the Optic Atrophy Gene Product: OPA1 Is Not Localized Exclusively in the Mammalian Retinal Ganglion Cell Layer. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1667–1673. [Google Scholar] [CrossRef] [Green Version]
- Ju, W.-K.; Misaka, T.; Kushnareva, Y.; Nakagomi, S.; Agarwal, N.; Kubo, Y.; Lipton, S.A.; Bossy-Wetzel, E. OPA1 Expression in the Normal Rat Retina and Optic Nerve. J. Comp. Neurol. 2005, 488, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesch, U.E.A.; Fries, J.E.; Bette, S.; Kalbacher, H.; Wissinger, B.; Alexander, C.; Kohler, K. OPA1, the Disease Gene for Autosomal Dominant Optic Atrophy, Is Specifically Expressed in Ganglion Cells and Intrinsic Neurons of the Retina. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4217–4225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.-G.; Fann, M.-J.; Yu, H.-Y.; Yen, M.-Y. OPA1 Expression in the Human Retina and Optic Nerve. Exp. Eye Res. 2006, 83, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Aung, T.; Ocaka, L.; Ebenezer, N.D.; Morris, A.G.; Brice, G.; Child, A.H.; Hitchings, R.A.; Lehmann, O.J.; Bhattacharya, S.S. Investigating the Association between OPA1 Polymorphisms and Glaucoma: Comparison between Normal Tension and High Tension Primary Open Angle Glaucoma. Hum. Genet. 2002, 110, 513–514. [Google Scholar] [CrossRef]
- Mabuchi, F.; Tang, S.; Kashiwagi, K.; Yamagata, Z.; Iijima, H.; Tsukahara, S. The OPA1 Gene Polymorphism Is Associated with Normal Tension and High Tension Glaucoma. Am. J. Ophthalmol. 2007, 143, 125–130. [Google Scholar] [CrossRef]
- Liu, Y.; Schmidt, S.; Qin, X.; Gibson, J.; Munro, D.; Wiggs, J.L.; Hauser, M.A.; Allingham, R.R. No Association between OPA1 Polymorphisms and Primary Open-Angle Glaucoma in Three Different Populations. Mol. Vis. 2007, 13, 2137–2141. [Google Scholar]
- Lavaris, A.; Gazouli, M.; Brouzas, D.; Moschos, M.M. Polymorphism Analysis of GSTM1 and OPA1 Genes in Greek Patients with Primary Open-Angle Glaucoma. Vivo 2016, 30, 473–477. [Google Scholar]
- Woo, S.J.; Kim, D.M.; Kim, J.Y.; Park, S.S.; Ko, H.S.; Yoo, T. Investigation of the Association between OPA1 Polymorphisms and Normal-Tension Glaucoma in Korea. J. Glaucoma 2004, 13, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Jiao, X.; Hejtmancik, J.F.; Leske, M.C.; Hennis, A.; Nemesure, B.; Barbados Family Study Group. Evaluation of the Association between OPA1 Polymorphisms and Primary Open-Angle Glaucoma in Barbados Families. Mol. Vis. 2006, 12, 649–654. [Google Scholar] [PubMed]
- Fan, B.J.; Liu, K.; Wang, D.Y.; Tham, C.C.Y.; Tam, P.O.S.; Lam, D.S.C.; Pang, C.P. Association of Polymorphisms of Tumor Necrosis Factor and Tumor Protein P53 with Primary Open-Angle Glaucoma. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4110–4116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buentello-Volante, B.; Elizondo-Olascoaga, C.; Miranda-Duarte, A.; Guadarrama-Vallejo, D.; Cabral-Macias, J.; Zenteno, J.C. Association Study of Multiple Gene Polymorphisms with the Risk of Adult-Onset Primary Open-Angle Glaucoma in a Mexican Population. Exp. Eye Res. 2013, 107, 59–64. [Google Scholar] [CrossRef]
- Kumaramanickavel, G.; Aung, T.; Sripriya, S.; Waseem, N.; Okada, K.; Seah, S.K.L.; Baskaran, M.; Mishima, H.K.; Vithana, E.N.; Bhattacharya, S. Lack of Association of IVS8+4 C/T and IVS8+32 T/C Polymorphisms in the OPA1 Gene With Normal Tension Glaucoma in Patients From Singapore, India and Japan. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3811. [Google Scholar]
- Gao, S.; Hu, J. Mitochondrial Fusion: The Machineries In and Out. Trends Cell Biol. 2021, 31, 62–74. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Musolino, C. Relationship between Mitofusin 2 and Cancer. Adv. Protein Chem. Struct. Biol. 2019, 116, 209–236. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 Coordinately Regulate Mitochondrial Fusion and Are Essential for Embryonic Development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Misko, A.; Jiang, S.; Wegorzewska, I.; Milbrandt, J.; Baloh, R.H. Mitofusin 2 Is Necessary for Transport of Axonal Mitochondria and Interacts with the Miro/Milton Complex. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 4232–4240. [Google Scholar] [CrossRef] [Green Version]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 Tethers Endoplasmic Reticulum to Mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-Mediated Coupling of Endoplasmic Reticulum and Mitochondrial Ca2+ Channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef]
- Zhao, T.; Huang, X.; Han, L.; Wang, X.; Cheng, H.; Zhao, Y.; Chen, Q.; Chen, J.; Cheng, H.; Xiao, R.; et al. Central Role of Mitofusin 2 in Autophagosome-Lysosome Fusion in Cardiomyocytes. J. Biol. Chem. 2012, 287, 23615–23625. [Google Scholar] [CrossRef] [Green Version]
- Sebastián, D.; Sorianello, E.; Segalés, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Muñoz, J.P.; Sánchez-Feutrie, M.; et al. Mfn2 Deficiency Links Age-Related Sarcopenia and Impaired Autophagy to Activation of an Adaptive Mitophagy Pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [Green Version]
- Escobar-Henriques, M.; Joaquim, M. Mitofusins: Disease Gatekeepers and Hubs in Mitochondrial Quality Control by E3 Ligases. Front. Physiol. 2019, 10, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.-Y.; Rojo, M.; Schapira, A.H.V.; Taanman, J.-W. Mitofusin 1 and Mitofusin 2 Are Ubiquitinated in a PINK1/Parkin-Dependent Manner upon Induction of Mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.-F.; Karbowski, M.; Youle, R.J. Proteasome and P97 Mediate Mitophagy and Degradation of Mitofusins Induced by Parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial Fission Facilitates the Selective Mitophagy of Protein Aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial Oxidative Stress Causes Mitochondrial Fragmentation via Differential Modulation of Mitochondrial Fission-Fusion Proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef]
- Nivison, M.P.; Ericson, N.G.; Green, V.M.; Bielas, J.H.; Campbell, J.S.; Horner, P.J. Age-Related Accumulation of Phosphorylated Mitofusin 2 Protein in Retinal Ganglion Cells Correlates with Glaucoma Progression. Exp. Neurol. 2017, 296, 49–61. [Google Scholar] [CrossRef]
- Fimia, G.M.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 Regulates Autophagy and Development of the Nervous System. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [Green Version]
- Van Humbeeck, C.; Cornelissen, T.; Hofkens, H.; Mandemakers, W.; Gevaert, K.; De Strooper, B.; Vandenberghe, W. Parkin Interacts with Ambra1 to Induce Mitophagy. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 10249–10261. [Google Scholar] [CrossRef] [Green Version]
- Strappazzon, F.; Nazio, F.; Corrado, M.; Cianfanelli, V.; Romagnoli, A.; Fimia, G.M.; Campello, S.; Nardacci, R.; Piacentini, M.; Campanella, M.; et al. AMBRA1 Is Able to Induce Mitophagy via LC3 Binding, Regardless of PARKIN and P62/SQSTM1. Cell Death Differ. 2015, 22, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 Regulates Mitophagy by Interacting with ATAD3A and Promoting PINK1 Stability. Autophagy 2022, 18, 1752–1762. [Google Scholar] [CrossRef] [PubMed]
- Hurlstone, A.F.; Reid, G.; Reeves, J.R.; Fraser, J.; Strathdee, G.; Rahilly, M.; Parkinson, E.K.; Black, D.M. Analysis of the CAVEOLIN-1 Gene at Human Chromosome 7q31.1 in Primary Tumours and Tumour-Derived Cell Lines. Oncogene 1999, 18, 1881–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nah, J.; Yoo, S.-M.; Jung, S.; Jeong, E.I.; Park, M.; Kaang, B.-K.; Jung, Y.-K. Phosphorylated CAV1 Activates Autophagy through an Interaction with BECN1 under Oxidative Stress. Cell Death Dis. 2017, 8, e2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Tan, S.-H.; Ng, S.; Zhou, J.; Yang, N.-D.; Koo, G.-B.; McMahon, K.-A.; Parton, R.G.; Hill, M.M.; Del Pozo, M.A.; et al. Critical Role of CAV1/Caveolin-1 in Cell Stress Responses in Human Breast Cancer Cells via Modulation of Lysosomal Function and Autophagy. Autophagy 2015, 11, 769–784. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Krantz, S.; Qin, X.; Li, S.; Gunasekara, H.; Kim, Y.-M.; Zimnicka, A.; Bae, M.; Ma, K.; Toth, P.T.; et al. Caveolin-1 Controls Mitochondrial Damage and ROS Production by Regulating Fission-Fusion Dynamics and Mitophagy. Redox Biol. 2022, 52, 102304. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Lack of Association of SNP Rs4236601 near CAV1 and CAV2 with POAG in a Saudi Cohort. Mol. Vis. 2012, 18, 1960–1965. [Google Scholar]
- Kuehn, M.H.; Wang, K.; Roos, B.; Stone, E.M.; Kwon, Y.H.; Alward, W.L.M.; Mullins, R.F.; Fingert, J.H. Chromosome 7q31 POAG Locus: Ocular Expression of Caveolins and Lack of Association with POAG in a US Cohort. Mol. Vis. 2011, 17, 430–435. [Google Scholar]
- Schwamborn, K.; Weil, R.; Courtois, G.; Whiteside, S.T.; Israël, A. Phorbol Esters and Cytokines Regulate the Expression of the NEMO-Related Protein, a Molecule Involved in a NF-Kappa B-Independent Pathway. J. Biol. Chem. 2000, 275, 22780–22789. [Google Scholar] [CrossRef] [Green Version]
- Montecalvo, A.; Watkins, S.C.; Orange, J.; Kane, L.P. Inducible Turnover of Optineurin Regulates T Cell Activation. Mol. Immunol. 2017, 85, 9–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef]
- Holm, E.; Holm, M.; Vilhelmsen, K.; Andorsdottir, G.; Vorum, H.; Simpson, A.; Roos, B.R.; Fingert, J.H.; Rosenberg, T. Prevalence of Open-Angle Glaucoma in the Faroese Population. J. Glaucoma 2022, 31, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Martinez, F.; Lopez-Garrido, M.-P.; Sanchez-Sanchez, F.; Campos-Mollo, E.; Coca-Prados, M.; Escribano, J. Role of MYOC and OPTN Sequence Variations in Spanish Patients with Primary Open-Angle Glaucoma. Mol. Vis. 2007, 13, 862–872. [Google Scholar]
- Yen, Y.-C.; Yang, J.-J.; Chou, M.-C.; Li, S.-Y. Absence of Optineurin (OPTN) Gene Mutations in Taiwanese Patients with Juvenile-Onset Open-Angle Glaucoma. Mol. Vis. 2008, 14, 487–494. [Google Scholar] [PubMed]
- Wu, Z.; Wu, J.; Zhao, Q.; Fu, S.; Jin, J. Emerging Roles of Aerobic Glycolysis in Breast Cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2020, 22, 631–646. [Google Scholar] [CrossRef] [PubMed]
- McCoy, M.K.; Kaganovich, A.; Rudenko, I.N.; Ding, J.; Cookson, M.R. Hexokinase Activity Is Required for Recruitment of Parkin to Depolarized Mitochondria. Hum. Mol. Genet. 2014, 23, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.-M.; Harper, N.J.; Paulo, J.A.; Li, M.; Xu, Q.; Coughlin, M.; Elledge, S.J.; Harper, J.W. Integrated Proteogenetic Analysis Reveals the Landscape of a Mitochondrial-Autophagosome Synapse during PARK2-Dependent Mitophagy. Sci. Adv. 2019, 5, eaay4624. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L.; Scorrano, L. A Cut Short to Death: Parl and Opa1 in the Regulation of Mitochondrial Morphology and Apoptosis. Cell Death Differ. 2007, 14, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial Rhomboid PARL Regulates Cytochrome c Release during Apoptosis via OPA1-Dependent Cristae Remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The Mitochondrial Intramembrane Protease PARL Cleaves Human Pink1 to Regulate Pink1 Trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane Protease PARL Defines a Negative Regulator of PINK1- and PARK2/Parkin-Dependent Mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, O.W.; Merry, D.; Givol, D. The Gene for Human P53 Cellular Tumor Antigen Is Located on Chromosome 17 Short Arm (17p13). Proc. Natl. Acad. Sci. USA 1986, 83, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the P53 Network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Zhang, J.; Dong, W.; Wu, J.; Wang, T.; Xie, Z. P53 and Parkin Co-Regulate Mitophagy in Bone Marrow Mesenchymal Stem Cells to Promote the Repair of Early Steroid-Induced Osteonecrosis of the Femoral Head. Cell Death Dis. 2020, 11, 42. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L.; Hewitt, A.W.; Fan, B.J.; Wang, D.Y.; Figueiredo Sena, D.R.; O’Brien, C.; Realini, A.; Craig, J.E.; Dimasi, D.P.; Mackey, D.A.; et al. The P53 Codon 72 PRO/PRO Genotype May Be Associated with Initial Central Visual Field Defects in Caucasians with Primary Open Angle Glaucoma. PLoS ONE 2012, 7, e45613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.; Przybylowska-Sygut, K.; Szymanek, K.; Szaflik, J.; Szaflik, J.; Majsterek, I. The Relationship of TP53 and GRIN2B Gene Polymorphisms with Risk of Occurrence and Progression of Primary Open-Angle Glaucoma in a Polish Population. Pol. J. Pathol. Off. J. Pol. Soc. Pathol. 2014, 65, 313–321. [Google Scholar] [CrossRef]
- Saglar, E.; Yucel, D.; Bozkurt, B.; Ozgul, R.K.; Irkec, M.; Ogus, A. Association of Polymorphisms in APOE, P53, and P21 with Primary Open-Angle Glaucoma in Turkish Patients. Mol. Vis. 2009, 15, 1270–1276. [Google Scholar]
- Mabuchi, F.; Sakurada, Y.; Kashiwagi, K.; Yamagata, Z.; Iijima, H.; Tsukahara, S. Lack of Association between P53 Gene Polymorphisms and Primary Open Angle Glaucoma in the Japanese Population. Mol. Vis. 2009, 15, 1045–1049. [Google Scholar]
- Dimasi, D.P.; Hewitt, A.W.; Green, C.M.; Mackey, D.A.; Craig, J.E. Lack of Association of P53 Polymorphisms and Haplotypes in High and Normal Tension Open Angle Glaucoma. J. Med. Genet. 2005, 42, e55. [Google Scholar] [CrossRef] [Green Version]
- Silva, R.E.; Arruda, J.T.; Rodrigues, F.W.; Moura, K.K.V.O. Primary Open Angle Glaucoma Was Not Found to Be Associated with P53 Codon 72 Polymorphism in a Brazilian Cohort. Genet. Mol. Res. GMR 2009, 8, 268–272. [Google Scholar] [CrossRef]
- Acharya, M.; Mitra, S.; Mukhopadhyay, A.; Khan, M.; Roychoudhury, S.; Ray, K. Distribution of P53 Codon 72 Polymorphism in Indian Primary Open Angle Glaucoma Patients. Mol. Vis. 2002, 8, 367–371. [Google Scholar]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct Roles for ROCK1 and ROCK2 in the Regulation of Cell Detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Cho, Y.-L.; Tang, Y.; Wang, J.; Park, J.-E.; Wu, Y.; Wang, C.; Tong, Y.; Chawla, R.; Zhang, J.; et al. PTEN-L Is a Novel Protein Phosphatase for Ubiquitin Dephosphorylation to Inhibit PINK1-Parkin-Mediated Mitophagy. Cell Res. 2018, 28, 787–802. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; Angus McQuibban, G. ROCK Inhibitors Upregulate the Neuroprotective Parkin-Mediated Mitophagy Pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, E.; Nakajima, T.; Minagawa, Y.; Shearer, T.R.; Azuma, M. Contribution of ROCK in Contraction of Trabecular Meshwork: Proposed Mechanism for Regulating Aqueous Outflow in Monkey and Human Eyes. J. Pharm. Sci. 2005, 94, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Demiryürek, S.; Okumus, S.; Bozgeyik, İ.; Oztuzcu, S.; Coskun, E.; Mat, E.; Durucu, E.; Tatar, M.G.; Erbagci, İ.; Gürler, B.; et al. Investigation of the Rho-Kinase Gene Polymorphism in Primary Open-Angle Glaucoma. Ophthalmic Genet. 2016, 37, 9–13. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y. Tumor Necrosis Factor and Cancer, Buddies or Foes? Acta Pharmacol. Sin. 2008, 29, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Tyciakova, S.; Valova, V.; Svitkova, B.; Matuskova, M. Overexpression of TNFα Induces Senescence, Autophagy and Mitochondrial Dysfunctions in Melanoma Cells. BMC Cancer 2021, 21, 507. [Google Scholar] [CrossRef]
- Bell, C.; English, L.; Boulais, J.; Chemali, M.; Caron-Lizotte, O.; Desjardins, M.; Thibault, P. Quantitative Proteomics Reveals the Induction of Mitophagy in Tumor Necrosis Factor-α-Activated (TNFα) Macrophages. Mol. Cell. Proteom. MCP 2013, 12, 2394–2407. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.-J.; Tsai, F.-J.; Chen, W.-C.; Shi, Y.-R.; Hsu, Y.; Tsai, S.-W. Association of Tumour Necrosis Factor Alpha -308 Gene Polymorphism with Primary Open-Angle Glaucoma in Chinese. Eye 2003, 17, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Mossböck, G.; Weger, M.; Moray, M.; Renner, W.; Haller-Schober, E.M.; Mattes, D.; Schmut, O.; Wegscheider, B.; El-Shabrawi, Y. TNF-Alpha Promoter Polymorphisms and Primary Open-Angle Glaucoma. Eye 2006, 20, 1040–1043. [Google Scholar] [CrossRef] [Green Version]
- Funayama, T.; Ishikawa, K.; Ohtake, Y.; Tanino, T.; Kurosaka, D.; Kimura, I.; Suzuki, K.; Ideta, H.; Nakamoto, K.; Yasuda, N.; et al. Variants in Optineurin Gene and Their Association with Tumor Necrosis Factor-Alpha Polymorphisms in Japanese Patients with Glaucoma. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4359–4367. [Google Scholar] [CrossRef]
- Sarraf, S.A.; Raman, M.; Guarani-Pereira, V.; Sowa, M.E.; Huttlin, E.L.; Gygi, S.P.; Harper, J.W. Landscape of the PARKIN-Dependent Ubiquitylome in Response to Mitochondrial Depolarization. Nature 2013, 496, 372–376. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin Is Recruited Selectively to Impaired Mitochondria and Promotes Their Autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. The Three ’P’s of Mitophagy: PARKIN, PINK1, and Post-Translational Modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Rothfuss, O.; Fischer, H.; Hasegawa, T.; Maisel, M.; Leitner, P.; Miesel, F.; Sharma, M.; Bornemann, A.; Berg, D.; Gasser, T.; et al. Parkin Protects Mitochondrial Genome Integrity and Supports Mitochondrial DNA Repair. Hum. Mol. Genet. 2009, 18, 3832–3850. [Google Scholar] [CrossRef] [Green Version]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 Enhancement Is Able to Compensate Mitophagy Alterations Found in Sporadic Alzheimer’s Disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef] [Green Version]
- Bonilla-Porras, A.R.; Arevalo-Arbelaez, A.; Alzate-Restrepo, J.F.; Velez-Pardo, C.; Jimenez-Del-Rio, M. PARKIN Overexpression in Human Mesenchymal Stromal Cells from Wharton’s Jelly Suppresses 6-Hydroxydopamine-Induced Apoptosis: Potential Therapeutic Strategy in Parkinson’s Disease. Cytotherapy 2018, 20, 45–61. [Google Scholar] [CrossRef]
- Fowler, P.C.; Byrne, D.J.; Blackstone, C.; O’Sullivan, N.C. Loss of the Mitochondrial Fission GTPase Drp1 Contributes to Neurodegeneration in a Drosophila Model of Hereditary Spastic Paraplegia. Brain Sci. 2020, 10, 646. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Li, H.; You, M.; Rong, R.; Xia, X. Dephosphorylation of ERK1/2 and DRP1 S585 Regulates Mitochondrial Dynamics in Glutamate Toxicity of Retinal Neurons in Vitro. Exp. Eye Res. 2022, 225, 109271. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-Related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.M.; Ryu, J.R.; Jo, Y.; Seo, T.W.; Choi, Y.N.; Kim, J.H.; Chung, J.M.; Cho, B.; Kang, H.C.; Yu, S.-W.; et al. Drp1-Zip1 Interaction Regulates Mitochondrial Quality Surveillance System. Mol. Cell 2019, 73, 364–376.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Lin, C.; Wu, K.; Jiang, L.; Wang, X.; Li, W.; Zhuang, H.; Zhang, X.; Chen, H.; Li, S.; et al. FUNDC1 Regulates Mitochondrial Dynamics at the ER-Mitochondrial Contact Site under Hypoxic Conditions. EMBO J. 2016, 35, 1368–1384. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Shirakabe, A.; Maejima, Y.; Zhai, P.; Sciarretta, S.; Toli, J.; Nomura, M.; Mihara, K.; Egashira, K.; Ohishi, M.; et al. Endogenous Drp1 Mediates Mitochondrial Autophagy and Protects the Heart against Energy Stress. Circ. Res. 2015, 116, 264–278. [Google Scholar] [CrossRef]
- Tian, X.Y.; Ma, S.; Tse, G.; Wong, W.T.; Huang, Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front. Physiol. 2018, 9, 1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, M.; Bianchi, F.; Cotugno, M.; Marchitti, S.; Stanzione, R.; Maglione, V.; Sciarretta, S.; Valenti, V.; Carnevale, R.; Versaci, F.; et al. An Interplay between UCP2 and ROS Protects Cells from High-Salt-Induced Injury through Autophagy Stimulation. Cell Death Dis. 2021, 12, 919. [Google Scholar] [CrossRef]
- Song, M.-Y.; Lee, D.-Y.; Chun, K.-S.; Kim, E.-H. The Role of NRF2/KEAP1 Signaling Pathway in Cancer Metabolism. Int. J. Mol. Sci. 2021, 22, 4376. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Zeb, A.; Choubey, V.; Gupta, R.; Kuum, M.; Safiulina, D.; Vaarmann, A.; Gogichaishvili, N.; Liiv, M.; Ilves, I.; Tämm, K.; et al. A Novel Role of KEAP1/PGAM5 Complex: ROS Sensor for Inducing Mitophagy. Redox Biol. 2021, 48, 102186. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Takamatsu, H.; Liu, S.; Kataoka, K.; Huh, N.-H.; Sakaguchi, M. NRF2 Regulates PINK1 Expression under Oxidative Stress Conditions. PLoS ONE 2015, 10, e0142438. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Kaufman, P.L. Effects of the Rho Kinase Inhibitor Y-27632 and the Phosphatase Inhibitor Calyculin A on Outflow Facility in Monkeys. Exp. Eye Res. 2005, 80, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Quadir, H.; Hakobyan, K.; Gaddam, M.; Ojinnaka, U.; Ahmed, Z.; Kannan, A.; Mostafa, J.A. Role of Rho-Associated Protein Kinase Inhibition As Therapeutic Strategy for Parkinson’s Disease: Dopaminergic Survival and Enhanced Mitophagy. Cureus 2021, 13, e16973. [Google Scholar] [CrossRef]
- Bertrand, J.; Winton, M.J.; Rodriguez-Hernandez, N.; Campenot, R.B.; McKerracher, L. Application of Rho Antagonist to Neuronal Cell Bodies Promotes Neurite Growth in Compartmented Cultures and Regeneration of Retinal Ganglion Cell Axons in the Optic Nerve of Adult Rats. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Koch, J.C.; Tönges, L.; Michel, U.; Bähr, M.; Lingor, P. Viral Vector-Mediated Downregulation of RhoA Increases Survival and Axonal Regeneration of Retinal Ganglion Cells. Front. Cell. Neurosci. 2014, 8, 273. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.X.; Sang, A.; Wang, Y.; Ho, D.; Douglas, C.; Dia, L.; Goldberg, J.L. Topical Administration of a Rock/Net Inhibitor Promotes Retinal Ganglion Cell Survival and Axon Regeneration after Optic Nerve Injury. Exp. Eye Res. 2017, 158, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Bacharach, J.; Dubiner, H.B.; Levy, B.; Kopczynski, C.C.; Novack, G.D.; AR-13324-CS202 Study Group. Double-Masked, Randomized, Dose-Response Study of AR-13324 versus Latanoprost in Patients with Elevated Intraocular Pressure. Ophthalmology 2015, 122, 302–307. [Google Scholar] [CrossRef]
- Batra, M.; Gupta, S.; Nair, A.B.; Dhanawat, M.; Sandal, S.; Morsy, M.A. Netarsudil: A New Ophthalmic Drug in the Treatment of Chronic Primary Open Angle Glaucoma and Ocular Hypertension. Eur. J. Ophthalmol. 2021, 31, 2237–2244. [Google Scholar] [CrossRef]
- Lian, W.; Hu, X.; Zhang, J.; Wu, Y.; Zhao, N.; Ma, H.; He, H.; Lu, Q. Fucoxanthin Protects Retinal Ganglion Cells and Promotes Parkin-Mediated Mitophagy against Glutamate Excitotoxicity. Neuroreport 2023, 34, 385–394. [Google Scholar] [CrossRef]
- Ma, H.; Hu, X.; Zhang, J.; Lian, W.; Wang, D.; Wu, Y.; Lu, Q. Fucoxanthin Protects Retinal Ganglion Cells and Regulates Parkin-Mediated Mitophagy in Rats with Chronic Ocular Hypertensive Glaucoma; SSRN; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Zhuang, D.; Zhang, R.; Liu, H.; Dai, Y. A Small Natural Molecule S3 Protects Retinal Ganglion Cells and Promotes Parkin-Mediated Mitophagy against Excitotoxicity. Molecules 2022, 27, 4957. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, W.K.; Lee, Y.-H.; Kang, E.S.; Cha, B.-S.; Lee, B.-W. Metformin Restores Parkin-Mediated Mitophagy, Suppressed by Cytosolic P53. Int. J. Mol. Sci. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.-C.; Stein, J.D.; Nan, B.; Childers, D.; Newman-Casey, P.A.; Thompson, D.A.; Richards, J.E. Association of Geroprotective Effects of Metformin and Risk of Open-Angle Glaucoma in Persons with Diabetes Mellitus. JAMA Ophthalmol. 2015, 133, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Vergroesen, J.; Stricker, B.H.; Ikram, A.M.; Kavousi, M.; Hofman, A.; Klaver, C.; Ramdas, W. Systemic Metformin Use Reduces Open-Angle Glaucoma Risk. Investig. Ophthalmol. Vis. Sci. 2020, 61, 656. [Google Scholar]
- Ju, W.-K.; Kim, K.-Y.; Angert, M.; Duong-Polk, K.X.; Lindsey, J.D.; Ellisman, M.H.; Weinreb, R.N. Memantine Blocks Mitochondrial OPA1 and Cytochrome c Release and Subsequent Apoptotic Cell Death in Glaucomatous Retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, R.N.; Liebmann, J.M.; Cioffi, G.A.; Goldberg, I.; Brandt, J.D.; Johnson, C.A.; Zangwill, L.M.; Schneider, S.; Badger, H.; Bejanian, M. Oral Memantine for the Treatment of Glaucoma: Design and Results of 2 Randomized, Placebo-Controlled, Phase 3 Studies. Ophthalmology 2018, 125, 1874–1885. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. P62/SQSTM1 Is a Target Gene for Transcription Factor NRF2 and Creates a Positive Feedback Loop by Inducing Antioxidant Response Element-Driven Gene Transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Tonner, H.; Hunn, S.; Auler, N.; Schmelter, C.; Pfeiffer, N.; Grus, F.H. Dynamin-like Protein 1 (DNML1) as a Molecular Target for Antibody-Based Immunotherapy to Treat Glaucoma. Int. J. Mol. Sci. 2022, 23, 13618. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, A.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gómez Izquierdo, L.; Cotán, D.; Navas, P.; Sánchez-Alcázar, J.A. Coenzyme Q Deficiency Triggers Mitochondria Degradation by Mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spanò, A.; Cavaliere, F.; Rombolà, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal Damage Caused by High Intraocular Pressure-Induced Transient Ischemia Is Prevented by Coenzyme Q10 in Rat. Int. Rev. Neurobiol. 2007, 82, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Alpogan, O. Effects of Topical Coenzyme Q10 on Retinal Nerve Fiber Layer and Ganglion Cell Complex in Patients with Open-Angle Glaucoma. Haydarpasa Numune Train. Res. Hosp. Med. J. 2022, 62, 190–194. [Google Scholar] [CrossRef]
- Parisi, V.; Centofanti, M.; Gandolfi, S.; Marangoni, D.; Rossetti, L.; Tanga, L.; Tardini, M.; Traina, S.; Ungaro, N.; Vetrugno, M.; et al. Effects of Coenzyme Q10 in Conjunction with Vitamin E on Retinal-Evoked and Cortical-Evoked Responses in Patients with Open-Angle Glaucoma. J. Glaucoma 2014, 23, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin Upregulates Mitophagy in Patients with T2DM: A Randomized Placebo-Controlled Study. J. Cell. Mol. Med. 2020, 24, 2832–2846. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Dhawan, V. Metformin Promotes Mitophagy in Mononuclear Cells: A Potential in Vitro Model for Unraveling Metformin’s Mechanism of Action. Ann. N. Y. Acad. Sci. 2020, 1463, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Q.; Wu, M.; Zhang, L.; Fu, W.; Yang, J.; Zhang, B.; Zheng, Z.; Zhang, H.; Lao, Y.; Xu, H. Gerontoxanthone I and Macluraxanthone Induce Mitophagy and Attenuate Ischemia/Reperfusion Injury. Front. Pharmacol. 2020, 11, 452. [Google Scholar] [CrossRef] [Green Version]
- Wiggs, J.L. Genetic Etiologies of Glaucoma. Arch. Ophthalmol. 2007, 125, 30–37. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Summary of mitochondrial quality control process and mitophagy pathways. Mitochondrial dynamics comprise constant cycles of fusion and fission. When a mitochondrion is depolarized or dysfunctional, it is subjected to elimination. Impaired mitochondria are removed from a cell through two main mitophagy pathways, the PTEN-induced kinase 1 (PINK1)/ Parkin RBR E3 ubiquitin protein ligase (PARKIN) pathway, which is a ubiquitin-mediated pathway, and the receptor-mediated pathway. At the beginning of the PINK1/PARKIN pathway, PINK1 is stabilized in the outer mitochondrial membrane (OMM) and recruits PARKIN, which is an E3 ubiquitin ligase. Consequently, PARKIN polyubiquitinates various OMM proteins. In turn, the polyubiquitin chains are identified by adaptor proteins such as optineurin (OPTN), which interact with microtubule-associated protein 1A/1B-light chain 3 (LC3), and as a result, the impaired mitochondrion gets isolated via a phagophore in the double-membraned autophagosome. During the receptor-mediated pathway, mitophagy receptors, such as BCL2-interacting protein 3 (BNIP3)/NIX or autophagy and beclin 1 regulator 1 (AMBRA1), attach to the interaction region of LC3, allowing them to interact directly with the phagophore. In the aftermath, the impaired mitochondrion will end up in the autophagosome where, after fusion with lysosomes, it will get degraded.
Figure 1.
Summary of mitochondrial quality control process and mitophagy pathways. Mitochondrial dynamics comprise constant cycles of fusion and fission. When a mitochondrion is depolarized or dysfunctional, it is subjected to elimination. Impaired mitochondria are removed from a cell through two main mitophagy pathways, the PTEN-induced kinase 1 (PINK1)/ Parkin RBR E3 ubiquitin protein ligase (PARKIN) pathway, which is a ubiquitin-mediated pathway, and the receptor-mediated pathway. At the beginning of the PINK1/PARKIN pathway, PINK1 is stabilized in the outer mitochondrial membrane (OMM) and recruits PARKIN, which is an E3 ubiquitin ligase. Consequently, PARKIN polyubiquitinates various OMM proteins. In turn, the polyubiquitin chains are identified by adaptor proteins such as optineurin (OPTN), which interact with microtubule-associated protein 1A/1B-light chain 3 (LC3), and as a result, the impaired mitochondrion gets isolated via a phagophore in the double-membraned autophagosome. During the receptor-mediated pathway, mitophagy receptors, such as BCL2-interacting protein 3 (BNIP3)/NIX or autophagy and beclin 1 regulator 1 (AMBRA1), attach to the interaction region of LC3, allowing them to interact directly with the phagophore. In the aftermath, the impaired mitochondrion will end up in the autophagosome where, after fusion with lysosomes, it will get degraded.

Figure 2.
Synopsis of the main effects that proteins derived from mitophagy-related genes have on mitophagy and mitophagy pathways.
Figure 2.
Synopsis of the main effects that proteins derived from mitophagy-related genes have on mitophagy and mitophagy pathways.


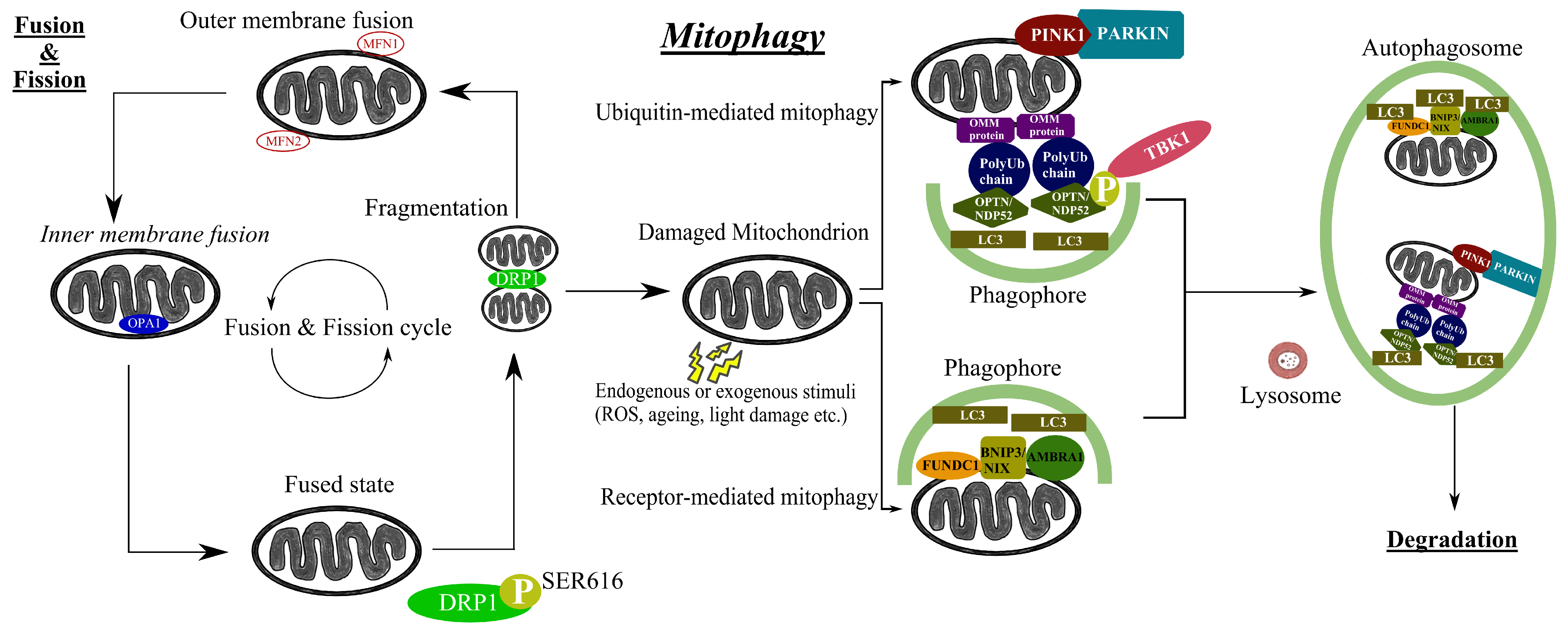{kind=link}
{kind=link}
Table 1.
Mitophagy-related genes linked to glaucoma.
| Gene Symbol | Gene Name | MIM ID | Chromosomal Location | References |
|---|---|---|---|---|
| TBK1 | TANK-binding kinase 1 | 604834 | 12q14.2 | [65,66,67,68,69,70,71] |
| OPA1 | Optic atrophy 1 | 605290 | 3q28-q29 | [72,73,74,75] |
| MFN1 | Mitofusin 1 | 608506 | 3q25-q26 | [75] |
| MFN2 | Mitofusin 2 | 608507 | 1p36.2 | [76] |
| AMBRA1 | Autophagy and beclin 1 regulator 1 | 611359 | 11p11.2 | [77,78] |
| CAV1 | Caveolin-1 | 601047 | 7q31.1 | [79,80,81,82,83,84] |
| OPTN | Optineurin | 602432 | 10p13 | [85,86,87,88,89,90,91,92,93,94,95,96] |
| HK2 | Hexokinase 2 | 601125 | 2p12 | [97,98] |
| PARL | Presenilin-associated rhomboid-like | 607858 | 3q27.1 | [99] |
| TP53 | Tumor protein p53 | 191170 | 17p13.1 | [100,101,102,103,104,105,106,107] |
| ROCK1 | Rho-associated coiled-coil containing protein kinase 1 | 601702 | 18q11.1 | [108] |
| TNFα | Tumor necrosis factor alpha | 191160 | 6p21.3 | [109,110,111] |
| PARKIN | Parkin RBR E3 ubiquitin protein ligase | 602544 | 6q26 | [112] |
| DRP1 | Dynamin-related protein 1 | 603850 | 12p11.21 | [113,114,115] |
| UCP2 | Uncoupling protein 2 | 601693 | 11q13.4 | [116,117] |
| KEAP1 | Kelch-like ECH-associated protein 1 | 606016 | 19p13.2 | [118] |
| NRF2 | NF-E2-related factor 2 | 600492 | 2q31.2 | [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stavropoulos, D.; Grewal, M.K.; Petriti, B.; Chau, K.-Y.; Hammond, C.J.; Garway-Heath, D.F.; Lascaratos, G. The Role of Mitophagy in Glaucomatous Neurodegeneration. Cells 2023, 12, 1969. https://doi.org/10.3390/cells12151969
AMA Style
Stavropoulos D, Grewal MK, Petriti B, Chau K-Y, Hammond CJ, Garway-Heath DF, Lascaratos G. The Role of Mitophagy in Glaucomatous Neurodegeneration. Cells. 2023; 12(15):1969. https://doi.org/10.3390/cells12151969
Chicago/Turabian StyleStavropoulos, Dimitrios, Manjot K. Grewal, Bledi Petriti, Kai-Yin Chau, Christopher J. Hammond, David F. Garway-Heath, and Gerassimos Lascaratos. 2023. "The Role of Mitophagy in Glaucomatous Neurodegeneration" Cells 12, no. 15: 1969. https://doi.org/10.3390/cells12151969
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.