
KLK6/PAR1 Axis Promotes Tumor Growth and Metastasis by Regulating Cross-Talk between Tumor Cells and Macrophages

 , , , , ,
, , , , ,
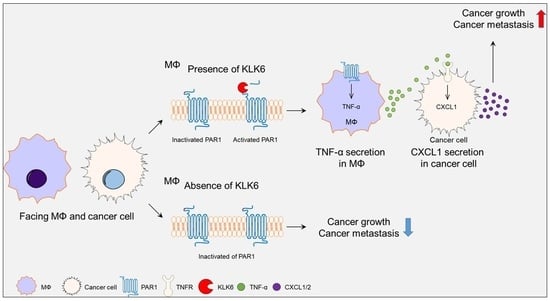
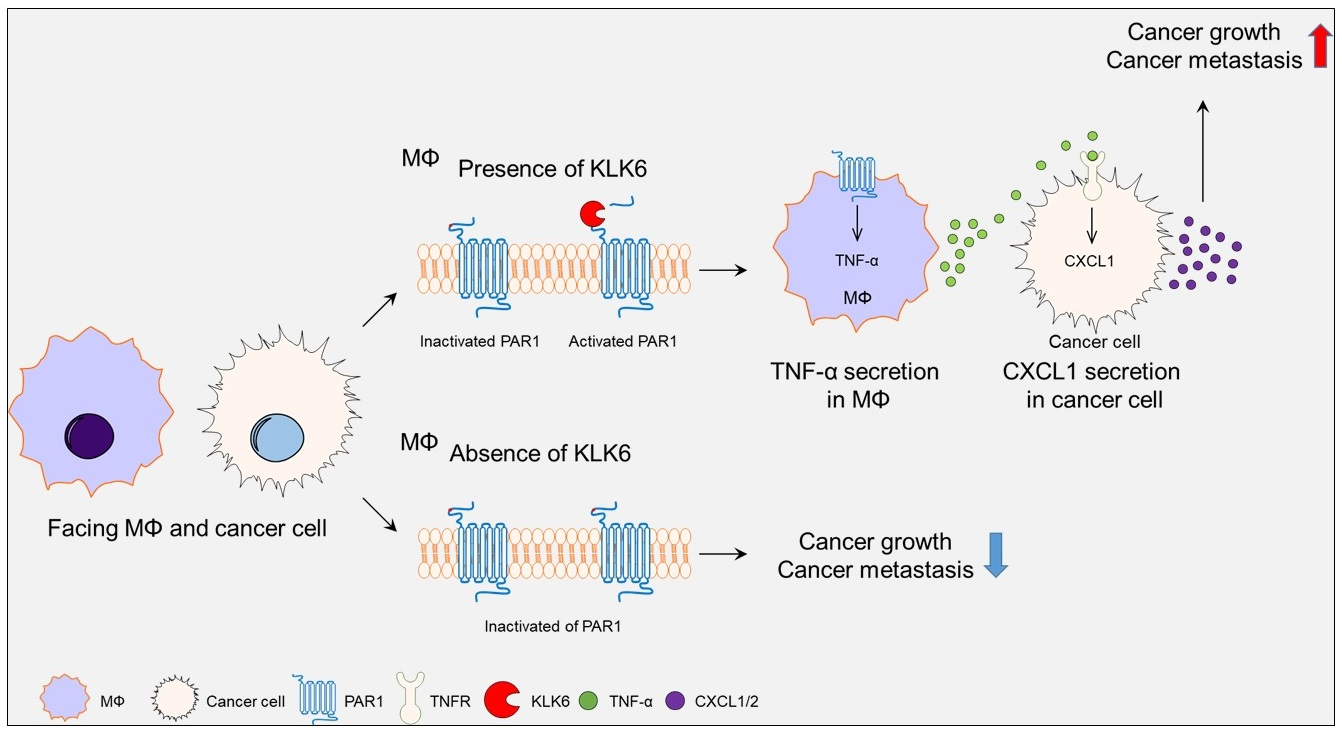
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Transfection and RNA Interference
2.4. Generation of KLK6-Deficient Mice
2.5. Animal Experiments
2.6. Bone Marrow–Derived Macrophage (BMDM) Isolation
2.7. In Vivo Macrophage Depletion
2.8. Coculture Assay and Conditioned Medium (CM) Production
2.9. Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.10. Western Blot Analysis
2.11. Cytokine Array and Enzyme-Linked Immunosorbent Assay (ELISA)
2.12. Flow Cytometry Analysis
2.13. Statistical Analysis
3. Results
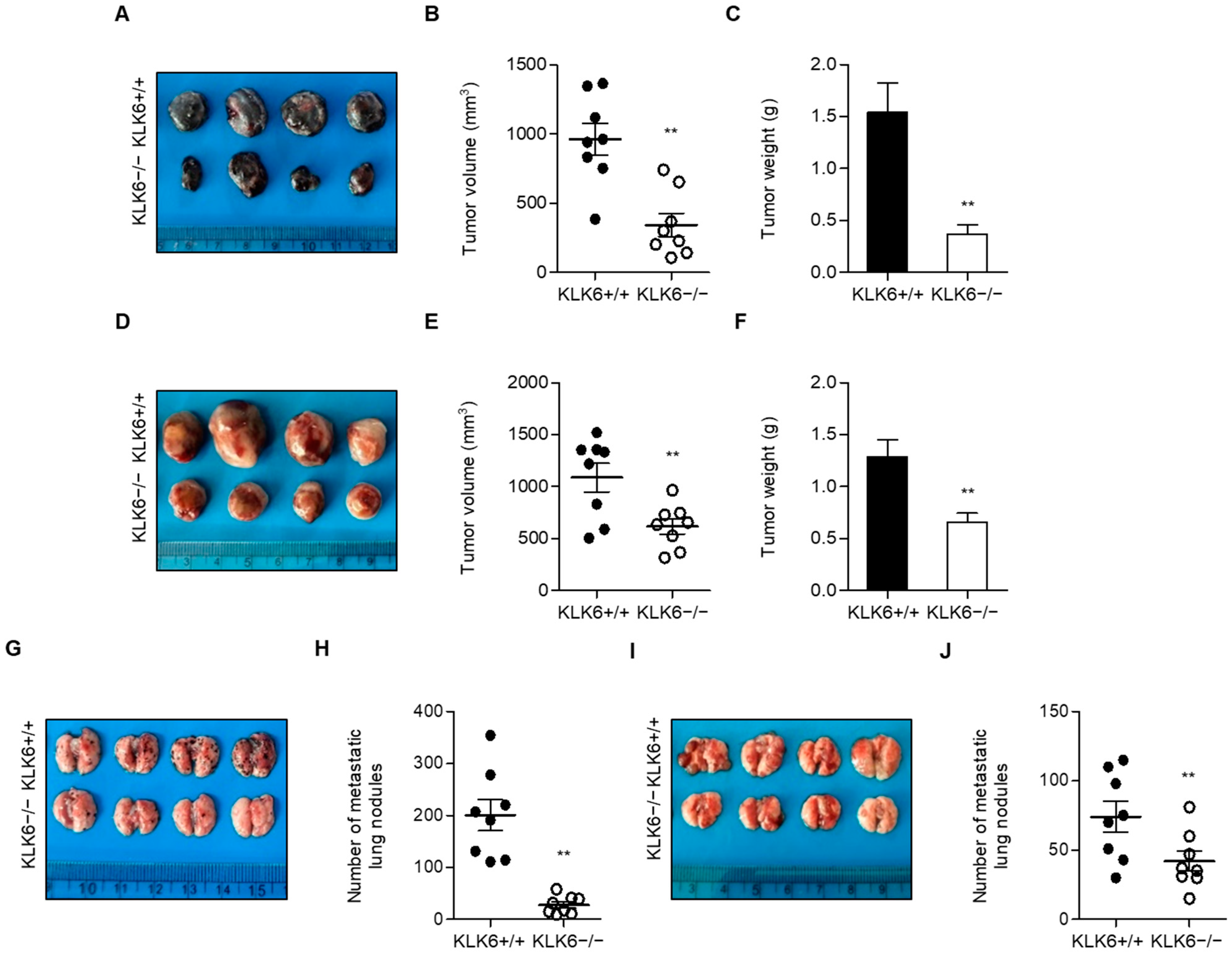
3.1. KLK6 Promoted Tumor Growth and Metastasis
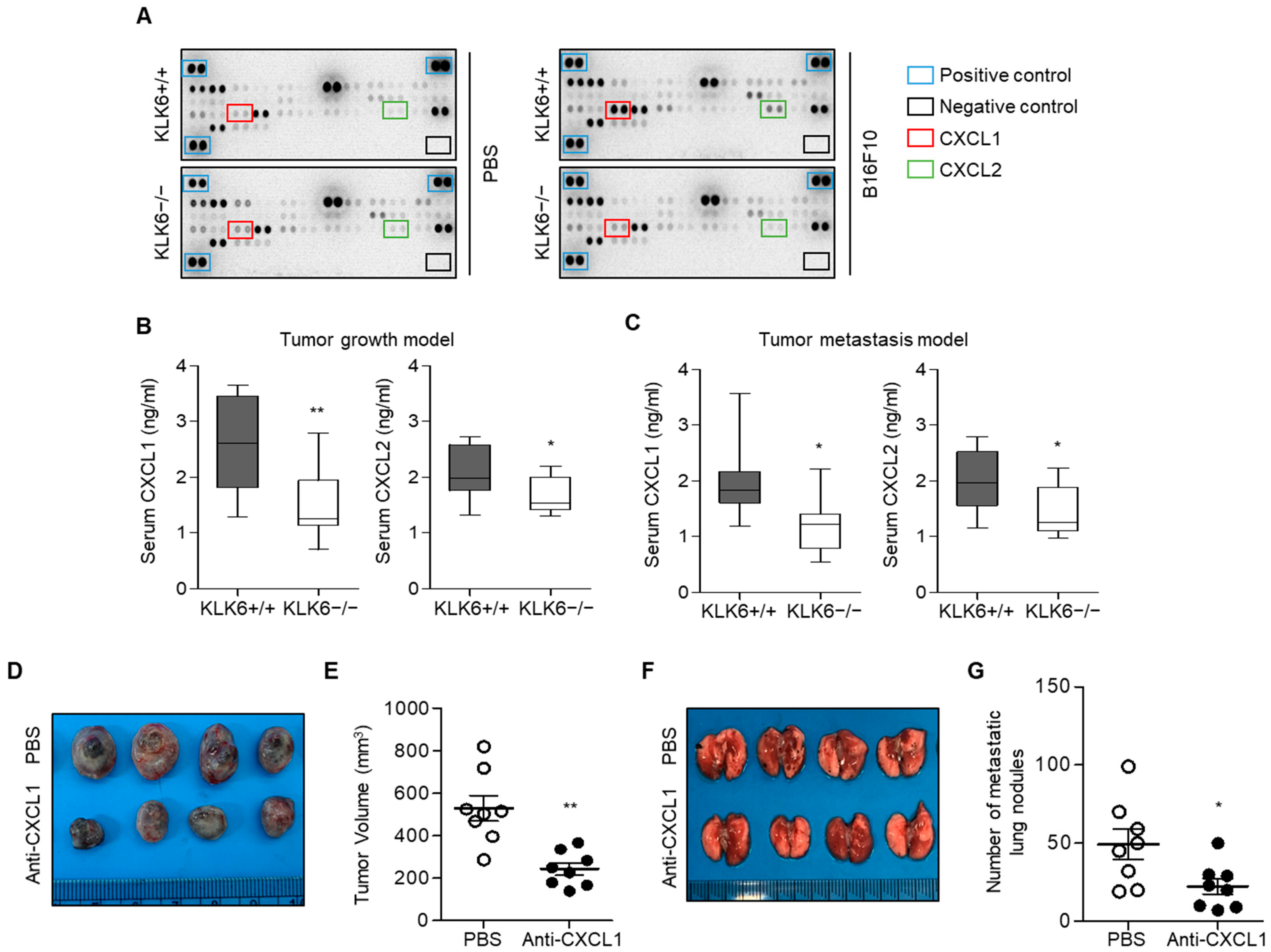
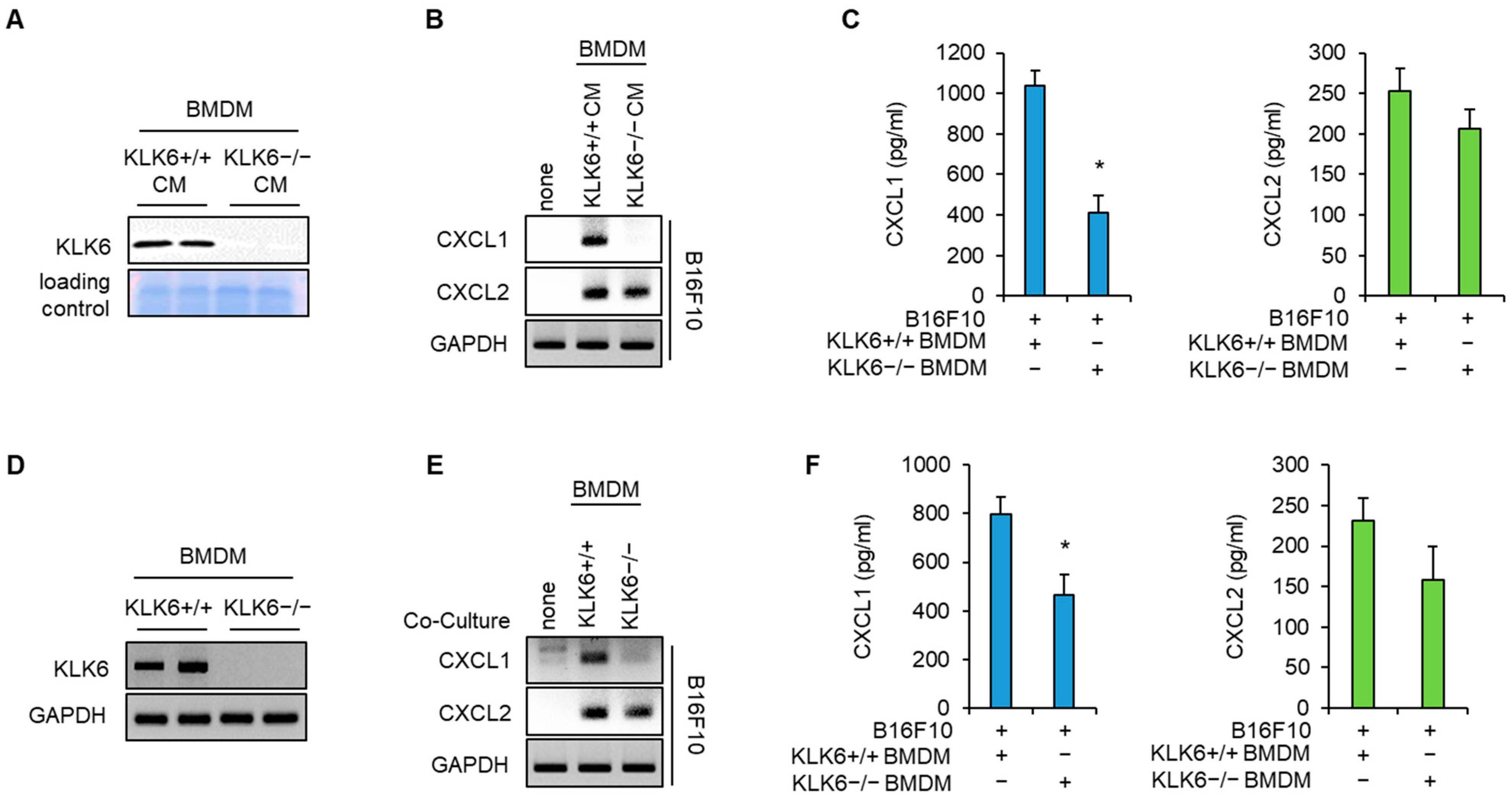
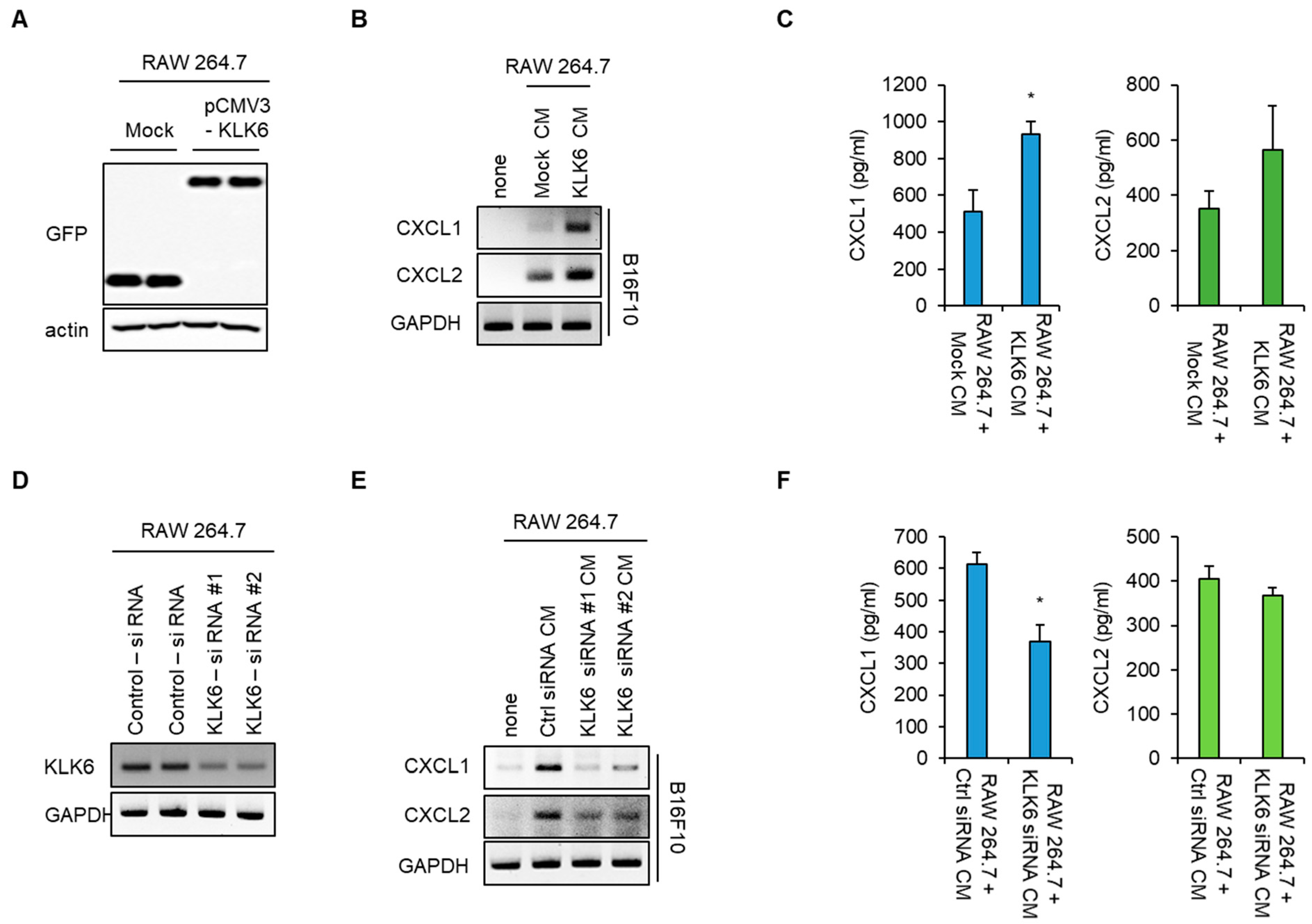
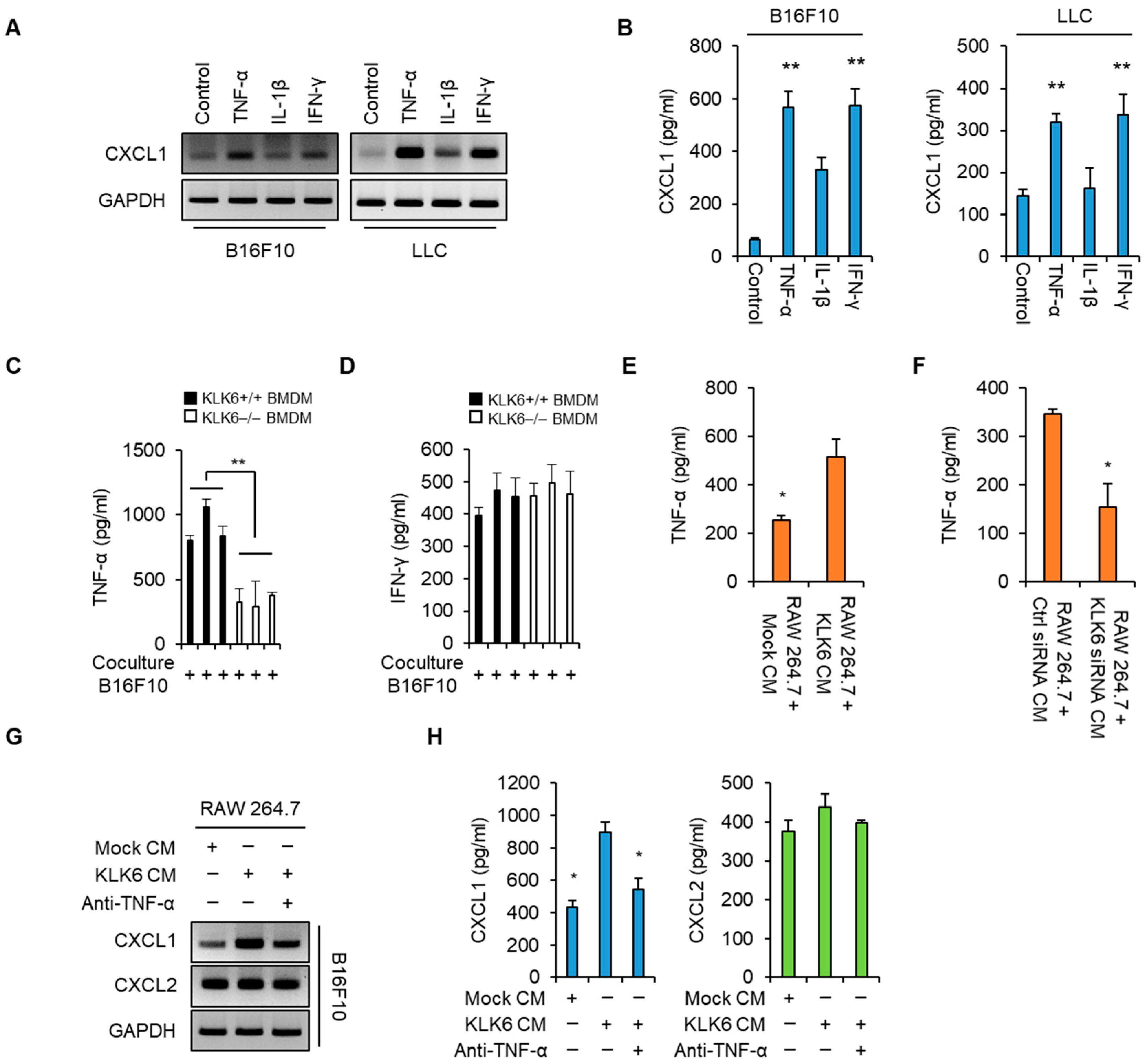
3.2. KLK6 Stimulated CXCL1 Expression in Cancer Cells by Promoting TNF-α Secretion in Macrophages
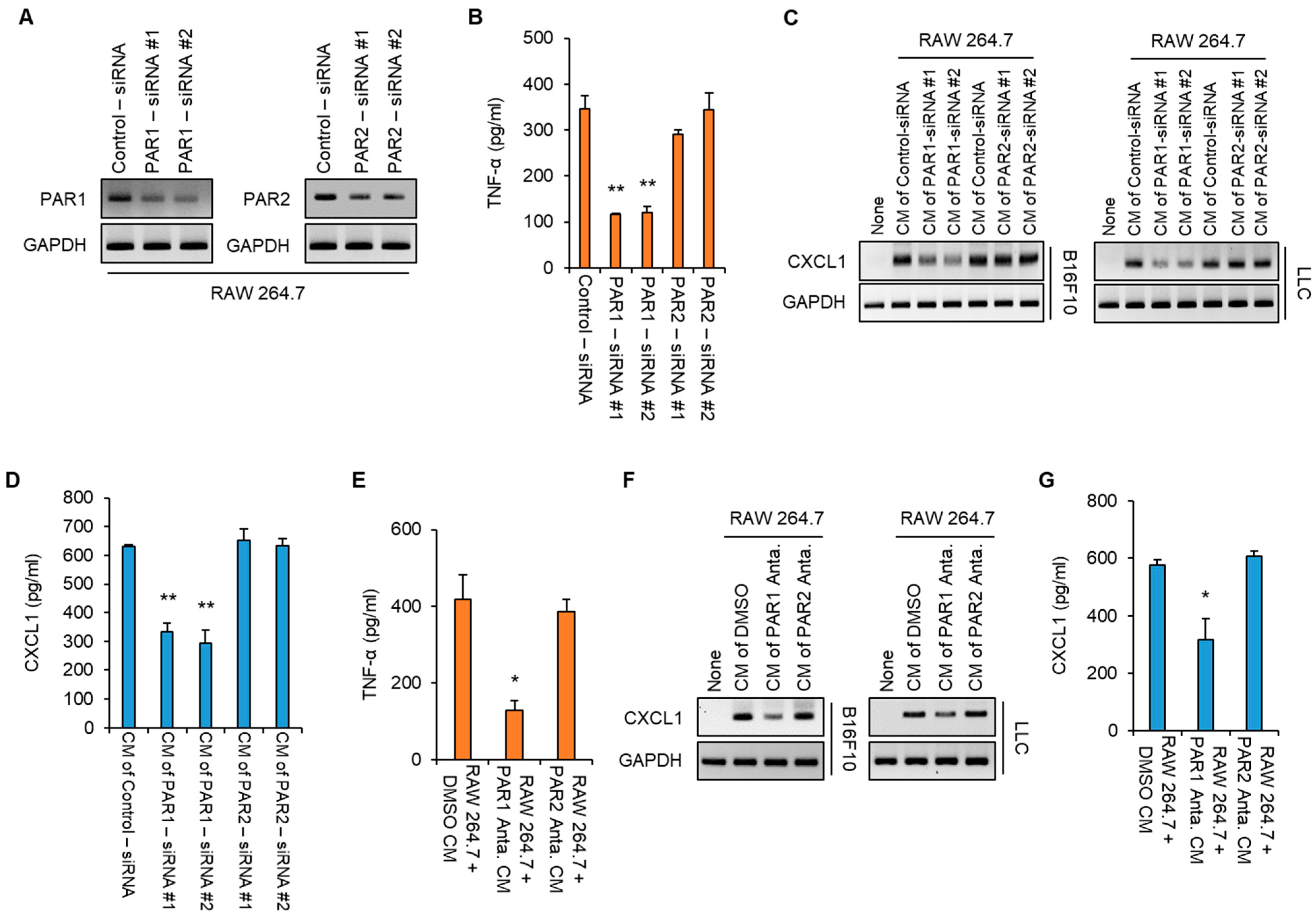
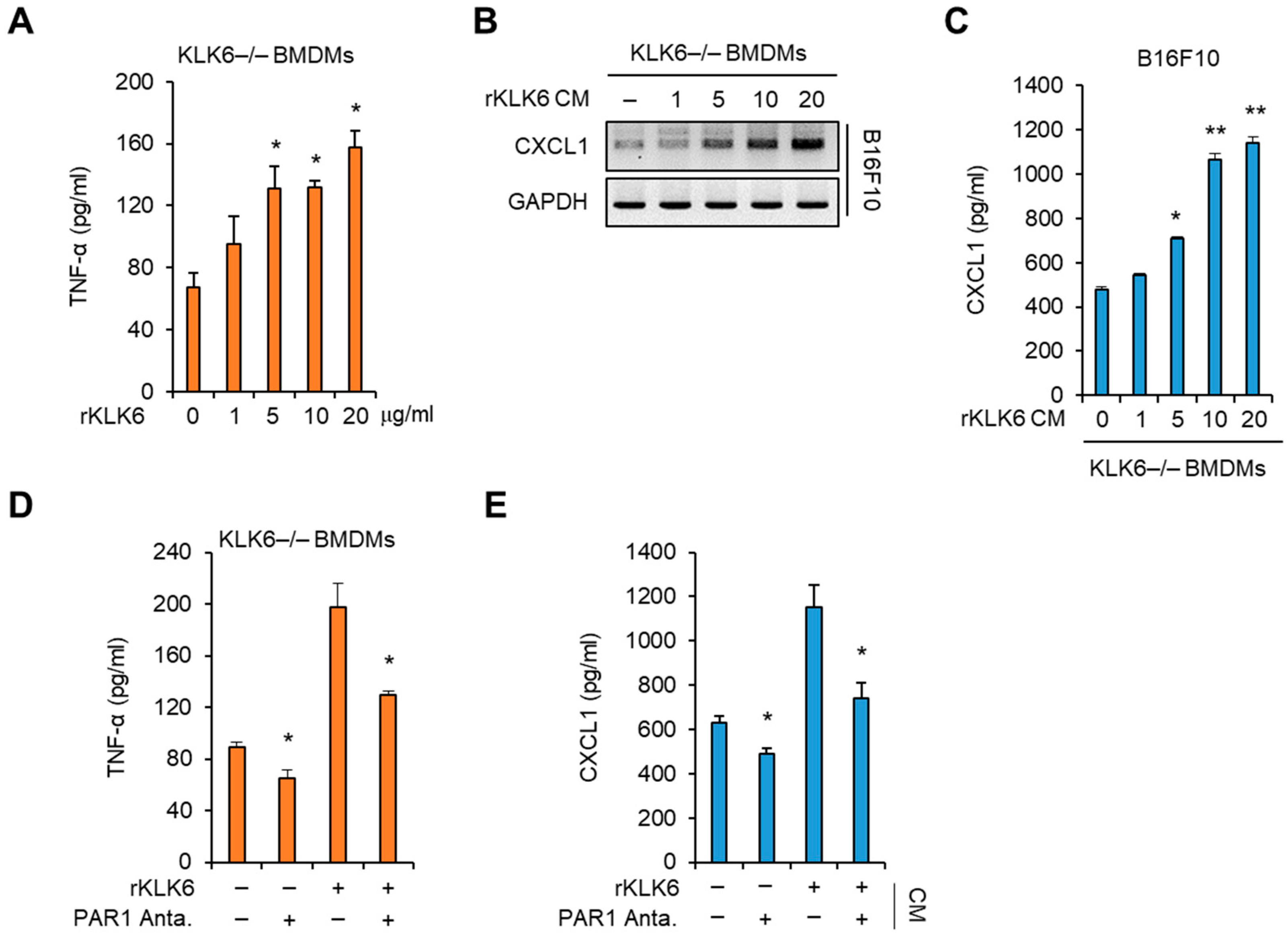
3.3. KLK6 Promoted TNF-α Production in Macrophages via PAR1
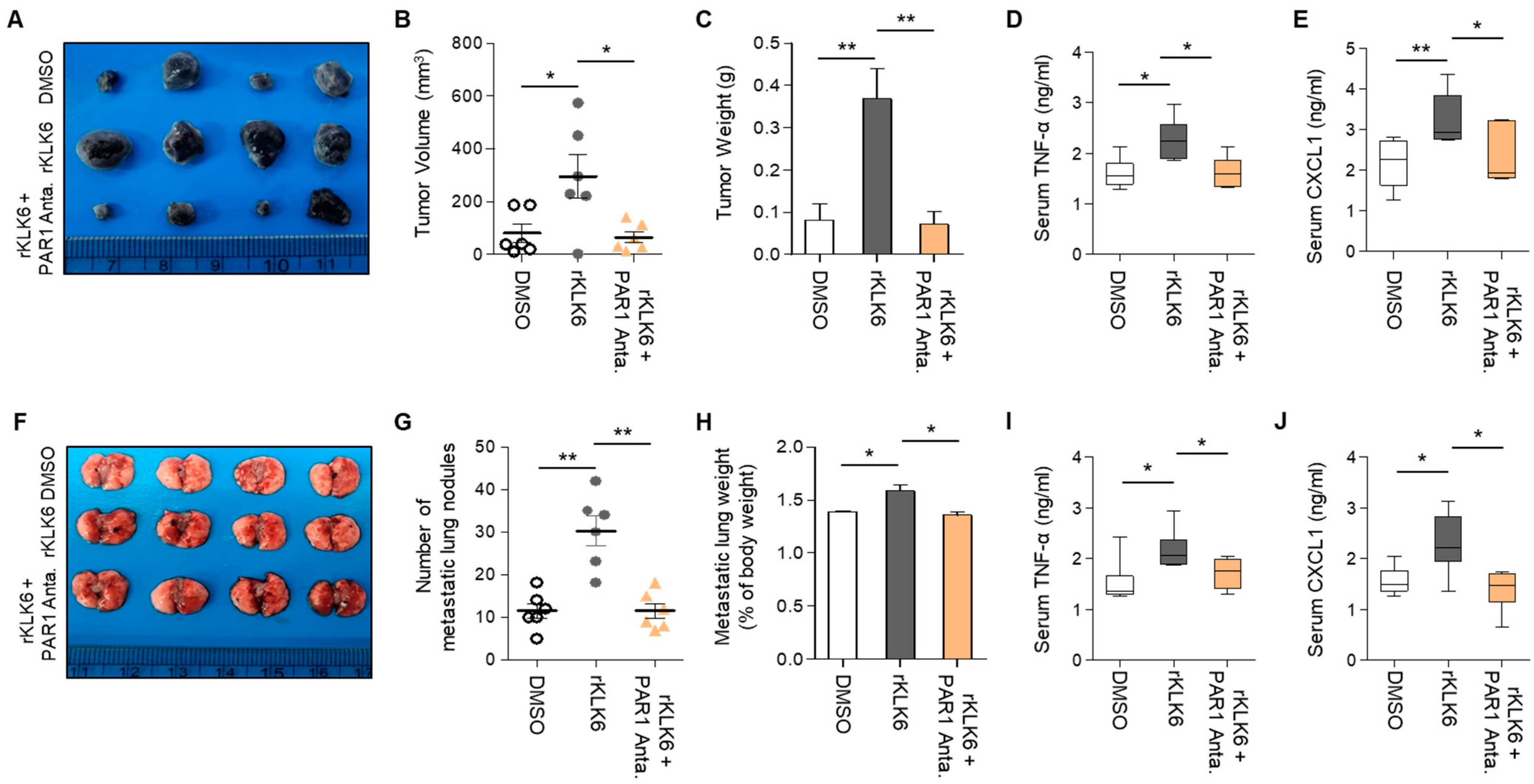
3.4. PAR1 Inhibitor Suppressed rKLK6-Mediated Tumor Growth and Lung Metastasis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [Green Version]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Borgono, C.A.; Diamandis, E.P. The emerging roles of human tissue kallikreins in cancer. Nat. Rev. Cancer 2004, 4, 876–890. [Google Scholar] [CrossRef]
- Bayani, J.; Diamandis, E.P. The physiology and pathobiology of human kallikrein-related peptidase 6 (KLK6). Clin. Chem. Lab. Med. 2011, 50, 211–233. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Maruyama, M.; Akagi, T.; Hashikawa, T.; Kanazawa, I.; Tsuji, S.; Nukina, N. Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 2003, 12, 2625–2635. [Google Scholar] [CrossRef] [Green Version]
- Ashby, E.L.; Kehoe, P.G.; Love, S. Kallikrein-related peptidase 6 in Alzheimer’s disease and vascular dementia. Brain Res. 2010, 1363, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vandell, A.G.; Larson, N.; Laxmikanthan, G.; Panos, M.; Blaber, S.I.; Blaber, M.; Scarisbrick, I.A. Protease-activated receptor dependent and independent signaling by kallikreins 1 and 6 in CNS neuron and astroglial cell lines. J. Neurochem. 2008, 107, 855–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtukiewicz, M.Z.; Hempel, D.; Sierko, E.; Tucker, S.C.; Honn, K.V. Protease-activated receptors (PARs)--biology and role in cancer invasion and metastasis. Cancer Metastasis Rev. 2015, 34, 775–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, S.R. On PAR with aPC to target inflammasomes. Blood 2017, 130, 2579–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattri, S.; Khattri, S.; Shemer, A.; Rozenblit, M.; Dhingra, N.; Czarnowicki, T.; Finney, R.; Gilleaudeau, P.; Whalen, M.S.; Zheng, X.; et al. Cyclosporine in patients with atopic dermatitis modulates activated inflammatory pathways and reverses epidermal pathology. J. Allergy Clin. Immunol. 2014, 133, 1626–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, J.G.; Fretzin, S.; Suárez-Fariñas, M.; Haslett, P.A.; Phipps, K.M.; Cameron, G.S.; McColm, J.; Katcherian, A.; Cueto, I.; White, T.; et al. IL-17A is essential for cell activation and inflammatory gene circuits in subjects with psoriasis. J. Allergy Clin. Immunol. 2012, 130, 145–154.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eissa, A.; Cretu, D.; Soosaipillai, A.; Thavaneswaran, A.; Pellett, F.; Diamandis, A.; Cevikbas, F.; Steinhoff, M.; Diamandis, E.P.; Gladman, D.; et al. Serum kallikrein-8 correlates with skin activity, but not psoriatic arthritis, in patients with psoriatic disease. Clin. Chem. Lab. Med. 2013, 51, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Stalmach, A.; Johnsson, H.; McInnes, I.B.; Husi, H.; Klein, J.; Dakna, M.; Mullen, W.; Mischak, H.; Porter, D. Identification of urinary peptide biomarkers associated with rheumatoid arthritis. PLoS ONE 2014, 9, e104625. [Google Scholar] [CrossRef] [PubMed]
- Billi, A.C.; Ludwig, J.E.; Fritz, Y.; Rozic, R.; Swindell, W.R.; Tsoi, L.C.; Gruzska, D.; Abdollahi-Roodsaz, S.; Xing, X.; Diaconu, D.; et al. KLK6 expression in skin induces PAR1-mediated psoriasiform dermatitis and inflammatory joint disease. J. Clin. Investig. 2020, 130, 3151–3157. [Google Scholar] [CrossRef]
- Yoon, H.; Radulovic, M.; Wu, J.; Blaber, S.I.; Blaber, M.; Fehlings, M.G.; Scarisbrick, I.A. Kallikrein 6 signals through PAR1 and PAR2 to promote neuron injury and exacerbate glutamate neurotoxicity. J. Neurochem. 2013, 127, 283–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pampalakis, G.; Zingkou, E.; Sidiropoulos, K.G.; Diamandis, E.P.; Zoumpourlis, V.; Yousef, G.M.; Sotiropoulou, G. Biochemical pathways mediated by KLK6 protease in breast cancer. Mol. Oncol. 2019, 13, 2329–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathalie, H.V.; Chris, P.; Serge, G.; Catherine, C.; Benjamin, B.; Claire, B.; Christelle, P.; Briollais, L.; Pascale, R.; Lise, J.M.; et al. High kallikrein-related peptidase 6 in non-small cell lung cancer cells: An indicator of tumour proliferation and poor prognosis. J. Cell. Mol. Med. 2009, 13, 4014–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckert, F.; Hennig, M.; Petraki, C.D.; Wehrum, D.; Distler, M.; Denz, A.; Schröder, M.; Dawelbait, G.; Kalthoff, H.; Saeger, H.D.; et al. Co-expression of KLK6 and KLK10 as prognostic factors for survival in pancreatic ductal adenocarcinoma. Br. J. Cancer 2008, 99, 1484–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; Kim, J.T.; Yoon, H.R.; Kang, M.A.; Kim, J.H.; Lee, Y.H.; Kim, J.W.; Lee, S.J.; Song, E.Y.; Myung, P.K.; et al. Upregulation and secretion of kallikrein-related peptidase 6 (KLK6) in gastric cancer. Tumor Biol. 2012, 33, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Song, E.Y.; Chung, K.S.; Kang, M.A.; Kim, J.W.; Kim, S.J.; Yeom, Y.I.; Kim, J.H.; Kim, K.H.; Lee, H.G. Up-regulation and clinical significance of serine protease kallikrein 6 in colon cancer. Cancer 2011, 117, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- Diamandis, E.P.; Yousef, G.M.; Soosaipillai, A.R.; Bunting, P. Human kallikrein 6 (zyme/protease M/neurosin): A new serum biomarker of ovarian carcinoma. Clin. Biochem. 2000, 33, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Klucky, B.; Mueller, R.; Vogt, I.; Teurich, S.; Hartenstein, B.; Breuhahn, K.; Flechtenmacher, C.; Angel, P.; Hess, J. Kallikrein 6 induces E-cadherin shedding and promotes cell proliferation, migration, and invasion. Cancer Res. 2007, 67, 8198–8206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenzer, S.; Peterziel, H.; Mauch, C.; Blaber, S.I.; Blaber, M.; Angel, P.; Hess, J. Expression and function of the kallikrein-related peptidase 6 in the human melanoma microenvironment. J. Investig. Dermatol. 2011, 131, 2281–2288. [Google Scholar] [CrossRef] [Green Version]
- Khoury, N.; Zingkou, E.; Pampalakis, G.; Sofopoulos, M.; Zoumpourlis, V.; Sotiropoulou, G. KLK6 protease accelerates skin tumor formation and progression. Carcinogenesis. 2018, 39, 1529–1536. [Google Scholar] [CrossRef]
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight. 2018, 3, e99364. [Google Scholar] [CrossRef]
- Sellau, J.; Groneberg, M.; Fehling, H.; Thye, T.; Hoenow, S.; Marggraff, C.; Weskamm, M.; Hansen, C.; Stanelle-Bertram, S.; Kuehl, S.; et al. Androgens predispose males to monocyte-mediated immunopathology by inducing the expression of leukocyte recruitment factor CXCL1. Nat. Commun. 2020, 11, 3459. [Google Scholar] [CrossRef] [PubMed]
- Gobbetti, T.; Cenac, N.; Motta, J.P.; Rolland, C.; Martin, L.; Gordon, P.A.; Steinhoff, M.; Barocelli, E.; Vergnolle, N. Serine protease inhibition reduces post-ischemic granulocyte recruitment in mouse intestine. Am. J. Pathol. 2012, 180, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.K., 2nd; Sheehan, J.P.; Bratton, B.P.; Moore, G.M.; Mateus, A.; Li, S.H.; Kim, H.; Rabinowitz, J.D.; Typas, A.; Savitski, M.M.; et al. A Dual-Mechanism Antibiotic Kills Gram-Negative Bacteria and Avoids Drug Resistance. Cell 2020, 181, 1518–1532.e14. [Google Scholar] [CrossRef]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Liu, W.; Zheng, Y.; Wang, S.; Yang, B.; Li, M.; Song, J.; Zhang, F.; Zhang, X.; Wang, Q.; et al. CXCL1 derived from tumor-associated macrophages promotes breast cancer metastasis via activating NF-kappaB/SOX4 signaling. Cell Death Dis. 2018, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Blaber, S.I.; Scarisbrick, I.A.; Bernett, M.J.; Dhanarajan, P.; Seavy, M.A.; Jin, Y.; Schwartz, M.A.; Rodriguez, M.; Blaber, M. Enzymatic properties of rat myelencephalon-specific protease. Biochemistry 2002, 41, 1165–1173. [Google Scholar] [CrossRef] [Green Version]
- Ooi, K.G.; Galatowicz, G.; Calder, V.L.; Lightman, S.L. Cytokines and chemokines in uveitis: Is there a correlation with clinical phenotype? Clin. Med. Res. 2006, 4, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Shieh, J.M.; Tsai, Y.J.; Tsou, C.J.; Wu, W.B. CXCL1 regulation in human pulmonary epithelial cells by tumor necrosis factor. Cell. Physiol. Biochem. 2014, 34, 1373–1384. [Google Scholar] [CrossRef]
- Lee, C.H.; Syu, S.H.; Liu, K.J.; Chu, P.Y.; Yang, W.C.; Lin, P.; Shieh, W.Y. Interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer. Oncotarget 2015, 6, 38866–38880. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Naruse, T.; Kato, H.; Ishida, Y.; Nakagawa, T.; Ono, S.; Shigeishi, H.; Takechi, M. Differential regulation by IFN-γ on TNF-α-induced chemokine expression in synovial fibroblasts from temporomandibular joint. Mol. Med. Rep. 2017, 16, 6850–6857. [Google Scholar] [CrossRef] [Green Version]
- Vakrakou, A.; Devetzi, M.; Papachristopoulou, G.; Malachias, A.; Scorilas, A.; Xynopoulos, D.; Talieri, M. Kallikrein-related peptidase 6 (KLK6) expression in the progression of colon adenoma to carcinoma. Biol. Chem. 2014, 395, 1105–1117. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, P.; Richmond, A. Role of CXCL1 in tumorigenesis of melanoma. J. Leukoc. Biol. 2002, 72, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Zambrano, M.; Rodriguez-Montesinos, J.; Crespo-Avilan, G.E.; Munoz-Vega, M.; Preissner, K.T. Thrombin Promotes Macrophage Polarization into M1-Like Phenotype to Induce Inflammatory Responses. Thromb. Haemost. 2020, 120, 658–670. [Google Scholar] [CrossRef]
- Kakoschky, B.; Pleli, T.; Schmithals, C.; Zeuzem, S.; Brüne, B.; Vogl, T.J.; Korf, H.W.; Weigert, A.; Piiper, A. Selective targeting of tumor associated macrophages in different tumor models. PLoS ONE 2018, 13, e0193015. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, L.C.; Rodriguez, E.; Degenhardt, F.; Baurecht, H.; Wehkamp, U.; Volks, N.; Szymczak, S.; Swindell, W.R.; Sarkar, M.K.; Raja, K.; et al. Atopic Dermatitis Is an IL-13-Dominant Disease with Greater Molecular Heterogeneity Compared to Psoriasis. J. Investig. Dermatol. 2019, 139, 1480–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covic, L.; Kuliopulos, A. Protease-Activated Receptor 1 as Therapeutic Target in Breast, Lung, and Ovarian Cancer: Pepducin Approach. Int. J. Mol. Sci. 2018, 19, 2237. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, Y.S.; Cho, H.J.; Park, E.S.; Lim, J.; Yoon, H.R.; Kim, J.-T.; Yoon, S.R.; Jung, H.; Choe, Y.-K.; Kim, Y.-H.; et al. KLK6/PAR1 Axis Promotes Tumor Growth and Metastasis by Regulating Cross-Talk between Tumor Cells and Macrophages. Cells 2022, 11, 4101. https://doi.org/10.3390/cells11244101
Hwang YS, Cho HJ, Park ES, Lim J, Yoon HR, Kim J-T, Yoon SR, Jung H, Choe Y-K, Kim Y-H, et al. KLK6/PAR1 Axis Promotes Tumor Growth and Metastasis by Regulating Cross-Talk between Tumor Cells and Macrophages. Cells. 2022; 11(24):4101. https://doi.org/10.3390/cells11244101
Chicago/Turabian StyleHwang, Yo Sep, Hee Jun Cho, Eun Sun Park, Jeewon Lim, Hyang Ran Yoon, Jong-Tae Kim, Suk Ran Yoon, Haiyoung Jung, Yong-Kyung Choe, Yong-Hoon Kim, and et al. 2022. "KLK6/PAR1 Axis Promotes Tumor Growth and Metastasis by Regulating Cross-Talk between Tumor Cells and Macrophages" Cells 11, no. 24: 4101. https://doi.org/10.3390/cells11244101