
PICALM and Alzheimer’s Disease: An Update and Perspectives

 , , , and
, , , and
Abstract
:1. Physiopathology of Alzheimer’s Disease and Genetic Risk Factors
2. Biology of PICALM
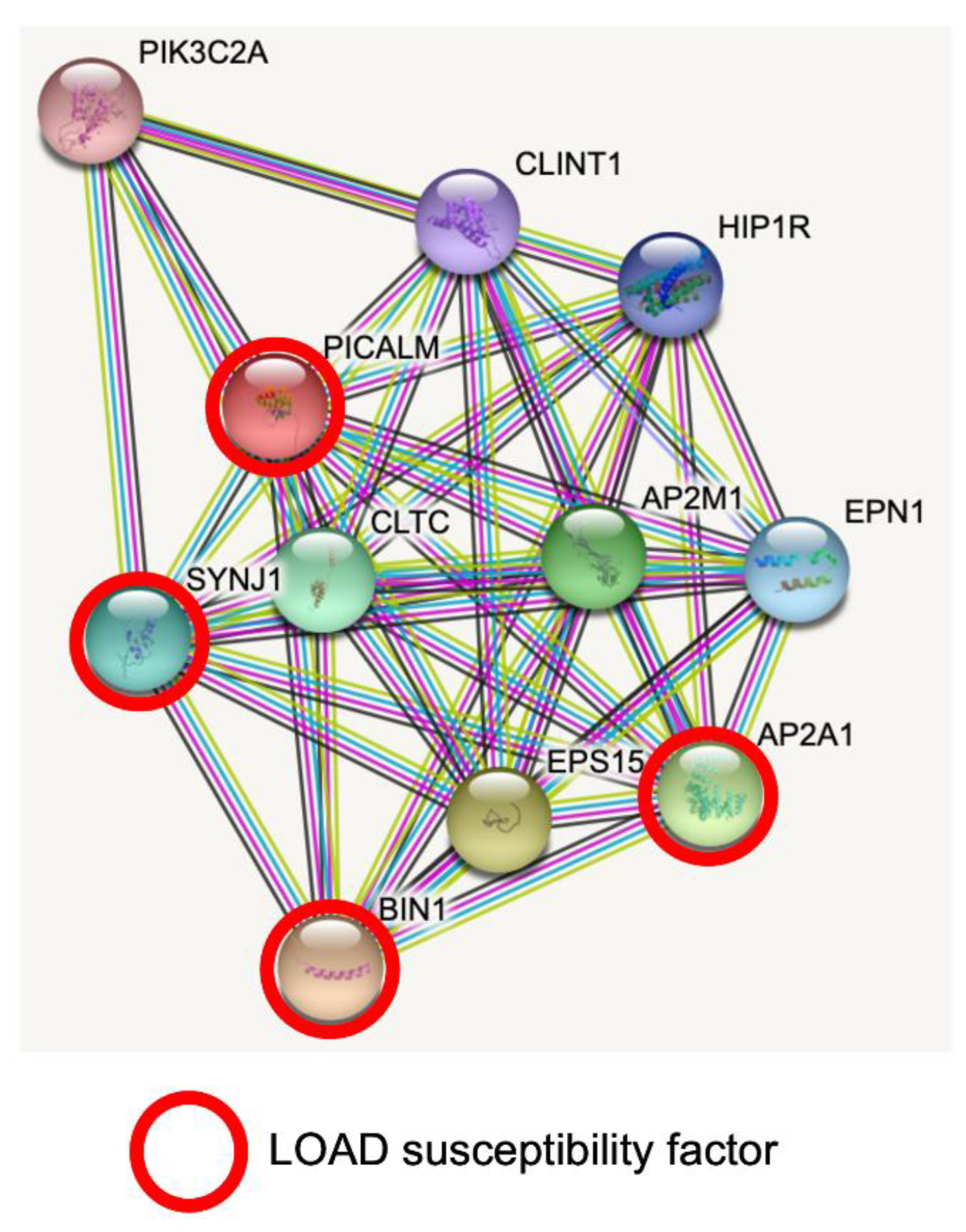
2.1. Tissue Expression, Cellular Functions and Protein Interaction of PICALM
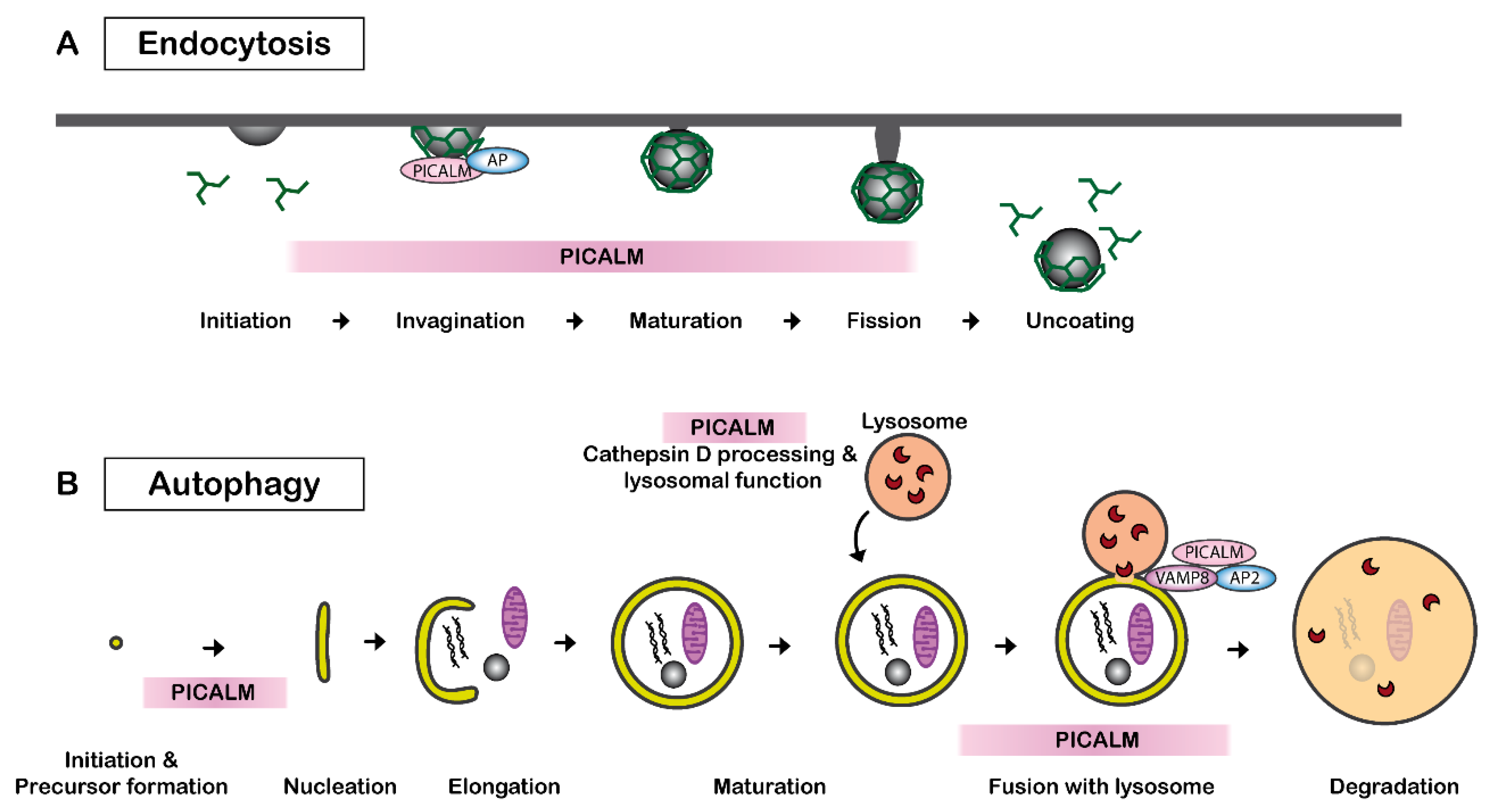
2.1.1. PICALM and Clathrin-Mediated Endocytosis
2.1.2. PICALM, Hematopoiesis, Transferrin-Uptake and Cholesterol Homeostasis
2.1.3. The Roles of PICALM in Neuronal Polarity and Synaptic Vesicle Sorting
2.1.4. PICALM and Autophagy
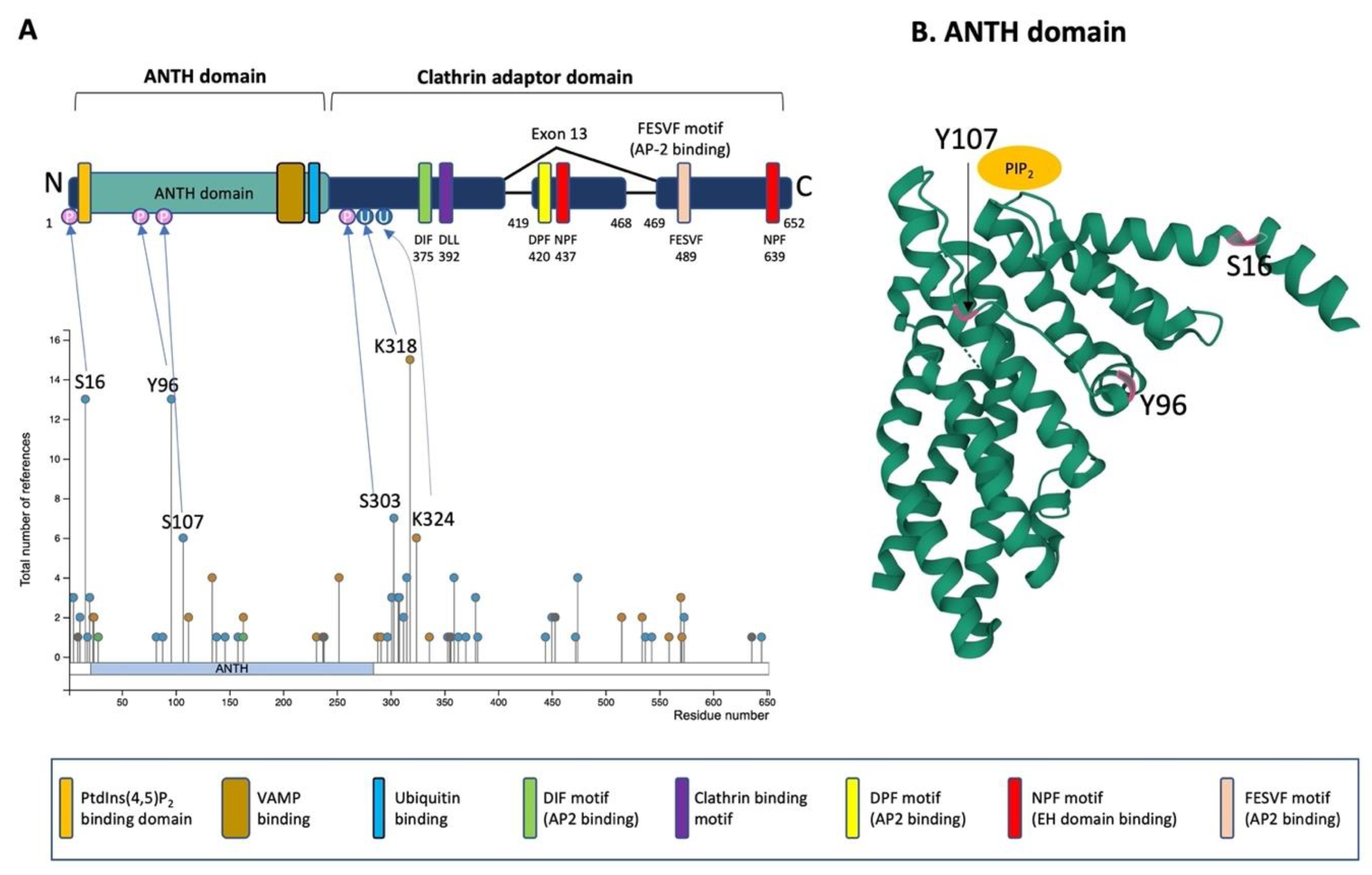
2.2. Protein Structure of PICALM
2.3. Post-Translational Modifications of PICALM
3. PICALM and Alzheimer’s Disease Pathology
3.1. PICALM Gene and AD
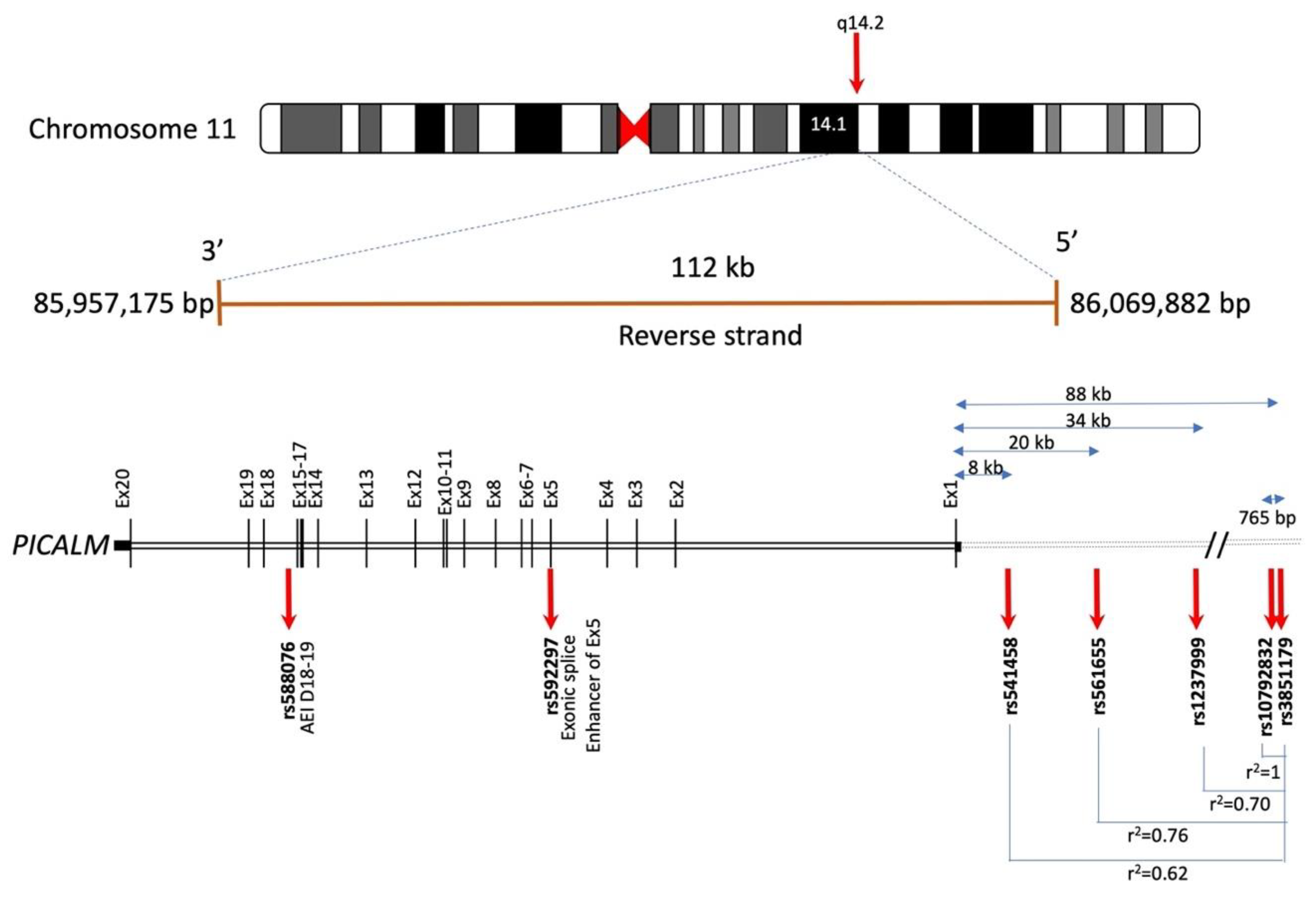
3.1.1. Gene Location and Genetic Variants of PICALM
3.1.2. PICALM and Quantitative Trait Loci (QTL)
3.1.3. PICALM Variants and Phenotypes Relevant to AD
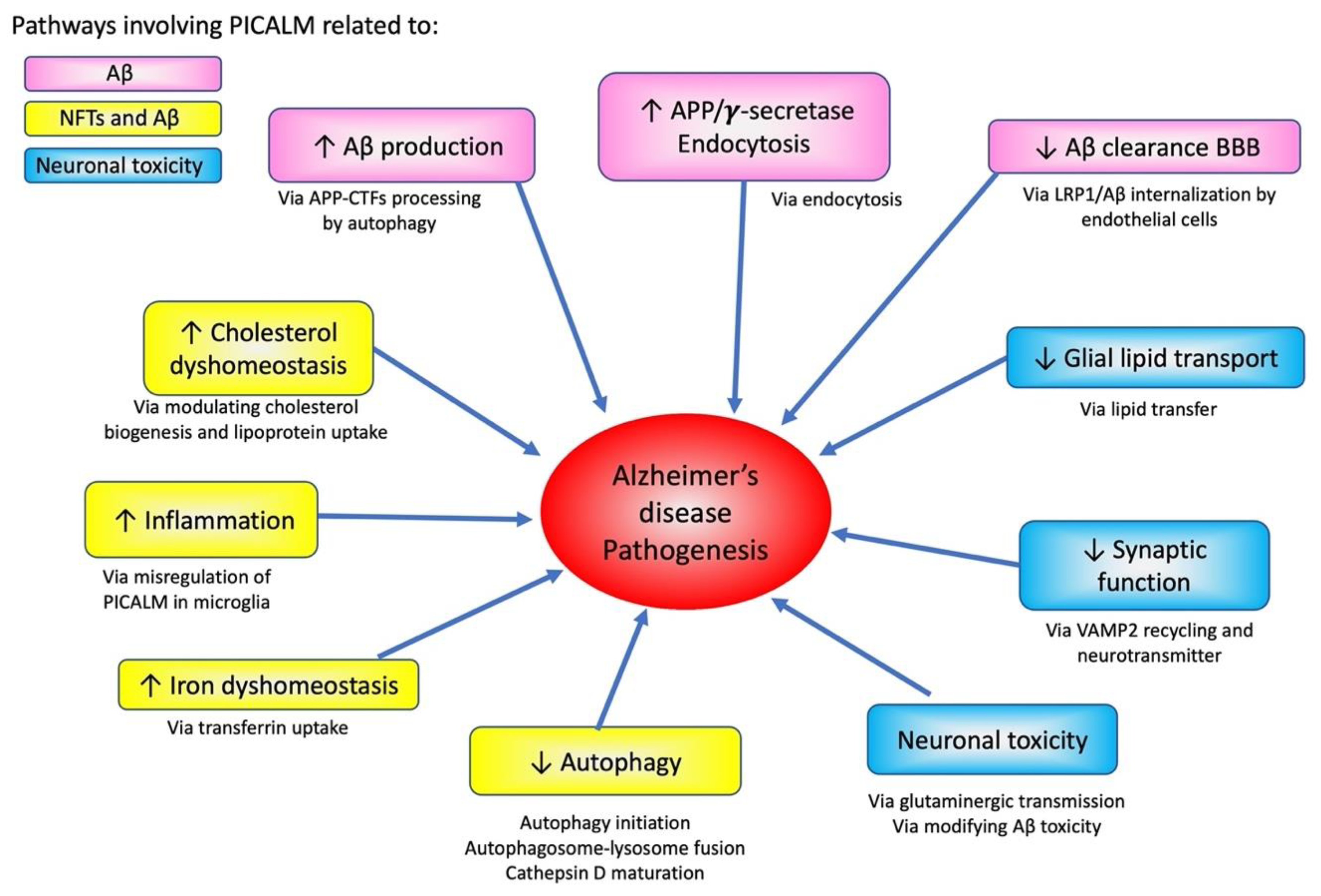
3.2. PICALM and Aβ Pathology
3.2.1. PICALM and APP Processing
3.2.2. PICALM and Aβ-Mediated Neurotoxicity via Glutaminergic Neurotransmission
3.3. PICALM and Tau
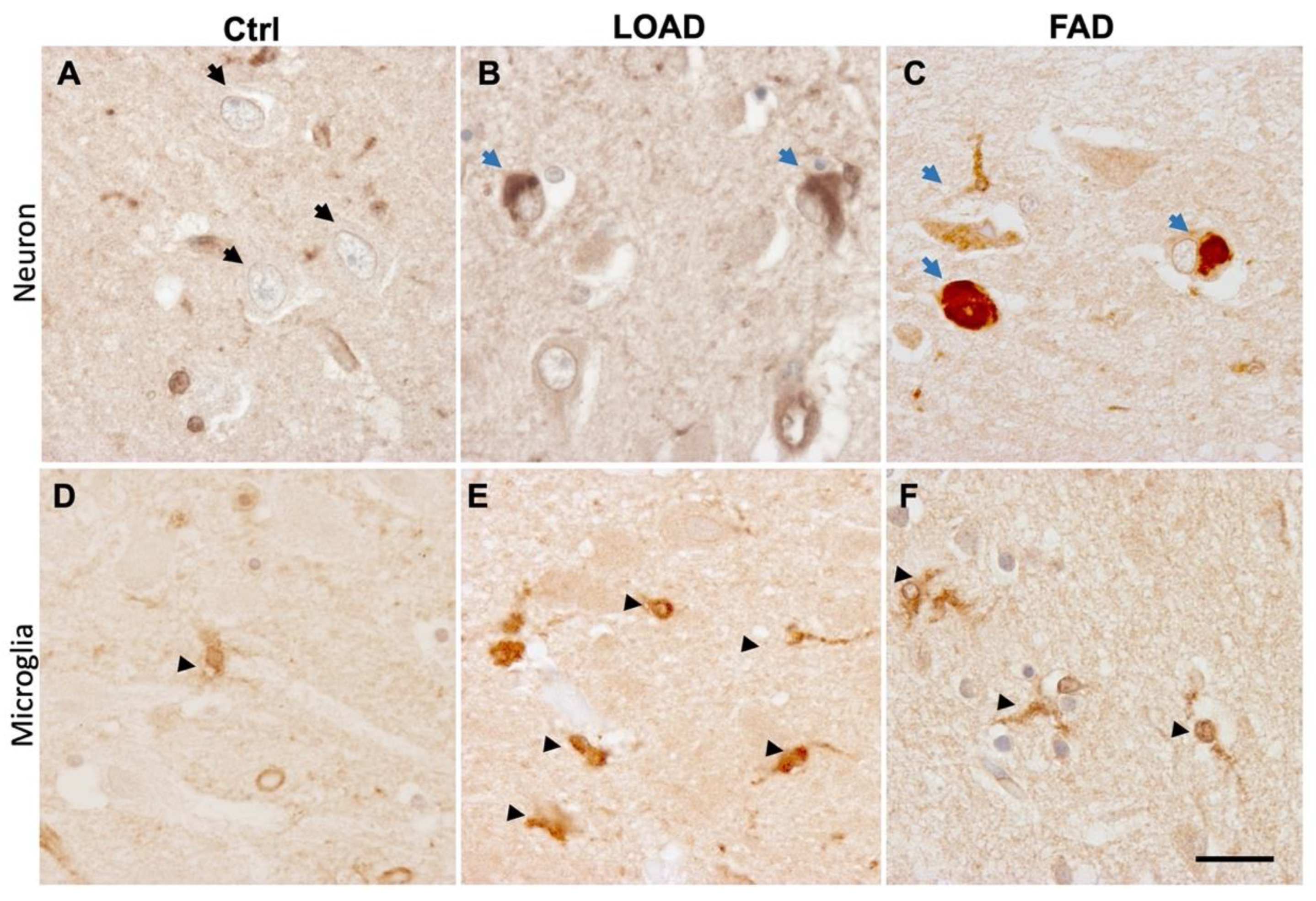
3.3.1. PICALM Expression and Localization in AD Brains
3.3.2. PICALM in Primary Tauopathy Brains
3.3.3. PICALM and Tau in Cellular and Animal Models
3.3.4. PICALM and Tau Pathology Propagation
3.4. PICALM and Glial Cells
4. Discussion
4.1. Difficulties in Interpreting Expression Levels of PICALM in AD Brains
4.2. Challenges in Deciphering the Effect of PICALM Variants on Its Expression
4.3. Potential Role of PICALM in Cellular Homeostasis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, C.W.; Quaranta, V.; Glenner, G.G. Neuritic Plaques and Cerebrovascular Amyloid in Alzheimer Disease Are Antigenically Related. Proc. Natl. Acad. Sci. USA 1985, 82, 8729–8732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brion, J.P.; Couck, A.M.; Passareiro, E.; Flament-Durand, J. Neurofibrillary Tangles of Alzheimer’s Disease: An Immunohistochemical Study. J. Submicrosc. Cytol. 1985, 17, 89–96. [Google Scholar]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and Basic Pathology of Alzheimer Disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Voytyuk, I.; De Strooper, B.; Chavez-Gutierrez, L. Modulation of Gamma- and Beta-Secretases as Early Prevention against Alzheimer’s Disease. Biol. Psychiatry 2018, 83, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. App Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Holtzman, D.M. Holtzman. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in Physiology and Pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef]
- Goedert, M. Tau Filaments in Neurodegenerative Diseases. FEBS Lett. 2018, 592, 2383–2391. [Google Scholar] [CrossRef] [Green Version]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Clavaguera, F.; Hench, J.; Goedert, M.; Tolnay, M. Prion-Like Transmission and Spreading of Tau Pathology. Neuropathol. Appl. Neurobiol. 2014, 41, 47–58. [Google Scholar] [CrossRef]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau Protein Isoforms, Phosphorylation and Role in Neurodegenerative Disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E. The Genetics of Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006296. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, R.; Bras, J. The Age Factor in Alzheimer’s Disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-Wide Association Study Identifies Variants at Clu and Picalm Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-Wide Association Study Identifies Variants at Clu and Cr1 Associated with Alzheimer’s Disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Bellenguez, C.; Kucukali, F.; Jansen, I.E.; Kleineidam, L.; Moreno-Grau, S.; Amin, N.; Naj, A.C.; Campos-Martin, R.; Grenier-Boley, B.; Andrade, V.; et al. New Insights into the Genetic Etiology of Alzheimer’s Disease and Related Dementias. Nat. Genet. 2022, 54, 412–436. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Dourlen, P.; Kilinc, D.; Malmanche, N.; Chapuis, J.; Lambert, J.C. The New Genetic Landscape of Alzheimer’s Disease: From Amyloid Cascade to Genetically Driven Synaptic Failure Hypothesis? Acta Neuropathol. 2019, 138, 221–236. [Google Scholar] [CrossRef] [Green Version]
- Van Acker, Z.P.; Bretou, M.; Annaert, W. Endo-Lysosomal Dysregulations and Late-Onset Alzheimer’s Disease: Impact of Genetic Risk Factors. Mol. Neurodegener. 2019, 14, 20. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, M.D.; Chen-Plotkin, A.S. The Post-Gwas Era: From Association to Function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-Wide Analysis of Genetic Loci Associated with Alzheimer Disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-Analysis of 74,046 Individuals Identifies 11 New Susceptibility Loci for Alzheimer’s Disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Li, L.X.; Zhang, L.; Yang, S.; Tian, Y.; Guo, S.S.; Zhang, W.; Zhao, Z.G. Correlation of Picalm Polymorphism Rs3851179 with Alzheimer’s Disease among Caucasian and Chinese Populations: A Meta-Analysis and Systematic Review. Metab. Brain Dis. 2018, 33, 1849–1857. [Google Scholar] [CrossRef]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic Meta-Analysis of Diagnosed Alzheimer’s Disease Identifies New Risk Loci and Implicates Abeta, Tau, Immunity and Lipid Processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-Wide Meta-Analysis Identifies New Loci and Functional Pathways Influencing Alzheimer’s Disease Risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Ensembl. November 2021. Available online: http://www.ensembl.org/ENSG00000073921 (accessed on 1 June 2022).
- Schnetz-Boutaud, N.C.; Hoffman, J.; Coe, J.E.; Murdock, D.G.; Pericak-Vance, M.A.; Haines, J.L. Identification and Confirmation of an Exonic Splicing Enhancer Variation in Exon 5 of the Alzheimer Disease Associated Picalm Gene. Ann. Hum. Genet. 2012, 76, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Parikh, I.; Fardo, D.W.; Estus, S. Genetics of Picalm Expression and Alzheimer’s Disease. PLoS ONE 2014, 9, e91242. [Google Scholar] [CrossRef]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org/ENSG00000073921-PICALM/tissue (accessed on 1 June 2022).
- Baig, S.; Joseph, S.A.; Tayler, H.; Abraham, R.; Owen, M.J.; Williams, J.; Kehoe, P.G.; Love, S. Distribution and Expression of Picalm in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2010, 69, 1071–1077. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; Brion, J.P.; Stygelbout, V.; Suain, V.; Authelet, M.; Dedecker, R.; Chanut, A.; Lacor, P.; Lavaur, J.; Sazdovitch, V.; et al. Clathrin Adaptor Calm/Picalm Is Associated with Neurofibrillary Tangles and Is Cleaved in Alzheimer’s Brains. Acta Neuropathol. 2013, 125, 861–878. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sagare, A.P.; Ma, Q.; Halliday, M.R.; Kong, P.; Kisler, K.; Winkler, E.A.; Ramanathan, A.; Kanekiyo, T.; Bu, G.; et al. Central Role for Picalm in Amyloid-Beta Blood-Brain Barrier Transcytosis and Clearance. Nat. Neurosci. 2015, 18, 978–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Gil, S.C.; Yan, P.; Wang, Y.; Han, S.; Gonzales, E.; Perez, R.; Cirrito, J.R.; Lee, J.M. Role of Phosphatidylinositol Clathrin Assembly Lymphoid-Myeloid Leukemia (Picalm) in Intracellular Amyloid Precursor Protein (App) Processing and Amyloid Plaque Pathogenesis. J. Biol. Chem. 2012, 287, 21279–21289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petralia, R.S.; Yao, P.J. Ap180 and Calm in the Developing Hippocampus: Expression at the Nascent Synapse and Localization to Trafficking Organelles. J. Comp. Neurol. 2007, 504, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Frigerio, C.S.; Corthout, N.; Qian, X.; Lalakova, J.; Kuhnemund, M.; Voytyuk, I.; et al. Spatial Transcriptomics and in Situ Sequencing to Study Alzheimer’s Disease. Cell 2020, 182, 976–991.e19. [Google Scholar] [CrossRef]
- Chen, W.T.; Lu, A.; Craessaerts, K.; Pavie, B.; Sala Frigerio, C.; Corthout, N.; Qian, X.; Lalakova, J.; Kuhnemund, M.; Voytyuk, I.; et al. Alzmap. Available online: https://alzmap.org/ (accessed on 29 April 2018).
- String. Available online: https://string-db.org/ (accessed on 1 June 2022).
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. String V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative Transcriptome Analyses of the Aging Brain Implicate Altered Splicing in Alzheimer’s Disease Susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef]
- Tebar, F.; Bohlander, S.K.; Sorkin, A. Clathrin Assembly Lymphoid Myeloid Leukemia (Calm) Protein: Localization in Endocytic-Coated Pits, Interactions with Clathrin, and the Impact of Overexpression on Clathrin-Mediated Traffic. Mol. Biol. Cell 1999, 10, 2687–2702. [Google Scholar] [CrossRef] [Green Version]
- Meyerholz, A.; Hinrichsen, L.; Groos, S.; Esk, P.C.; Brandes, G.; Ungewickell, E.J. Effect of Clathrin Assembly Lymphoid Myeloid Leukemia Protein Depletion on Clathrin Coat Formation. Traffic 2005, 6, 1225–1234. [Google Scholar] [CrossRef]
- Xu, W.; Tan, L.; Yu, J.T. The Role of Picalm in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 399–413. [Google Scholar] [CrossRef]
- Miranda, A.M.; Herman, M.; Cheng, R.; Nahmani, E.; Barrett, G.; Micevska, E.; Fontaine, G.; Potier, M.C.; Head, E.; Schmitt, F.A.; et al. Excess Synaptojanin 1 Contributes to Place Cell Dysfunction and Memory Deficits in the Aging Hippocampus in Three Types of Alzheimer’s Disease. Cell Rep. 2018, 23, 2967–2975. [Google Scholar] [CrossRef]
- Petralia, R.S.; Wang, Y.X.; Indig, F.E.; Bushlin, I.; Wu, F.; Mattson, M.P.; Yao, P.J. Reduction of Ap180 and Calm Produces Defects in Synaptic Vesicle Size and Density. Neuromol. Med. 2013, 15, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.E.; Mathiasen, S.; Bright, N.A.; Pierre, F.; Kelly, B.T.; Kladt, N.; Schauss, A.; Merrifield, C.J.; Stamou, D.; Honing, S.; et al. Calm Regulates Clathrin-Coated Vesicle Size and Maturation by Directly Sensing and Driving Membrane Curvature. Dev. Cell 2015, 33, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattersley, K.J.; Carosi, J.M.; Hein, L.K.; Bensalem, J.; Sargeant, T.J. Picalm Regulates Cathepsin D Processing and Lysosomal Function. Biochem. Biophys. Res. Commun. 2021, 570, 103–109. [Google Scholar] [CrossRef]
- Nonet, M.L.; Holgado, A.M.; Brewer, F.; Serpe, C.J.; Norbeck, B.A.; Holleran, J.; Wei, L.; Hartwieg, E.; Jorgensen, E.M.; Alfonso, A. Unc-11, a Caenorhabditis Elegans Ap180 Homologue, Regulates the Size and Protein Composition of Synaptic Vesicles. Mol. Biol. Cell 1999, 10, 2343–2360. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, Y.; Maeda, M.; Pasham, M.; Aguet, F.; Tacheva-Grigorova, S.K.; Masuda, T.; Yi, H.; Lee, S.U.; Xu, J.; Teruya-Feldstein, J.; et al. Role of the Clathrin Adaptor Picalm in Normal Hematopoiesis and Polycythemia Vera Pathophysiology. Haematologica 2015, 100, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, K.; Fleming, A.; Imarisio, S.; Ramirez, A.L.; Mercer, J.L.; Jimenez-Sanchez, M.; Bento, C.F.; Puri, C.; Zavodszky, E.; Siddiqi, F.; et al. Picalm Modulates Autophagy Activity and Tau Accumulation. Nat. Commun. 2014, 5, 4998. [Google Scholar] [CrossRef] [Green Version]
- Dreyling, M.H.; Martinez-Climent, J.A.; Zheng, M.; Mao, J.; Rowley, J.D.; Bohlander, S.K. The T(10;11)(P13;Q14) in the U937 Cell Line Results in the Fusion of the Af10 Gene and Calm, Encoding a New Member of the Ap-3 Clathrin Assembly Protein Family. Proc. Natl. Acad. Sci. USA 1996, 93, 4804–4809. [Google Scholar] [CrossRef] [Green Version]
- Klebig, M.L.; Wall, M.D.; Potter, M.D.; Rowe, E.L.; Carpenter, D.A.; Rinchik, E.M. Mutations in the Clathrin-Assembly Gene Picalm Are Responsible for the Hematopoietic and Iron Metabolism Abnormalities in Fit1 Mice. Proc. Natl. Acad. Sci. USA 2003, 100, 8360–8365. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Tanaka, H.; Tanimura, A.; Tanabe, K.; Oe, N.; Rai, S.; Kon, S.; Fukumoto, M.; Takei, K.; Abe, T.; et al. The Clathrin Assembly Protein Picalm Is Required for Erythroid Maturation and Transferrin Internalization in Mice. PLoS ONE 2012, 7, e31854. [Google Scholar] [CrossRef]
- Huang, F.; Khvorova, A.; Marshall, W.; Sorkin, A. Analysis of Clathrin-Mediated Endocytosis of Epidermal Growth Factor Receptor by Rna Interference. J. Biol. Chem. 2004, 279, 16657–16661. [Google Scholar] [CrossRef] [PubMed]
- Harel, A.; Wu, F.; Mattson, M.P.; Morris, C.M.; Yao, P.J. Evidence for Calm in Directing Vamp2 Trafficking. Traffic 2008, 9, 417–429. [Google Scholar] [CrossRef]
- Mercer, J.L.; Argus, J.P.; Crabtree, D.M.; Keenan, M.M.; Wilks, M.Q.; Chi, J.T.; Bensinger, S.J.; Lavau, C.P.; Wechsler, D.S. Modulation of Picalm Levels Perturbs Cellular Cholesterol Homeostasis. PLoS ONE 2015, 10, e0129776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushlin, I.; Petralia, R.S.; Wu, F.; Harel, A.; Mughal, M.R.; Mattson, M.P.; Yao, P.J. Clathrin Assembly Protein Ap180 and Calm Differentially Control Axogenesis and Dendrite Outgrowth in Embryonic Hippocampal Neurons. J. Neurosci. 2008, 28, 10257–10271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koo, S.J.; Markovic, S.; Puchkov, D.; Mahrenholz, C.C.; Beceren-Braun, F.; Maritzen, T.; Dernedde, J.; Volkmer, R.; Oschkinat, H.; Haucke, V. Snare Motif-Mediated Sorting of Synaptobrevin by the Endocytic Adaptors Clathrin Assembly Lymphoid Myeloid Leukemia (Calm) and Ap180 at Synapses. Proc. Natl. Acad. Sci. USA 2012, 108, 13540–13545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.E.; Sahlender, D.A.; Graham, S.C.; Honing, S.; Robinson, M.S.; Peden, A.A.; Owen, D.J. The Molecular Basis for the Endocytosis of Small R-Snares by the Clathrin Adaptor Calm. Cell 2011, 147, 1118–1131. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; Ma, Y.Y. Calcium Permeable-Ampa Receptors and Excitotoxicity in Neurological Disorders. Front. Neural Circuits 2021, 15, 711564. [Google Scholar] [CrossRef]
- Azarnia Tehran, D.; Kochlamazashvili, G.; Pampaloni, N.P.; Sposini, S.; Shergill, J.K.; Lehmann, M.; Pashkova, N.; Schmidt, C.; Lowe, D.; Napieczynska, H.; et al. Selective Endocytosis of Ca2+-Permeable Ampars by the Alzheimer’s Disease Risk Factor Calm Bidirectionally Controls Synaptic Plasticity. Sci. Adv. 2022, 8, eabl5032. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised Autophagy and Neurodegenerative Diseases. Nat. Rev. Neurosci. 2015, 16, 345–357. [Google Scholar] [CrossRef]
- Ford, M.G.; Pearse, B.M.; Higgins, M.K.; Vallis, Y.; Owen, D.J.; Gibson, A.; Hopkins, C.R.; Evans, P.R.; McMahon, H.T. Simultaneous Binding of Ptdins(4,5)P2 and Clathrin by Ap180 in the Nucleation of Clathrin Lattices on Membranes. Science 2001, 291, 1051–1055. [Google Scholar] [CrossRef] [Green Version]
- Phoshositeplus. Available online: https://www.phosphosite.org/ (accessed on 1 June 2022).
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. Phosphositeplus: A Comprehensive Resource for Investigating the Structure and Function of Experimentally Determined Post-Translational Modifications in Man and Mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef] [PubMed]
- Pashkova, N.; Gakhar, L.; Yu, L.; Schnicker, N.J.; Minard, A.Y.; Winistorfer, S.; Johnson, I.E.; Piper, R.C. Anth Domains within Calm, Hip1r, and Sla2 Recognize Ubiquitin Internalization Signals. eLife 2021, 10, e72583. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.C.; Sylvestersen, K.B.; Mund, A.; Lyon, D.; Mullari, M.; Madsen, M.V.; Daniel, J.A.; Jensen, L.J.; Nielsen, M.L. Proteome-Wide Analysis of Arginine Monomethylation Reveals Widespread Occurrence in Human Cells. Sci. Signal 2016, 9, rs9. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics Connects Somatic Mutations to Signalling in Breast Cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Schweppe, D.K.; Rigas, J.R.; Gerber, S.A. Quantitative Phosphoproteomic Profiling of Human Non-Small Cell Lung Cancer Tumors. J. Proteom. 2013, 91, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Sharma, K.; D’Souza, R.C.; Tyanova, S.; Schaab, C.; Wisniewski, J.R.; Cox, J.; Mann, M. Ultradeep Human Phosphoproteome Reveals a Distinct Regulatory Nature of Tyr and Ser/Thr-Based Signaling. Cell Rep. 2014, 8, 1583–1594. [Google Scholar] [CrossRef] [Green Version]
- Shiromizu, T.; Adachi, J.; Watanabe, S.; Murakami, T.; Kuga, T.; Muraoka, S.; Tomonaga, T. Identification of Missing Proteins in the Nextprot Database and Unregistered Phosphopeptides in the Phosphositeplus Database as Part of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2013, 12, 2414–2421. [Google Scholar] [CrossRef]
- Zhou, H.; di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a Comprehensive Characterization of a Human Cancer Cell Phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Mertins, P.; Qiao, J.W.; Patel, J.; Udeshi, N.D.; Clauser, K.R.; Mani, D.R.; Burgess, M.W.; Gillette, M.A.; Jaffe, J.D.; Carr, S.A. Integrated Proteomic Analysis of Post-Translational Modifications by Serial Enrichment. Nat. Methods 2013, 10, 634–637. [Google Scholar] [CrossRef]
- Henriques, A.G.; Muller, T.; Oliveira, J.M.; Cova, M.; da Cruz, E.S.C.B.; da Cruz, E.S.O.A. Altered Protein Phosphorylation as a Resource for Potential Ad Biomarkers. Sci. Rep. 2016, 6, 30319. [Google Scholar] [CrossRef] [Green Version]
- Kettenbach, A.N.; Schweppe, D.K.; Faherty, B.K.; Pechenick, D.; Pletnev, A.A.; Gerber, S.A. Quantitative Phosphoproteomics Identifies Substrates and Functional Modules of Aurora and Polo-Like Kinase Activities in Mitotic Cells. Sci. Signal 2011, 4, rs5. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in Tumors Induces Early and Sustained Phosphorylation Changes in Stress Kinase Pathways but Does Not Affect Global Protein Levels. Mol. Cell Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative Phosphoproteomics Reveals Widespread Full Phosphorylation Site Occupancy During Mitosis. Sci. Signal 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Kim, H.L. Cleavage of Purified Neuronal Clathrin Assembly Protein (Calm) by Caspase 3 and Calpain. Exp. Mol. Med. 2001, 33, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudinskiy, N.; Grishchuk, Y.; Vaslin, A.; Puyal, J.; Delacourte, A.; Hirling, H.; Clarke, P.G.; Luthi-Carter, R. Calpain Hydrolysis of Alpha- and Beta2-Adaptins Decreases Clathrin-Dependent Endocytosis and May Promote Neurodegeneration. J. Biol. Chem. 2009, 284, 12447–12458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Tan, C.C.; Cao, X.P.; Tan, L.; Initiative Alzheimer’s Disease Neuroimaging. Association of Alzheimer’s Disease Risk Variants on the Picalm Gene with Picalm Expression, Core Biomarkers, and Feature Neurodegeneration. Aging 2020, 12, 21202–21219. [Google Scholar] [CrossRef]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Borte, S.; Winsvold, B.S.; Drange, O.K.; et al. A Genome-Wide Association Study with 1,126,563 Individuals Identifies New Risk Loci for Alzheimer’s Disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef]
- Parikh, I.; Medway, C.; Younkin, S.; Fardo, D.W.; Estus, S. An Intronic Picalm Polymorphism, Rs588076, Is Associated with Allelic Expression of a Picalm Isoform. Mol. Neurodegener. 2014, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Lutz, M.W.; Chiba-Falek, O. Bioinformatics Pipeline to Guide Late-Onset Alzheimer’s Disease (Load) Post-Gwas Studies: Prioritizing Transcription Regulatory Variants within Load-Associated Regions. Alzheimers Dement. 2022, 8, e12244. [Google Scholar] [CrossRef]
- Lowy-Gallego, E.; Fairley, S.; Zheng-Bradley, X.; Ruffier, M.; Clarke, L.; Flicek, P.; Consortium Genomes Project. Variant Calling on the Grch38 Assembly with the Data from Phase Three of the 1000 Genomes Project. Wellcome Open Res. 2019, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants at Ms4a4/Ms4a6e, Cd2ap, Cd33 and Epha1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Kamboh, M.I.; Minster, R.L.; Demirci, F.Y.; Ganguli, M.; Dekosky, S.T.; Lopez, O.L.; Barmada, M.M. Association of Clu and Picalm Variants with Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vardarajan, B.N.; Ghani, M.; Kahn, A.; Sheikh, S.; Sato, C.; Barral, S.; Lee, J.H.; Cheng, R.; Reitz, C.; Lantigua, R.; et al. Rare Coding Mutations Identified by Sequencing of Alzheimer Disease Genome-Wide Association Studies Loci. Ann. Neurol. 2015, 78, 487–498. [Google Scholar] [CrossRef]
- Moreno-Grau, S.; de Rojas, I.; Hernandez, I.; Quintela, I.; Montrreal, L.; Alegret, M.; Hernandez-Olasagarre, B.; Madrid, L.; Gonzalez-Perez, A.; Maronas, O.; et al. Genome-Wide Association Analysis of Dementia and Its Clinical Endophenotypes Reveal Novel Loci Associated with Alzheimer’s Disease and Three Causality Networks: The Gr@Ace Project. Alzheimers Dement. 2019, 15, 1333–1347. [Google Scholar] [CrossRef] [PubMed]
- Bryois, J.; Calini, D.; Macnair, W.; Foo, L.; Urich, E.; Ortmann, W.; Iglesias, V.A.; Selvaraj, S.; Nutma, E.; Marzin, M.; et al. Cell-Type-Specific Cis-Eqtls in Eight Human Brain Cell Types Identify Novel Risk Genes for Psychiatric and Neurological Disorders. Nat. Neurosci. 2022, 25, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Kosoy, R.; Fullard, J.F.; Zeng, B.; Bendl, J.; Dong, P.; Rahman, S.; Kleopoulos, S.P.; Shao, Z.; Girdhar, K.; Humphrey, J.; et al. Genetics of the Human Microglia Regulome Refines Alzheimer’s Disease Risk Loci. Nat. Genet. 2022, 54, 1145–1154. [Google Scholar] [CrossRef]
- Young, A.M.H.; Kumasaka, N.; Calvert, F.; Hammond, T.R.; Knights, A.; Panousis, N.; Park, J.S.; Schwartzentruber, J.; Liu, J.; Kundu, K.; et al. A Map of Transcriptional Heterogeneity and Regulatory Variation in Human Microglia. Nat. Genet. 2021, 53, 861–868. [Google Scholar] [CrossRef]
- Lopes, K.P.; Snijders, G.J.L.; Humphrey, J.; Allan, A.; Sneeboer, M.A.M.; Navarro, E.; Schilder, B.M.; Vialle, R.A.; Parks, M.; Missall, R.; et al. Genetic Analysis of the Human Microglial Transcriptome across Brain Regions, Aging and Disease Pathologies. Nat. Genet. 2022, 54, 4–17. [Google Scholar] [CrossRef]
- Thomas, R.S.; Henson, A.; Gerrish, A.; Jones, L.; Williams, J.; Kidd, E.J. Decreasing the Expression of Picalm Reduces Endocytosis and the Activity of Beta-Secretase: Implications for Alzheimer’s Disease. BMC Neurosci. 2016, 17, 50. [Google Scholar] [CrossRef] [Green Version]
- Gockley, J.; Montgomery, K.S.; Poehlman, W.L.; Wiley, J.C.; Liu, Y.; Gerasimov, E.; Greenwood, A.K.; Sieberts, S.K.; Wingo, A.P.; Wingo, T.S.; et al. Multi-Tissue Neocortical Transcriptome-Wide Association Study Implicates 8 Genes across 6 Genomic Loci in Alzheimer’s Disease. Genome Med. 2021, 13, 76. [Google Scholar] [CrossRef]
- Melville, S.A.; Buros, J.; Parrado, A.R.; Vardarajan, B.; Logue, M.W.; Shen, L.; Risacher, S.L.; Kim, S.; Jun, G.; DeCarli, C.; et al. Multiple Loci Influencing Hippocampal Degeneration Identified by Genome Scan. Ann. Neurol. 2012, 72, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Wang, H.F.; Tan, L.; Tan, M.S.; Tan, C.C.; Zhu, X.C.; Miao, D.; Yu, W.J.; Jiang, T.; Tan, L.; et al. The Impact of Picalm Genetic Variations on Reserve Capacity of Posterior Cingulate in Ad Continuum. Sci. Rep. 2016, 6, 24480. [Google Scholar] [CrossRef] [PubMed]
- Schjeide, B.M.; Schnack, C.; Lambert, J.C.; Lill, C.M.; Kirchheiner, J.; Tumani, H.; Otto, M.; Tanzi, R.E.; Lehrach, H.; Amouyel, P.; et al. The Role of Clusterin, Complement Receptor 1, and Phosphatidylinositol Binding Clathrin Assembly Protein in Alzheimer Disease Risk and Cerebrospinal Fluid Biomarker Levels. Arch. Gen. Psychiatry 2011, 68, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, L.; Liu, X.; Shi, Y.; Liu, X.; Luo, B. Genetic Variants of Picalm Rs541458 Modulate Brain Spontaneous Activity in Older Adults with Amnestic Mild Cognitive Impairment. Front. Neurol. 2019, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- Sweet, R.A.; Seltman, H.; Emanuel, J.E.; Lopez, O.L.; Becker, J.T.; Bis, J.C.; Weamer, E.A.; DeMichele-Sweet, M.A.; Kuller, L.H. Effect of Alzheimer’s Disease Risk Genes on Trajectories of Cognitive Function in the Cardiovascular Health Study. Am. J. Psychiatry 2012, 169, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Jones, E.L.; Mok, K.; Hanney, M.; Harold, D.; Sims, R.; Williams, J.; Ballard, C. Evidence That Picalm Affects Age at Onset of Alzheimer’s Dementia in Down Syndrome. Neurobiol. Aging 2013, 34, 2441.e1–2441.e5. [Google Scholar] [CrossRef] [Green Version]
- Chibnik, L.B.; Shulman, J.M.; Leurgans, S.E.; Schneider, J.A.; Wilson, R.S.; Tran, D.; Aubin, C.; Buchman, A.S.; Heward, C.B.; Myers, A.J.; et al. Cr1 Is Associated with Amyloid Plaque Burden and Age-Related Cognitive Decline. Ann. Neurol. 2011, 69, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Biffi, A.; Anderson, C.D.; Desikan, R.S.; Sabuncu, M.; Cortellini, L.; Schmansky, N.; Salat, D.; Rosand, J.; Initiative Alzheimer’s Disease Neuroimaging. Genetic Variation and Neuroimaging Measures in Alzheimer Disease. Arch. Neurol. 2010, 67, 677–685. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.S.; Yang, Y.X.; Xu, W.; Wang, H.F.; Tan, L.; Zuo, C.T.; Dong, Q.; Tan, L.; Suckling, J.; Yu, J.T.; et al. Associations of Alzheimer’s Disease Risk Variants with Gene Expression, Amyloidosis, Tauopathy, and Neurodegeneration. Alzheimers Res. Ther. 2021, 13, 15. [Google Scholar] [CrossRef]
- Thambisetty, M.; An, Y.; Tanaka, T. Alzheimer’s Disease Risk Genes and the Age-at-Onset Phenotype. Neurobiol. Aging 2013, 34, 2696.e1–2696.e5. [Google Scholar] [CrossRef] [Green Version]
- Mengel-From, J.; Christensen, K.; McGue, M.; Christiansen, L. Genetic Variations in the Clu and Picalm Genes Are Associated with Cognitive Function in the Oldest Old. Neurobiol. Aging 2011, 32, 554.e7–554.e11. [Google Scholar] [CrossRef] [PubMed]
- Ponomareva, N.V.; Andreeva, T.V.; Protasova, M.A.; Filippova, Y.V.; Kolesnikova, E.P.; Fokin, V.F.; Illarioshkin, S.N.; Rogaev, E.I. Genetic Association between Alzheimer’s Disease Risk Variant of the Picalm Gene and Auditory Event-Related Potentials in Aging. Biochemistry 2018, 83, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Furney, S.J.; Simmons, A.; Breen, G.; Pedroso, I.; Lunnon, K.; Proitsi, P.; Hodges, A.; Powell, J.; Wahlund, L.O.; Kloszewska, I.; et al. Genome-Wide Association with Mri Atrophy Measures as a Quantitative Trait Locus for Alzheimer’s Disease. Mol. Psychiatry 2011, 16, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Dai, X.; Zhang, J.; Li, X.; Chen, Y.; Ma, C.; Chen, K.; Peng, D.; Zhang, Z. The Interactive Effects of Age and Picalm Rs541458 Polymorphism on Cognitive Performance, Brain Structure, and Function in Non-Demented Elderly. Mol. Neurobiol. 2018, 55, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Huang, C.W.; Huang, S.H.; Hsu, S.W.; Chang, W.N.; Lee, J.J.; Chang, C.C. Genetic Interaction Is Associated with Lower Metabolic Connectivity and Memory Impairment in Clinically Mild Alzheimer’s Disease. Genes Brain Behav. 2019, 18, e12490. [Google Scholar] [CrossRef]
- Sun, D.M.; Chen, H.F.; Zuo, Q.L.; Su, F.; Bai, F.; Liu, C.F. Effect of Picalm Rs3851179 Polymorphism on the Default Mode Network Function in Mild Cognitive Impairment. Behav Brain Res. 2017, 331, 225–232. [Google Scholar] [CrossRef]
- Ponomareva, N.V.; Andreeva, T.V.; Protasova, M.S.; Shagam, L.I.; Malina, D.D.; Goltsov, A.Y.; Fokin, V.F.; Illarioshkin, S.N.; Rogaev, E.I. Quantitative Eeg During Normal Aging: Association with the Alzheimer’s Disease Genetic Risk Variant in Picalm Gene. Neurobiol. Aging 2017, 51, 177.e1–177.e8. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu, K.; Morohashi, Y.; Suzuki, M.; Kuroda, H.; Watanabe, T.; Tomita, T.; Iwatsubo, T. Decreased Calm Expression Reduces Abeta42 to Total Abeta Ratio through Clathrin-Mediated Endocytosis of Gamma-Secretase. Nat. Commun. 2014, 5, 3386. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Chang, J.C.; Fan, E.Y.; Flajolet, M.; Greengard, P. Adaptor Complex Ap2/Picalm, through Interaction with Lc3, Targets Alzheimer’s App-Ctf for Terminal Degradation Via Autophagy. Proc. Natl. Acad. Sci. USA 2013, 110, 17071–17076. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Chang, J.C.; Greengard, P.; Flajolet, M. The Convergence of Endosomal and Autophagosomal Pathways: Implications for App-Ctf Degradation. Autophagy 2014, 10, 694–696. [Google Scholar] [CrossRef] [Green Version]
- Kanatsu, K.; Hori, Y.; Takatori, S.; Watanabe, T.; Iwatsubo, T.; Tomita, T. Partial Loss of Calm Function Reduces Abeta42 Production and Amyloid Deposition in Vivo. Hum. Mol. Genet. 2016, 25, 3988–3997. [Google Scholar] [CrossRef]
- Yu, Y.; Niccoli, T.; Ren, Z.; Woodling, N.S.; Aleyakpo, B.; Szabadkai, G.; Partridge, L. Picalm Rescues Glutamatergic Neurotransmission, Behavioural Function and Survival in a Drosophila Model of Abeta42 Toxicity. Hum. Mol. Genet. 2020, 29, 2420–2434. [Google Scholar] [CrossRef]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional Links between Abeta Toxicity, Endocytic Trafficking, and Alzheimer’s Disease Risk Factors in Yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Angelo, F.; Vignaud, H.; di Martino, J.; Salin, B.; Devin, A.; Cullin, C.; Marchal, C. A Yeast Model for Amyloid-Beta Aggregation Exemplifies the Role of Membrane Trafficking and Picalm in Cytotoxicity. Dis. Model. Mech. 2013, 6, 206–216. [Google Scholar] [PubMed] [Green Version]
- Li, S.; Hong, S.; Shepardson, N.E.; Walsh, D.M.; Shankar, G.M.; Selkoe, D. Soluble Oligomers of Amyloid Beta Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron 2009, 62, 788–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Abeta Induces Astrocytic Glutamate Release, Extrasynaptic Nmda Receptor Activation, and Synaptic Loss. Proc. Natl. Acad. Sci. USA 2013, 110, E2518–E2527. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of Glutamate and Nmda Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Takahashi, T.; Nakamori, M.; Hosomi, N.; Maruyama, H.; Miyazaki, Y.; Izumi, Y.; Matsumoto, M. The Identification of Raft-Derived Tau-Associated Vesicles That Are Incorporated into Immature Tangles and Paired Helical Filaments. Neuropathol. Appl. Neurobiol. 2016, 42, 639–653. [Google Scholar] [CrossRef] [Green Version]
- Tasaki, S.; Xu, J.; Avey, D.R.; Johnson, L.; Petyuk, V.A.; Dawe, R.J.; Bennett, D.A.; Wang, Y.; Gaiteri, C. Inferring Protein Expression Changes from Mrna in Alzheimer’s Dementia Using Deep Neural Networks. Nat. Commun. 2022, 13, 655. [Google Scholar] [CrossRef]
- Kumon, H.; Yoshino, Y.; Funahashi, Y.; Mori, H.; Ueno, M.; Ozaki, Y.; Yamazaki, K.; Ochi, S.; Mori, T.; Iga, J.I.; et al. Picalm Mrna Expression in the Blood of Patients with Neurodegenerative Diseases and Geriatric Depression. J. Alzheimers Dis. 2021, 79, 1055–1062. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-Cell Transcriptomic Analysis of Alzheimer’s Disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin Is a Component of Paired Helical Filaments in Alzheimer’s Disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Takahashi, T.; Nakamori, M.; Yamazaki, Y.; Kurashige, T.; Nagano, Y.; Nishida, Y.; Izumi, Y.; Matsumoto, M. Phosphatidylinositol-4,5-Bisphosphate Is Enriched in Granulovacuolar Degeneration Bodies and Neurofibrillary Tangles. Neuropathol. Appl. Neurobiol. 2014, 40, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Gal, J.; Bachstetter, A.; Nelson, P.T. Alpha Adaptins Show Isoform-Specific Association with Neurofibrillary Tangles in Alzheimer’s Disease. Neuropathol. Appl. Neurobiol. 2022, 48, e12776. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, X.Q.; Wyss-Coray, T.; Brecht, W.J.; Sanan, D.A.; Mahley, R.W. Apolipoprotein E Fragments Present in Alzheimer’s Disease Brains Induce Neurofibrillary Tangle-Like Intracellular Inclusions in Neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 8838–8843. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hayashi, I.; Wong, J.; Tugusheva, K.; Renger, J.J.; Zerbinatti, C. Intracellular Clusterin Interacts with Brain Isoforms of the Bridging Integrator 1 and with the Microtubule-Associated Protein Tau in Alzheimer’s Disease. PLoS ONE 2014, 9, e103187. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; van Cauwenberghe, C.; Kolen, K.V.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased Expression of Bin1 Mediates Alzheimer Genetic Risk by Modulating Tau Pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Dourlen, P.; Fernandez-Gomez, F.J.; Dupont, C.; Grenier-Boley, B.; Bellenguez, C.; Obriot, H.; Caillierez, R.; Sottejeau, Y.; Chapuis, J.; Bretteville, A.; et al. Functional Screening of Alzheimer Risk Loci Identifies Ptk2b as an in Vivo Modulator and Early Marker of Tau Pathology. Mol. Psychiatry 2017, 22, 874–883. [Google Scholar] [CrossRef]
- Ando, K.; Tomimura, K.; Sazdovitch, V.; Suain, V.; Yilmaz, Z.; Authelet, M.; Ndjim, M.; Vergara, C.; Belkouch, M.; Potier, M.C.; et al. Level of Picalm, a Key Component of Clathrin-Mediated Endocytosis, Is Correlated with Levels of Phosphotau and Autophagy-Related Proteins and Is Associated with Tau Inclusions in Ad, Psp and Pick Disease. Neurobiol. Dis. 2016, 94, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Ando, K.; de Decker, R.; Vergara, C.; Yilmaz, Z.; Mansour, S.; Suain, V.; Sleegers, K.; de Fisenne, M.A.; Houben, S.; Potier, M.C.; et al. Picalm Reduction Exacerbates Tau Pathology in a Murine Tauopathy Model. Acta Neuropathol. 2020, 139, 773–789. [Google Scholar] [CrossRef] [Green Version]
- Adamec, E.; Mohan, P.; Vonsattel, J.P.; Nixon, R.A. Calpain Activation in Neurodegenerative Diseases: Confocal Immunofluorescence Study with Antibodies Specifically Recognizing the Active Form of Calpain 2. Acta Neuropathol. 2002, 104, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Grynspan, F.; Griffin, W.R.; Cataldo, A.; Katayama, S.; Nixon, R.A. Active Site-Directed Antibodies Identify Calpain Ii as an Early-Appearing and Pervasive Component of Neurofibrillary Pathology in Alzheimer’s Disease. Brain Res. 1997, 763, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Elce, J.S.; Hamos, J.E.; Nixon, R.A. Widespread Activation of Calcium-Activated Neutral Proteinase (Calpain) in the Brain in Alzheimer Disease: A Potential Molecular Basis for Neuronal Degeneration. Proc. Natl. Acad. Sci. USA 1993, 90, 2628–2632. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated Caspase-3 Expression in Alzheimer’s and Aged Control Brain: Correlation with Alzheimer Pathology. Brain Res. 2001, 898, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, T.; Shimohama, S.; Kimura, J.; Shimizu, K. M-Calpain (Calcium-Activated Neutral Proteinase) in Alzheimer’s Disease Brains. Neurosci. Lett. 1998, 248, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Bretteville, A.; Schindowski, K.; Gilissen, E.; Authelet, M.; de Decker, R.; Yilmaz, Z.; Buee, L.; Brion, J.P. Early Axonopathy Preceding Neurofibrillary Tangles in Mutant Tau Transgenic Mice. Am J Pathol. 2007, 171, 976–992. [Google Scholar] [CrossRef] [Green Version]
- Braak, E.; Braak, H.; Mandelkow, E.M. A Sequence of Cytoskeleton Changes Related to the Formation of Neurofibrillary Tangles and Neuropil Threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef]
- Wu, J.W.; Herman, M.; Liu, L.; Simoes, S.; Acker, C.M.; Figueroa, H.; Steinberg, J.I.; Margittai, M.; Kayed, R.; Zurzolo, C.; et al. Small Misfolded Tau Species Are Internalized Via Bulk Endocytosis and Anterogradely and Retrogradely Transported in Neurons. J. Biol. Chem. 2013, 288, 1856–1870. [Google Scholar] [CrossRef] [Green Version]
- Kanekiyo, T.; Cirrito, J.R.; Liu, C.C.; Shinohara, M.; Li, J.; Schuler, D.R.; Shinohara, M.; Holtzman, D.M.; Bu, G. Neuronal Clearance of Amyloid-Beta by Endocytic Receptor Lrp1. J. Neurosci. 2013, 33, 19276–19283. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Luna, G.; Guzman, E.; Audouard, M.; Challis, C.; Sibih, Y.E.; Leshuk, C.; Hernandez, I.; Wegmann, S.; Hyman, B.T.; et al. Lrp1 Is a Master Regulator of Tau Uptake and Spread. Nature 2020, 580, 381–385. [Google Scholar] [CrossRef]
- Auderset, L.; Cullen, C.L.; Young, K.M. Low Density Lipoprotein-Receptor Related Protein 1 Is Differentially Expressed by Neuronal and Glial Populations in the Developing and Mature Mouse Central Nervous System. PLoS ONE 2016, 11, e0155878. [Google Scholar]
- Cooper, J.M.; Lathuiliere, A.; Migliorini, M.; Arai, A.L.; Wani, M.M.; Dujardin, S.; Muratoglu, S.C.; Hyman, B.T.; Strickland, D.K. Regulation of Tau Internalization, Degradation, and Seeding by Lrp1 Reveals Multiple Pathways for Tau Catabolism. J. Biol. Chem. 2021, 296, 100715. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, G.; Kim, T.W. Linking Lipids to Alzheimer’s Disease: Cholesterol and Beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef] [Green Version]
- Panchal, M.; Loeper, J.; Cossec, J.C.; Perruchini, C.; Lazar, A.; Pompon, D.; Duyckaerts, C. Enrichment of Cholesterol in Microdissected Alzheimer’s Disease Senile Plaques as Assessed by Mass Spectrometry. J. Lipid Res. 2010, 51, 598–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuck, B.J.; Miller, L.V.C.; Katsinelos, T.; Smith, A.E.; Wilson, E.L.; Keeling, S.; Cheng, S.; Vaysburd, M.J.; Knox, C.; Tredgett, L.; et al. Cholesterol Determines the Cytosolic Entry and Seeded Aggregation of Tau. Cell Rep. 2022, 39, 110776. [Google Scholar] [CrossRef]
- Good, P.F.; Perl, D.P.; Bierer, L.M.; Schmeidler, J. Selective Accumulation of Aluminum and Iron in the Neurofibrillary Tangles of Alzheimer’s Disease: A Laser Microprobe (Lamma) Study. Ann. Neurol. 1992, 31, 286–292. [Google Scholar] [CrossRef]
- Rao, S.S.; Adlard, P.A. Untangling Tau and Iron: Exploring the Interaction between Iron and Tau in Neurodegeneration. Front. Mol. Neurosci. 2018, 11, 276. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.T.; Chen, W.Y.; Huang, X.T.; Xu, Y.C.; Zhang, H.Y. Iron Dysregulates App Processing Accompanying with Sappalpha Cellular Retention and Beta-Secretase Inhibition in Rat Cortical Neurons. Acta Pharmacol. Sin. 2018, 39, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic Tau Seeding Predicts Tauopathy in Vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [Green Version]
- Kolay, S.; Diamond, M.I. Alzheimer’s Disease Risk Modifier Genes Do Not Affect Tau Aggregate Uptake, Seeding or Maintenance in Cell Models. FEBS Open Bio 2020, 10, 1912–1920. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and Microglial Activation in Alzheimer Disease: Where Do We Go from Here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic Pathway Abnormalities Precede Amyloid Beta Deposition in Sporadic Alzheimer’s Disease and Down Syndrome: Differential Effects of Apoe Genotype and Presenilin Mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Narayan, P.; Sienski, G.; Bonner, J.M.; Lin, Y.T.; Seo, J.; Baru, V.; Haque, A.; Milo, B.; Akay, L.A.; Graziosi, A.; et al. Picalm Rescues Endocytic Defects Caused by the Alzheimer’s Disease Risk Factor Apoe4. Cell Rep. 2020, 33, 108224. [Google Scholar] [CrossRef] [PubMed]
- Moulton, M.J.; Barish, S.; Ralhan, I.; Chang, J.; Goodman, L.D.; Harland, J.G.; Marcogliese, P.C.; Johansson, J.O.; Ioannou, M.S.; Bellen, H.J. Neuronal Ros-Induced Glial Lipid Droplet Formation Is Altered by Loss of Alzheimer’s Disease-Associated Genes. Proc. Natl. Acad. Sci. USA 2021, 118, e2112095118. [Google Scholar] [CrossRef] [PubMed]
- Pachter, J.S.; Yen, T.J.; Cleveland, D.W. Autoregulation of Tubulin Expression Is Achieved through Specific Degradation of Polysomal Tubulin Mrnas. Cell 1987, 51, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan Micrornas. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [Green Version]
- Zingale, V.D.; Gugliandolo, A.; Mazzon, E. Mir-155: An Important Regulator of Neuroinflammation. Int. J. Mol. Sci. 2021, 23, 90. [Google Scholar] [CrossRef]
- Sierksma, A.; Lu, A.; Salta, E.; Eynden, E.V.; Callaerts-Vegh, Z.; D’Hooge, R.; Blum, D.; Buee, L.; Fiers, M.; de Strooper, B. Deregulation of Neuronal Mirnas Induced by Amyloid-Beta or Tau Pathology. Mol. Neurodegener. 2018, 13, 54. [Google Scholar] [CrossRef] [Green Version]
- Lukiw, W.J.; Alexandrov, P.N. Regulation of Complement Factor H (Cfh) by Multiple Mirnas in Alzheimer’s Disease (Ad) Brain. Mol. Neurobiol. 2012, 46, 11–19. [Google Scholar] [CrossRef]
- Wu, J.; Gu, J.; Shen, L.; Fang, D.; Zou, X.; Cao, Y.; Wang, S.; Mao, L. Exosomal Microrna-155 Inhibits Enterovirus A71 Infection by Targeting Picalm. Int. J. Biol. Sci. 2019, 15, 2925–2935. [Google Scholar] [CrossRef] [Green Version]
- Readhead, B.; Haure-Mirande, J.V.; Mastroeni, D.; Audrain, M.; Fanutza, T.; Kim, S.H.; Blitzer, R.D.; Gandy, S.; Dudley, J.T.; Ehrlich, M.E. Mir155 Regulation of Behavior, Neuropathology, and Cortical Transcriptomics in Alzheimer’s Disease. Acta Neuropathol. 2020, 140, 295–315. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, C.B.; Herrmann, A.M.; Bock, S.; Vogelsang, A.; Eichler, S.; Albrecht, P.; Meuth, S.G.; Ruck, T. One Brain-All Cells: A Comprehensive Protocol to Isolate All Principal Cns-Resident Cell Types from Brain and Spinal Cord of Adult Healthy and Eae Mice. Cells 2021, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, K.; Erneux, C.; Homa, M.; Houben, S.; de Fisenne, M.A.; Brion, J.P.; Leroy, K. Dysregulation of Phosphoinositide 5-Phosphatases and Phosphoinositides in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 614855. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
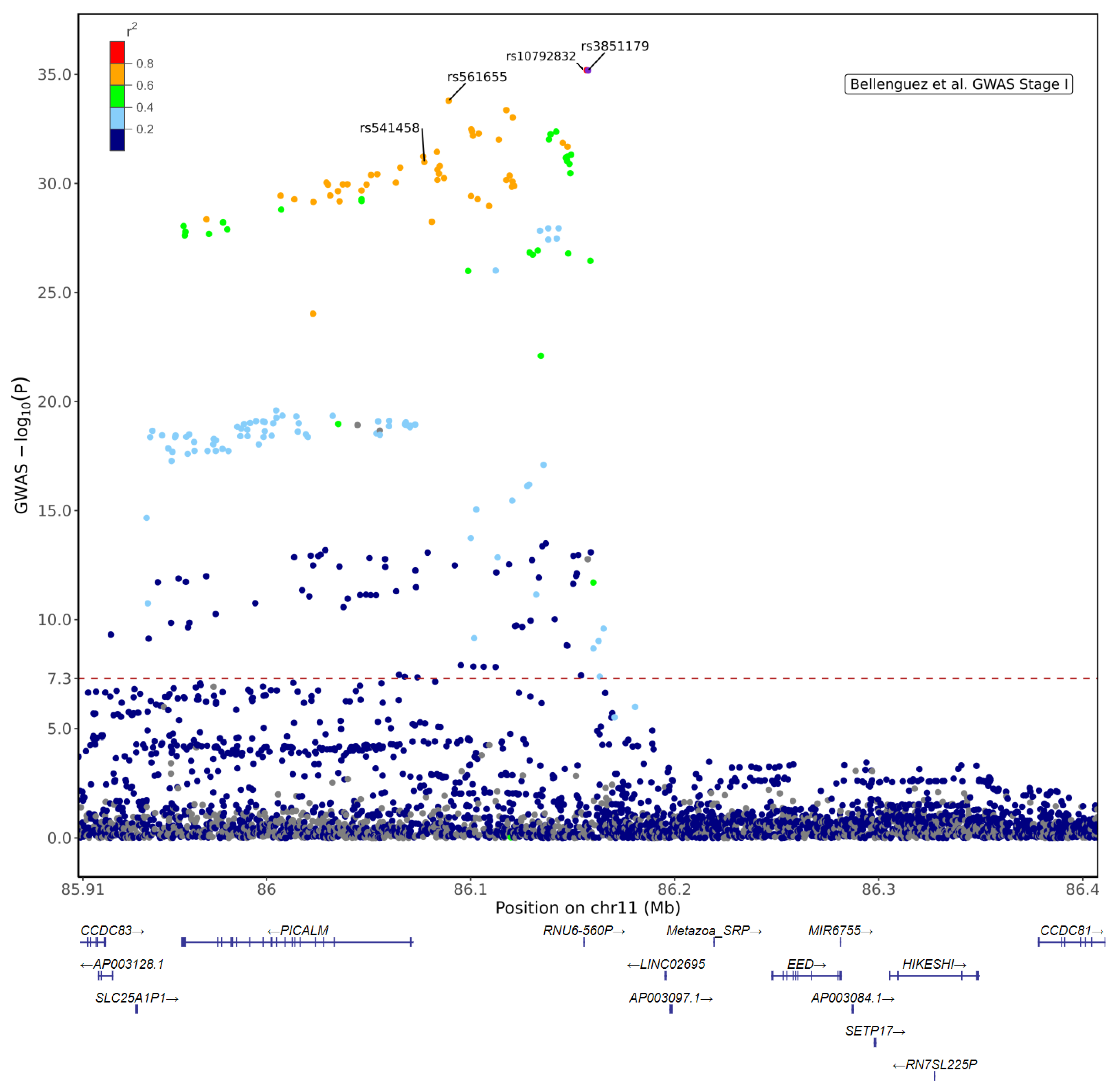
| SNP | Location | GWAS p | r2 | Phenotype | Sample Size | Effect Size (SE) | p-Value | Reference |
|---|---|---|---|---|---|---|---|---|
| rs510566 | 85966796 | 5.669 × 10−05 | 0.042 | Risk G allele is associated with lower CSF Aβ42, higher pTau and pTau/Aβ42 ratio | AD: n = 114, MCI: n = 395, Ctrl: n = 203 | NA | p = 0.048, p = 0.0006, p = 0.0006 | [81] |
| rs510566 | 85966796 | 5.669 × 10−05 | 0.042 | Risk G allele is associated with hippocampal atrophy | AD: n = 141, MCI: n = 461, Ctrl: n = 235 | NA | p < 0.0001 | [81] |
| rs592297 | 86014894 | NA | NA | C allele was associated with faster atrophy rate of hippocampus | AD: n = 141, MCI: n = 461, Ctrl: n = 235 | NA | p < 0.0045 | [81] |
| rs592297 | 86014894 | NA | NA | C allele was associated with slower atrophy rate of posterior cingulate in MCI | AD n = 39, MCI n = 422, Ctrl n = 257 | NA | p < 0.05 | [97] |
| rs17148741 | 86054749 | 0.07777 | 0.017 | C allele is associated with hippocampal volume | n = 1400 | NA | p = 9.4 × 10−5 | [96] |
| rs541458 | 86077309 | 1.032 × 10−31 | 0.622 | Risk T allele is associated with decreased CSF Aβ | n = 412 | NA | p = 0.002 | [98] |
| rs541458 | 86077309 | 1.032 × 10−31 | 0.622 | Differential modulation of spontaneous brain activity (C vs. TT) | MCI n = 35, Ctrl n = 26 | NA | p < 0.05 | [99] |
| rs541458 | 86077309 | 1.032 × 10−31 | 0.622 | Risk T allele is associated with an earlier age at midpoint of cognitive decline | n = 1831 | NA | NA | [100] |
| rs1237999 | 86103988 | 5.091 × 10−33 | 0.696 | A allele is associated with lower CSF Aβ42, higher Tau, pTau, pTau/Aβ42 ratio | AD: n = 114, MCI: n = 395, Ctrl: n = 203 | NA | p = 0.042, p = 0.030, p = 0.011, p = 0.045 | [81] |
| rs543293 | 86109035 | 1.07 × 10−29 | 0.637 | A allele is associated with atrophy rate of posterior cingulate in MCI | AD n = 39, MCI n = 422, Ctrl n = 257 | NA | p < 0.05 | [97] |
| rs10501610 | 86133444 | 1.186 × 10−12 | 0.082 | T allele is associated with slower rise of CSF pTau and pTau/Aβ42 ratio | AD: n = 114, MCI: n = 395, Ctrl: n = 203 | NA | p = 0.0001 | [81] |
| rs2888903 | 86143134 | 1.14 × 10−28 | 0.396 | Major allele is associated with earlier age of onset of AD in individuals with down syndrome | n = 67 | 3.31 | p = 0.011 | [101] |
| rs7110631 | 86145145 | 1.367 × 10−32 | 0.740 | Risk G allele is associated with an age-dependent cognitive decline | n = 1564 | NA | p = 0.03 | [102] |
| rs7941541 | 86147496 | 2.052 × 10−32 | 0.740 | Major allele is associated with earlier age of onset of AD in individuals with down syndrome | n = 67 | Beta: 3.92 | p = 0.016 | [101] |
| rs10751134 | 86147845 | 1.618 × 10−27 | 0.483 | Major allele is associated with earlier age of onset of AD in individuals with down syndrome | n = 67 | Beta: 2.78 | p = 0.040 | [101] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Protective T allele is associated with increased volume of hippocampus and entorhinal cortex | AD n = 168, MCI n = 357, Ctrl n= 215 | Beta: Hip: 0.061 (0.029), EC: 0.066 (0.021) | Hip p = 0.04, EC n = 0.01 | [103] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Protective T allele is associated with slower atrophy rate of hippocampus | AD n = 205, MCI n = 639, Ctrl n = 339 | NA | p < 0.05 | [104] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Non-protective C allele is associated with earlier age of onset of AD | n = 2569 | NA | p = 0.0086 (1 side) | [105] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Protective T allele is associated with better cognitive function in the oldest old | n = 1369 | Beta: 0.60 | p = 0.024 | [106] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Protective T allele is associated with faster information processing speed | n = 87 | NA | p < 0.05 | [107] |
| rs3851179 | 86157598 | 6.496 × 10−36 | 1 | Protective T allele is associated with larger entorhinal cortical thickness | AD n = 245, MCI n = 434, Ctrl n = 284 | Beta: 0.043 | p = 0.034 | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ando, K.; Nagaraj, S.; Küçükali, F.; de Fisenne, M.-A.; Kosa, A.-C.; Doeraene, E.; Lopez Gutierrez, L.; Brion, J.-P.; Leroy, K. PICALM and Alzheimer’s Disease: An Update and Perspectives. Cells 2022, 11, 3994. https://doi.org/10.3390/cells11243994
Ando K, Nagaraj S, Küçükali F, de Fisenne M-A, Kosa A-C, Doeraene E, Lopez Gutierrez L, Brion J-P, Leroy K. PICALM and Alzheimer’s Disease: An Update and Perspectives. Cells. 2022; 11(24):3994. https://doi.org/10.3390/cells11243994
Chicago/Turabian StyleAndo, Kunie, Siranjeevi Nagaraj, Fahri Küçükali, Marie-Ange de Fisenne, Andreea-Claudia Kosa, Emilie Doeraene, Lidia Lopez Gutierrez, Jean-Pierre Brion, and Karelle Leroy. 2022. "PICALM and Alzheimer’s Disease: An Update and Perspectives" Cells 11, no. 24: 3994. https://doi.org/10.3390/cells11243994