
Insights into Regulators of p53 Acetylation
by
 , , and
, , and
Mai Nagasaka
1,
Chiharu Miyajima
1,
Hiromasa Aoki
2,
Mineyoshi Aoyama
2,
Daisuke Morishita
1,3,
Yasumichi Inoue
1,* and
Hidetoshi Hayashi
1,* 1
Department of Cell Signaling, Graduate School of Pharmaceutical Sciences, Nagoya City University, Nagoya 467-8603, Japan
2
Department of Pathobiology, Graduate School of Pharmaceutical Sciences, Nagoya City University, Nagoya 467-8603, Japan
3
Chordia Therapeutics Inc., Kanagawa 251-0012, Japan
*
Authors to whom correspondence should be addressed.
Cells 2022, 11(23), 3825; https://doi.org/10.3390/cells11233825
Submission received: 8 November 2022
/
Revised: 25 November 2022
/
Accepted: 25 November 2022
/
Published: 29 November 2022
(This article belongs to the Special Issue p53 Signaling and Cancer)
Abstract
:The tumor suppressor p53 is a transcription factor that regulates the expression of dozens of target genes and diverse physiological processes. To precisely regulate the p53 network, p53 undergoes various post-translational modifications and alters the selectivity of target genes. Acetylation plays an essential role in cell fate determination through the activation of p53. Although the acetylation of p53 has been examined, the underlying regulatory mechanisms remain unclear and, thus, have attracted the interest of researchers. We herein discuss the role of acetylation in the p53 pathway, with a focus on p53 acetyltransferases and deacetylases. We also review recent findings on the regulators of these enzymes to understand the mode of p53 acetylation from a broader perspective.
1. p53: Guardian of the Genome
p53 is the most well-known tumor suppressor in the human genome. The gene encoding p53 (also known as TP53) is frequently mutated in human cancers, including colorectal, lung, brain, liver, bladder, and esophageal cancers [1]. Germ-line mutations in TP53 have been reported in Li–Fraumeni syndrome, an inherited disorder that increases the risk of developing certain cancers. Furthermore, a dysfunctional p53 pathway has also been detected in cancers that retain wild-type p53 due to various causes [2,3].
p53 is a transcription factor that regulates the expression of dozens of target genes with diverse biological functions, including cell cycle arrest, apoptosis, senescence, DNA repair, cellular metabolism, and autophagy [4,5,6]. p53 and its downstream genes consist of a complex gene network, and post-translational modifications in specific p53 residues may regulate its fine-tuned responses [7]. Approximately 50 different amino acids of p53 may be modified, and many different forms of post-translational modifications have been reported, including phosphorylation, ubiquitination, acetylation, methylation, sumoylation, neddylation, glycosylation, and poly-ribosylation [8].
We herein focus on p53 acetylation, which plays an important role in the regulation of p53. p53 was the first non-histone protein shown to be acetylated [9], and with the accumulation of evidence, acetylation has been established as one of the indicators of p53 activation. While there have been several excellent reviews on p53 acetylation [10,11,12], we discuss the role of acetylation in the regulation of p53, with a focus on not only p53 acetyltransferases and deacetylases, but also the regulators of these enzymes.
2. p53 Acetylation
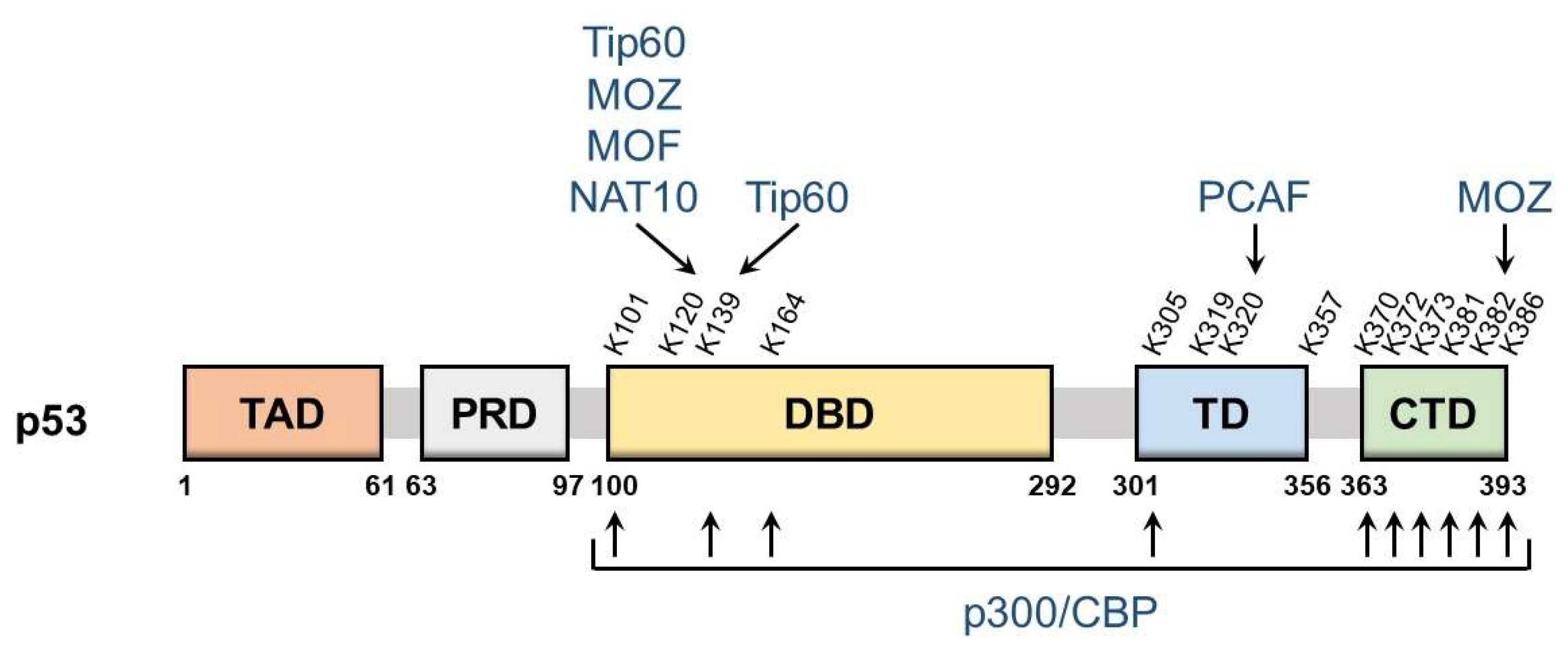
p53 is a nuclear protein that comprises 393 amino acids and has the domain structure shown in Figure 1. It contains an N-terminal transactivation domain, proline-rich domain, a centrally located sequence-specific DNA-binding domain (DBD), and a C-terminal tetramerization domain followed by the extreme C-terminal domain (CTD). p53 was the first non-histone protein reported to be acetylated [9]. The acetylation site on p53 has been detected in the following three regions: CTD, DBD, and between these two domains.
The lysine residues K370, K372, K373, K381, K382, and K386 in CTD may be acetylated, which promotes the DNA binding of p53 and enhances its transcriptional activity. Mechanistically, acetylation in CTD forms antagonistic crosstalk with ubiquitination. In unstressed cells, low p53 levels are maintained by a feedback interaction with mouse double minute 2 homolog (MDM2), an E3 ubiquitin ligase. MDM2 binds and ubiquitinates p53, leading to its degradation via the ubiquitin–proteasome system. The major residues for p53 ubiquitination by MDM2 are located in CTD and, thus, the acetylation of these sites blocks the degradation of p53 and induces its accumulation.
DBD also has the acetylation sites K101, K120, K139, and K164. The acetylation of these sites is responsible for the target selectivity of p53. K101 acetylation is essential for the regulation of p53 metabolic targets. Mutations in K101 result in the failure of p53-mediated ferroptosis [13]. K139 was recently revealed as a novel acetylated site; however, its contribution to the selectivity of p53 target genes remains unknown [14]. K120 acetylation is crucial for the selective induction of apoptosis. The acetylation of this site up-regulates the expression of pro-apoptotic genes such as BCL2 binding component 3 (also known as PUMA), but it is dispensable for the induction of Cyclin Dependent Kinase Inhibitor 1A (also known as p21), a cell cycle arrest gene [15]. K164 acetylation is critical for the induction of p53-mediated cell cycle arrest [16]. K120 and K164 are located in DBD, and both of these lysine residues are highly mutated in human cancer [6]. Mutations in K120 and K164 along with six C-terminal lysine residues (p53-8KR) were previously shown to completely suppress the p53-dependent induction of p21 [16]. Importantly, the p53-8KR mutant retains its DNA-binding ability and may induce a p53-MDM2 feedback loop. The p53-8KR mutant is unable to promote cell cycle and apoptotic regulators, suggesting that K120 and K164 are essential for the tumor suppressor function of p53.
The acetylation of the lysine residues K305, K319, K320, and K357 has been detected between CTD and DBD. The acetylation of K320 was found to suppress p53 pro-apoptotic activities after DNA damage [17,18]. K305, K319, and K357 may also be acetylated [19,20]; however, the characteristics of these residues remain unknown and the enzymes catalyzing the acetylation of K319 and K357 have not yet been identified.
3. p53 Acetyltransferase
3.1. p300/CBP
p300 and cyclic AMP response element-binding protein-binding protein (CBP) were originally identified as binding factors of the adenoviral E1A oncoprotein and cyclic AMP response element-binding protein, respectively [21,22]. These two proteins have highly conserved regions and show 63% homology at the amino-acid level [23]. p300/CBP acts as an acetyltransferase or transcriptional coactivator, and is involved in a broad spectrum of biological processes, including cell proliferation and differentiation [24]. Aberrations have been detected in p300/CBP in many cancers. In the CBP gene, approximately 15% of non–small cell and small cell lung cancers harbor loss-of-function aberrations [25,26]. These abnormalities have also been observed in multiple cancers, including lymphoma, leukemia, and bladder cancer [27,28,29]. Missense mutations in CBP are frequently detected in G1411, W1472, and H1487, which are important residues for its histone acetyltransferase (HAT) and transcriptional coactivation activities. Furthermore, gross deletions and protein-truncating mutations in CBP frequently occur [26,30,31]. Protein-truncating mutations accompanied by the inactivation of the second allele in the p300 gene were identified in colorectal cancer and breast cancer [32,33], while in-frame deletions that result in the loss of HAT activity have also been reported [34]. A previous study demonstrated that a proportion of breast and colorectal carcinomas expressed p300 at extremely low levels (Table 1) [24].
p300 and CBP directly bind to p53 and acetylate multiple C-terminal lysine residues, namely, K370, K372, K373, K381, K382, and K386 [9,35,36]. Moreover, four additional lysine residues, K101, K139, K164, and K305, may be acetylated by p300/CBP [13,16]. The acetylation of these sites has been suggested to enhance the DNA-binding ability of p53 and promote its transcription, resulting in growth arrest or apoptosis.

{kind=link}
{kind=link}
{kind=link}
Table 1.
List of cancer types with low-expressed p53 acetyltransferases.
| p53 Acetyltransferases | Cancer Types | Genetic Evidence | References |
|---|---|---|---|
| p300 | Breast carcinoma | N/A 1 | [24] |
| Colorectal carcinoma | |||
| PCAF | Colorectal carcinoma | N/A | [37] |
| Gastric carcinoma | PCAF suppresses tumorigenesis and tumor growth both in vitro and in vivo | [38] | |
| Tip60 | Breast cancer | Tip60-silenced cells are resistant to cisplatin-induced cell death | [39] |
| Colorectal carcinoma | N/A | [40] | |
| Lung carcinoma | The knockdown of Tip60 promotes cell proliferation and invasion | [40,41] | |
| MOZ | Hepatocellular carcinoma | The knockdown of MOZ promotes cell proliferation and colony formation | [42] |
| MOF | Endometrial carcinoma | N/A | [43] |
| Lung squamous cell carcinoma | |||
| Stomach adenocarcinoma | |||
| NAT10 | N/A | ||
1 N/A, no data were available.
3.2. PCAF
p300/CBP-associating factor (PCAF) was originally identified as a competitive factor with E1A for binding to p300/CBP [44]. It exhibits HAT activity and plays an important role in transcriptional regulation associated with DNA repair [45,46]. The down-regulation of PCAF was detected in colorectal and gastric carcinoma (Table 1) [37,38]. A few missense alterations in the PCAF gene have been detected in several cancers; however, these alterations showed rare inactivating mutations [47].
3.3. Tip60
HIV-1 Tat-interacting protein of 60 kDa (Tip60) is a member of the MOZ, Ybf2/Sas3, Sas2, and TIP60 (MYST) family of HATs. It is ubiquitously expressed and has been reported to play a role in various cellular processes, including gene transcription and DNA damage responses [48,49]. Low expression levels of the Tip60 gene have been detected in breast cancer, colorectal carcinoma, and lung carcinoma (Table 1) [39,40,41].
3.4. MOZ
The monocytic leukemia zinc finger protein (MOZ) belongs to the MYST family of HATs. It was initially characterized as a fusion partner of CBP in acute myeloid leukemia (AML) [50]. MOZ is involved in various cellular functions, such as cellular senescence [51]. MOZ gene alterations are often detected in solid tumors. Whole-genome sequencing revealed mutations in the MOZ gene in esophageal adenocarcinoma [52]. The expression of MOZ is significantly down-regulated in hepatocellular carcinoma (Table 1) [42]. The abnormal expression of mouse MOZ promoted the metastasis of medulloblastoma [53].
3.5. MOF
Males absent on the first (MOF) is a member of the MYST family of HATs. It was originally identified as a crucial component of the X chromosome dosage compensation male-specific lethal complex in Drosophila [55]. MOF plays an important role in various cellular functions, including cell cycle progression and DNA damage responses [56,57]. The down-regulation of MOF was detected in various cancers, including endometrial carcinoma, lung squamous cell carcinoma, and stomach adenocarcinoma (Table 1) [43].
MOF rapidly acetylates p53 at K120 after DNA damage, which leads to p53-mediated gene-specific responses [58].
3.6. NAT10
N-acetyltransferase 10 (NAT10) belongs to the Gcn5-related N-acetyltransferase family of HATs. It is a nucleolar protein that is involved in a wide range of cellular biological processes, including the regulation of telomerase activity and RNA polymerase I transcription [59,60]. Contrary to expectations, limited information is currently available on the repressive aberration of the NAT10 gene. Instead, the up-regulation of NAT10 has been detected in soft tissue sarcomas [61]. Moreover, the overexpression of NAT10 in hepatocellular carcinoma and its positive correlation with the tumor stage has been suggested [62].
NAT10 directly binds to p53 and MDM2 [63]. It acetylates p53 at K120 and up-regulates PUMA upon DNA damage. The knockdown of NAT10 decreases UV-induced cell death in a p53-dependent manner. In addition, NAT10 functions as a E3 ligase for MDM2 and promotes its degradation. NAT10 affects both p53 and MDM2, resulting in the synergistical stabilization of p53.
4. Regulators of p53 Acetyltransferase
As shown in Figure 2, multiple factors are involved in the modulation of p53 acetyltransferases. We herein list regulators that affect the p53 network through their effects on HAT activity.
4.1. p300/CBP
Various regulatory modes of p300/CBP activity towards p53 have been reported. Several viral proteins are known to regulate p300/CBP. The E6 protein is one of the oncoproteins encoded by oncogenic human papillomavirus 16 and 18 [64]. E6 stimulates the degradation of p53 via a well-known mechanism in which E6 interacts with p53 and promotes its degradation via the ubiquitin–proteasome system. E6 also binds to p300 and CBP and inhibits their co-activation, which blocks p300/CBP-mediated p53 acetylation and down-regulates p53 transcriptional activity [65,66]. E1A inhibits p300-mediated p53 acetylation [67]. It also directly interacts with the HAT domain of p300 and suppresses its HAT activity. In addition, E1A reportedly reduced the p53-dependent transactivation of p21 and Bcl-2-associated X (BAX), leading to the disruption of p53-mediated cell cycle arrest and apoptosis [35].
Some proteins related to E3 ubiquitin ligases have been suggested to inhibit p300/CBP activity. MDM2 directly binds to p300/CBP and suppresses its acetyltransferase activity. It was also shown to attenuate p53 acetylation by p300/CBP, which impaired p53 transcriptional activity [68,69]. MDMX, a homolog of MDM2, was found to inhibit the acetylation of p53 by p300/CBP [70]. S phase kinase-associated protein 2 (Skp2), a substrate recognition factor in the SKP-cullin-F-box (SCF) ubiquitin ligase complex, inhibits p300 and suppresses the transcriptional activity of p53 [71]. Skp2 directly binds to p300 via the cysteine–histidine-rich regions, CH1 and CH3, which are also p53-binding regions. As Skp2 antagonizes p300-p53 binding, it represses p300-mediated p53 acetylation at K382 as well as the p53-dependent transactivation of both cell cycle arrest genes and pro-apoptotic genes.
Some members of the inhibitor of growth proteins (ING) family have also been shown to modulate p300/CBP. p29ING4 and p28ING5 both interact with p300 and promote p53 acetylation at K382, resulting in the p53-mediated transactivation of the p21 gene [72]. p33ING2 and p300 form a complex with p53, which enhances p53 acetylation at K382 [73,74]. p33ING2 suppresses cell proliferation through the activation of p53 via p300-mediated acetylation.
Multiple nucleolar proteins, including Myb-binding protein 1a (MYBBP1A), ribosomal protein L11 (RPL11), and ribosomal protein S26 (RPS26), are involved in the regulation of p300/CBP. MYBBP1A directly binds to p53 and strengthens the p53-p300 interaction to promote p53 acetylation in response to ribosomal stress, thereby enhancing p53-mediated transcription [75]. RPL11 plays a crucial role in the enhancement of p53-p300 binding and subsequent p300-mediated p53 acetylation at K382 upon nucleolar stress [76]. RPS26 forms a complex with p53 and p300 in response to DNA damage and may facilitate p300-mediated p53 acetylation [77].
The involvement of long non-coding RNA (lncRNA) in the regulation of p300/CBP has been suggested. lncRNA induced by p53 (lnc-Ip53) interacts with p300 and suppresses p300 activity, leading to the abrogation of p53 acetylation [78]. Since lnc-Ip53 is a direct transcriptional target of p53, p53/lnc-Ip53/p300 may form a negative feedback loop in the regulation of p53 activity.
Other proteins are also known to positively regulate p300/CBP. Homeodomain interacting protein-kinase 2 (HIPK2), a nuclear serine/threonine kinase, directly binds to p300 and phosphorylates p300, which enhances its HAT activity [79]. Moreover, HIPK2 promotes p300-mediated p53 acetylation at K382 and influences the selective transactivation of pro-apoptotic p53 target genes [80,81]. To trigger the HIPK2-mediated induction of p53 pro-apoptotic genes, p53 needs to be modified by both S46 phosphorylation and K382 acetylation. Interferon regulatory factor 1 (IRF-1) binds to p300 and strengthens the interaction between p300 and the LXXLL transactivation domain of p53 [82]. IRF-1 enhances p300-mediated p53 acetylation at K373 and K382, and stimulates the transactivation of p21. Promyelocytic leukemia (PML), a nuclear protein, forms a complex with p53-CBP and promotes p53 acetylation at K382 [83]. Moreover, PML inhibits the SCFFbx3-mediated degradation of p300 [84]. The PML protein localizes to discrete nuclear speckles called PML nuclear bodies (PML-NBs) [85]. When p300 is located within PML-NBs, it is stabilized by PML. However, when p300 is located outside of PML-NBs, it is degraded via the ubiquitin–proteasome pathway. Sex Determining Region Y-Box Transcription Factor 4 (SOX4), a transcriptional factor involved in the regulation of embryonic development and cell fate determination, enhances p53 acetylation at K373 and K382 by interacting with p300/CBP and promoting the formation of the p300/CBP/p53 complex [86]. SOX4 may also interact with p53, which inhibits MDM2-dependent p53 ubiquitination and degradation. p85α, a regulatory subunit of phosphatidylinositol-3-kinase, interacts with p300 and augments p300-p53 binding, which leads to p53 transactivation under UVB exposure [87]. p85α is required for the induction of mouse p53 acetylation at K370, followed by K373 of human p53. Wilms tumor gene on X chromosome (WTX), which is frequently inactivated in Wilms tumors, augments p53-CBP binding, thereby promoting CBP-mediated p53 acetylation at K373 and K382 and its transcriptional activation [88]. HLA-B-associated transcript 3 (BAT3), a nucleo-cytoplasmic shuttling protein, regulates the p300-mediated acetylation of p53 during autophagy [89]. During starvation, BAT3 facilitates the nuclear translocation of p300 and promotes p300-dependent p53 acetylation at K373, which leads to the transactivation of pro-autophagic p53 target genes such as SESTRIN1.
Several proteins have been reported to function as negative regulators of p300/CBP. Scaffold/matrix attachment region-binding protein 1 (SMAR1), a chromatin remodeling protein, was found to repress p300 expression and inhibit p53 acetylation [90]. SMAR1 may associate with the p53-p300 complex and antagonize the interaction of p300 with p53. DEAD (Asp-Glu-Ala-Asp) box RNA helicase 24 (DDX24), a family member of DEAD-box RNA helicases, negatively regulated the transcriptional ability of p53 by suppressing the HAT activity of p300 [91]. DDX24 competes with p53 in its interaction with p300 and inhibits the p300-mediated acetylation of p53 at K164 and K382. DDX24 suppresses p53-dependent cell cycle arrest and senescence. Transcriptional coactivator with a PDZ-binding motif (TAZ), a transcriptional coactivator of the Hippo pathway, inhibits p300-mediated p53 acetylation at K382, resulting in the attenuation of p53 transcriptional activity [92]. TAZ also interacts with p53 to inhibit the p53-p300 interaction and subsequent p53 acetylation. The knockdown of TAZ has been shown to promote p53-mediated cellular senescence.
4.2. PCAF
Multiple viral oncoproteins have been implicated in the regulation of PCAF and the subsequent inactivation of p53. E1A directly binds to PCAF via its HAT domain and diminishes its acetyltransferase activity towards p53 [67]. E1A was previously shown to repress p53-induced cell cycle arrest and apoptosis [35]. Furthermore, E1B interacted with PCAF and competed for PCAF-p53 binding [93]. It also inhibited PCAF-mediated p53 acetylation both in vitro and in vivo and reduced the sequence-specific DNA-binding activity of p53.
Some E3 ubiquitin ligases also contribute to the suppression of PCAF. MDM2 was previously shown to inhibit PCAF-mediated p53 acetylation at K320 [94,95]. MDM2 directly bound to PCAF and promoted its degradation via the ubiquitin–proteasome pathway, which repressed the PCAF-mediated transactivation of p53 target genes. Seven in absentia homolog 2 (SIAH2), which is a member of the SIAH E3 ubiquitin ligases family, also reportedly binds to PCAF and promotes its degradation via the ubiquitin–proteasome pathway [96]. The ubiquitination ability of SIAH2 is needed for the attenuation of p53 acetylation and its transcriptional activity. Since SIAH2 is p53 target genes, p53 and SIAH2 may form a feedback loop for the regulation of p53 activity [97].
As an example of a positive regulator of PCAF, HIPK2 cooperates with PCAF and drives p53 to selectively transactivate the p21 gene [98]. Under apoptotic conditions, HIPK2 is activated in response to severe DNA damage and directly phosphorylates p53 at S46, which induces the expression of pro-apoptotic genes [80,99]. On the other hand, under non-apoptotic conditions, HIPK2 enhances PCAF-mediated p53 acetylation at K320 by promoting the nuclear localization of PCAF, leading to p53 activation and the selective induction of the p21 gene, but not other pro-apoptotic target genes.
4.3. Tip60
p53 acetylation activity of Tip60 is mainly regulated by post-translational modifications, including ubiquitination, phosphorylation, and sumoylation. Regarding ubiquitination, various E3 ubiquitin ligases are involved in the inhibition of Tip60. Tripartite motif 29 (TRIM29) interacts with Tip60 and induces its ubiquitination, resulting in the proteasome-mediated degradation of Tip60 [100]. TRIM29 reduces Tip60-mediated acetylation of p53 at K120. SIAH2 binds to Tip60 and promotes its degradation via the ubiquitin–proteasome pathway, which abrogates Tip60-mediated p53 acetylation [96]. Ubiquitin-like with PHD and RING finger domains 1 (UHRF1) have also been reported to negatively regulate p53 acetylation activity of Tip60 [101]. UHRF1 interacts with Tip60 and promotes its proteasomal degradation. UHRF1 suppresses TIP60-mediated p53 acetylation at K120. Furthermore, the deubiquitinase participates in the regulation of Tip60. Ubiquitin-specific protease 7 (USP7) directly interacts with Tip60 and removes its ubiquitin chains. USP7 enhances Tip60 acetyltransferase activity towards p53 at K120 and promotes p53-mediated pro-apoptotic gene expression [102].
Phosphatases act as regulators of Tip60. A previous study demonstrated that glycogen synthase kinase-3 (GSK-3) phosphorylated Tip60 on S86 and enhanced its p53K120 acetyltransferase activity. Moreover, the phosphorylation of S86 promoted the induction of p53-mediated PUMA and apoptosis [103].
Sumoylation is associated with Tip60 activity. PIASy, a member of the PIAS family of SUMO E3 ligases, positively regulates Tip60. PIASy induced Tip60 sumoylation and promoted the acetylation of p53 at K120, thereby stimulating p53-mediated apoptosis [104].
Other factors related to the regulation of Tip60 are as follows. Programmed cell death 5 (PDCD5), an apoptosis-related protein, interacts with Tip60 and promotes its stabilization. PDCD5 enhances the p53 acetylation activity of Tip60, thereby inducing pro-apoptotic gene expression in a p53-dependent manner [105]. Moreover, p90, ING5, and nuclear interactor of ARF and Mdm2 (NIAM) regulate Tip60-mediated p53 acetylation at K120 [106,107,108].
4.4. MOZ
The mode of regulation of MOZ activity towards p53 remains unclear. PML directly binds to MOZ, which is then recruited into PML-NBs [51]. Under cellular stress conditions, MOZ colocalizes with p53 in PML-NBs and promotes p53 acetylation at K120 and K382. These modifications stimulate the p53-mediated transactivation of p21. In contrast, Akt, a serine/threonine kinase, phosphorylates MOZ at T369 and suppresses the interaction between PML and MOZ [51]. The phosphorylation of T369 is essential for the negative regulation of MOZ-mediated p53 acetylation.
4.5. MOF
Regarding the p53 acetylation activity of MOF, its regulatory mode has not yet been examined in as much detail as those of other p53 acetyltransferases. Male-specific lethal 1v1 (MSL1v1) may form a complex with MOF and markedly enhance p53-dependent transcriptional activity [109]. MOF-MSL1v1 is recruited to the promoter region through interactions with p53 and other cofactors and then acetylates p53 at K120, which enhances the p53-mediated transcription of PUMA and BAX.
4.6. NAT10
Limited information is currently available on the regulatory mode of the acetylation activity of NAT10 towards p53. Acetyltransferases generally regulate enzymatic activity by autoacetylation [110]. NAT10 also appears to be regulated by autoacetylation [111]. Several enzymes have been reported to deacetylate NAT10. For example, SIRT1 deacetylates NAT10 and inhibits its rRNA biogenesis function [112]. It currently remains unclear whether SIRT1 affects the p53 acetylation activity of NAT10 and, thus, further studies are needed.
5. p53 Deacetylase
5.1. HDAC1
HDAC1 is a member of the class I HDAC enzyme family, which are Zn+-dependent proteases, and regulates a wide range of biological processes by removing acetyl moieties from the lysine residues of histones and non-histone proteins. The overexpression of HDAC1 has been detected in many solid tumors, including colorectal carcinoma, hepatocellular carcinoma, prostate cancer, and renal cancer [113,114,115,116,117]. HDAC1 has also been reported to be highly expressed in some lymphomas, including diffuse large B-cell lymphoma (DLBCL), peripheral T-cell lymphoma (PTCL), and Hodgkin’s lymphoma (HL) (Table 2). The high expression of HDAC1 is associated with a poor prognosis in multiple myeloma [118].
5.2. HDAC2
HDAC2 is a member of the class I HDAC enzyme family. It modulates various physiological processes, such as cell cycle progression, embryonic development, and cytokine signaling [121]. HDAC2 is frequently dysregulated in colorectal cancer [122]. In addition, the high expression of HDAC2 has been detected in several solid and hematologic tumors (Table 2) [114,115,116,117,118,123,124,125,126,127].
HDAC2 is a site-specific deacetylase for p53. It removes the acetyl moiety from K320, and suppresses p53-mediated cell cycle control and apoptosis [128].
5.3. HDAC6
HDAC6 is classified as a Class IIb HDAC enzyme that functions as a Zn+-dependent protease. It contains 1215 amino acids and, thus, is the largest member of the HDAC family [129]. It plays a role in multiple cellular pathways, including autophagy and apoptosis [130]. The overexpression of HDAC6 has been detected in pancreatic cancer and various hematologic tumors (Table 2) [118,125,126,131,132].
HDAC6 interacts with the C-terminal region of p53 and deacetylates it at K381 and K382 [133]. A452, an HDAC6-selective inhibitor, was shown to inhibit the nuclear localization of HDAC6, which enhanced the acetylation of K381 and K382 as well as the pro-apoptotic activity of p53. Furthermore, K120 of p53 was reportedly modified by HDAC6 [134]. HDAC6 contributes to silencing the pro-apoptotic function of p53 by deacetylating K120.
5.4. HDAC8
HDAC8 is a class I HDAC enzyme that is involved in various biological processes, such as the regulation of telomerase activity and cohesion dynamics [135,136]. The expression levels of HDAC8 were previously shown to be significantly elevated in various tumors, including urothelial cancer, acute lymphoblastic leukemia (ALL), and multiple myeloma (MM) (Table 2) [118,124,125].
Table 2.
List of cancer types with highly expressed p53 deacetylases.
| p53 Deacetylases | Cancer Types | Genetic Evidence | References | |
|---|---|---|---|---|
| HDAC1 | Solid tumors | Colorectal carcinoma | The knockdown of HDAC1 suppresses growth of cancer cells | [114] |
| Hepatocellular carcinoma | The knockdown of HDAC1 leads to increased apoptosis and decreased cell proliferation | [115] | ||
| Prostate cancer | N/A | [116] | ||
| Renal cancer | [117] | |||
| Hematologic tumors | DLBCL and PTCL | N/A | [126] | |
| HL | [127] | |||
| HDAC2 | Solid tumors | Colorectal carcinoma | The knockdown of HDAC2 suppresses growth of cancer cells | [114] |
| Hepatocellular carcinoma | The knockdown of HDAC2 leads to increased apoptosis and decreased cell proliferation | [115] | ||
| Pancreatic ductal adenocarcinoma | HDAC2 confers resistance towards etoposide | [123] | ||
| Prostate cancer | N/A | [116] | ||
| Renal cancer | [117] | |||
| Urothelial cancer | [124] | |||
| Hematologic tumors | ALL | N/A | [125] | |
| DLBCL and PTCL | [126] | |||
| HL | [127] | |||
| MM | [118] | |||
| HDAC6 | Solid tumors | Pancreatic cancer | The knockdown of HDAC6 impairs the motility of cancer cells | [131] |
| Hematologic tumors | ALL | N/A | [125] | |
| AML | [132] | |||
| DLBCL and PTCL | [126] | |||
| MM | [118] | |||
| HDAC8 | Solid tumors | Urothelial cancer | N/A | [124] |
| Hematologic tumors | ALL | N/A | [125] | |
| MM | [118] | |||
| SIRT1 | Solid tumors | Colorectal cancer | The knockdown of SIRT1 leads to inhibition of cell proliferation | [137] |
| Prostate cancer | N/A | [138] | ||
| Hematologic tumors | AML | N/A | [132] | |
| SIRT3 | Solid tumors | Melanoma | The knockdown of SIRT3 leads to decreased cell proliferation, colony formation, and cellular migration | [139] |
HDAC8 interacts with p53 and regulates its transcriptional activity via deacetylation. Its deletion was shown to promote the p53-dependent apoptotic pathway in response to DNA damage [140].
5.5. SIRT1
SIRT1 is a member of the class III HDAC family, which functions through a NAD+-dependent mechanism. It is widely known as a regulator of aging [141]. It also targets various proteins for acetylation and participates in a wide range of cellular functions, including apoptosis and glucose metabolism [142,143]. The overexpression of SIRT1 has been reported in prostate cancer, AML, and colorectal cancer (Table 2) [132,137,138].
Previous studies demonstrated that SIRT1 directly bound to p53 and specifically deacetylated the K382 residue [143,144]. The deacetylation of p53 by SIRT1 represses p53-dependent p21 transactivation. SIRT1 also inhibits the p53-mediated apoptotic pathway. SIRT1-deficient cells show p53 hyperacetylation and enhanced apoptotic responses [145]. Collectively, these findings demonstrate that SIRT1 acts as a negative regulator of p53.
5.6. SIRT3
SIRT3 is classified as a Class III HDAC enzyme. It is involved in several biological processes, including cellular metabolism and oxidative stress [146]. SIRT3 is significantly upregulated in melanoma (Table 2) [139]. The high expression of SIRT3 correlates with a poor clinical prognosis in several cancers, including colorectal and gastric cancers [147]. In contrast, SIRT3 levels are down-regulated in breast cancer and decrease further in advanced stages [148,149].
Cancer-promoting and inhibitory effects have been implicated for SIRT3 [150]. It has been shown to deacetylate p53 at K320 and K382, which promotes the degradation of p53 via the ubiquitin–proteasome pathway [151]. On the other hand, a tumor suppressor role has been suggested for SIRT3 in several solid tumors based on findings showing that it prevented the reprogramming of cancer cell metabolism via the destabilization of hypoxia-inducible factor-1α [148]. Moreover, SIRT3 inhibited hepatocellular carcinoma cell proliferation by reducing MDM2-mediated p53 degradation [152]. Further studies are warranted on the dual role of SIRT3 in the regulation of p53.
6. Regulators of p53 Deacetylase
Various factors, including cellular proteins, micro-RNAs (miRNAs), and lncRNAs, have been reported to alter p53 deacetylase activity, thereby regulating p53-mediated biological effects (Figure 3).
6.1. HDAC1
MDM2 promotes p53 deacetylation by recruiting the HDAC1 complex [119]. Acetylation and ubiquitination occur at a common set of lysine residues. The acetylation of p53 prevents MDM2-dependent ubiquitination, which enhances the stability of p53. The HDAC1 complex interacts with MDM2 in a p53-independent manner and promotes ubiquitin-mediated degradation by removing acetyl moieties on p53. HDAC1 and MDM2 then cooperatively establish a complete p53 negative feedback loop.
Other factors that modulate HDAC1 have also been reported. Tribbles 1 (TRB1) suppresses the transcriptional activity of p53 in a HDAC1-dependent manner [153]. TRB1 interacts with HDAC1 and promotes HDAC1-mediated p53 deacetylation at K382, which attenuates the DNA-binding ability of p53. N-arginine dibasic convertase (NRDC), a zinc peptidase of the M16 family, promotes the development of colorectal cancer through the HDAC1/p53 pathway [154]. Nuclear NRDC directly interacts with HDAC1 and modulates its recruitment to the promoter region of p53 target genes, which, in turn, regulates p53 acetylation levels. NRDC controls p53-mediated apoptosis and chemosensitivity via the regulation of HDAC1.
A previous study reported that lnc-Ip53 promoted p53 deacetylation at K382 by stabilizing HDAC1 [78]. It directly interacted with HDAC1 via 2-150 amino acids and has been suggested to mask K74 of HDAC1, a target residue for ubiquitination. Therefore, lnc-Ip53 binding inhibits the ubiquitin-dependent degradation of HDAC1.
6.2. HDAC2
Regarding the p53 deacetylation activity of HDAC2, its regulatory mode has not yet been examined in great detail. USP4 has been identified as a deubiquitinase of HDAC2 [155]. It directly binds to HDAC2 and removes its polyubiquitin chains, which stabilizes HDAC2. USP4 inhibits p53 acetylation and its transcriptional activity. The USP4-mediated accumulation of HDAC2 suppresses p53-dependent pro-apoptotic responses upon DNA damage.
6.3. HDAC6
Limited information is currently available on the regulation of HDAC6 related to the p53 pathway. ARID1A, a subunit of the SWI/SNF chromatin remodeling complex, contributes to the activation of the apoptosis-promoting function of p53 by inhibiting HDAC6 [134]. ARID1A directly represses the transcription of HDAC6. ARID1A mutations up-regulate HDAC6, which deacetylates p53 at K120, resulting in the inactivation of p53 pro-apoptotic functions.
6.4. HDAC8
Regulators of the HDAC8/p53 axis have not yet been identified. A previous study demonstrated that the inhibition of the FMS-like receptor tyrosine kinase-3 (FLT3) up-regulated HDAC8, which promoted p53 acetylation in AML cells with FLT3 internal tandem duplication (ITD) mutations [156]. FLT3 signaling is downregulated by FLT3 inhibitors, which activate forkhead box protein O1 (FOXO1) and FOXO3. Activated FOXO1 and FOXO3 bind to the HDAC8 promoter and enhance its transcription, which attenuates p53 activity via deacetylation at K382. The up-regulation of HDAC8 promotes resistance to FLT3 tyrosine kinase inhibitors (TKIs) and the maintenance of leukemia cells. Therefore, the combination of a HDAC8 inhibitor and FLT3 TKI may be effective in the treatment of FLT3-ITD+ AML.
6.5. SIRT1
Various proteins have been implicated in the positive regulation of SIRT1. Kim et al. identified active regulator of SIRT1 (AROS) as the first direct SIRT1 regulator [157]. AROS directly binds to SIRT1 and enhances SIRT1-mediated p53 deacetylation at K382. It also inhibits p53-dependent transcriptional activation, thereby promoting cell survival in response to DNA damage. PML physically interacts with SIRT1 and promotes SIRT1-mediated p53 deacetylation [83,158]. It also makes a context-dependent contribution to both tumorigenesis and tumor suppression [159]. In this case, PML exerts tumor-promoting effects by modulating SIRT1. When PML is up-regulated, SIRT1 is recruited to PML-NBs and colocalizes with p53, resulting in the suppression of p53-dependent transcription and subsequent cellular senescence. Ski, the transforming protein of the avian Sloan–Kettering virus, cooperates with SIRT1 to suppress the biological functions of p53 [160]. Ski binds to SIRT1 and stabilizes the p53-SIRT interaction, which enhances p53 deacetylation at K382. Ski negatively controls the DNA-binding activity of p53 and confers resistance to DNA-damaging agents. USP22 also acts as a positive regulator of SIRT1 [161]. USP22 interacts with SIRT1 and removes its polyubiquitin chains, which stabilizes SIRT1. The USP22-mediated accumulation of SIRT1 inhibits p53 acetylation and its transcriptional activity. USP22 suppresses cell apoptosis by inhibiting p53 functions in a SIRT1-dependent manner, while its anti-apoptotic effects are required for mouse embryonic development. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) regulates p53 activity by enhancing the deacetylation activity of SIRT1 [162]. It interacts with deleted in breast cancer-1 (DBC1) and prevents the binding of SIRT1-DBC1. The up-regulation of MALAT1 promotes the deacetylation of p53 at K382, thereby inhibiting the transactivation of p53-inducible genes, including p21, BAX, and PUMA. Phospholipase D2 (PLD2) positively modulates SIRT1 and suppresses p53-dependent apoptosis [163]. L173 and L174 in the LXXLL motif of PLD2 interact with the C-terminal region of SIRT1, thereby stimulating the deacetylase activity of SIRT1. Since proteins that bind to the LXXLL motif have been proposed to induce structural changes [164], PLD2 may also induce conformational changes in SIRT1 that enhance its deacetylase activity. PLD2 promotes the SIRT1-mediated deacetylation of p53 and suppresses the p53-dependent transactivation of BAX and NOXA via SIRT1. Moreover, it inhibits etoposide-induced apoptosis in a SIRT1-mediated manner. La Ribonucleoprotein 7 (LARP7), a 7SK RNA-binding protein, enhances SIRT1 deacetylase activity and promotes p53 transactivation [165]. LARP7 directly binds to the N-terminal activation domain of SIRT1, resulting in allosteric and specific increases in its deacetylase activity. LARP1-mediated SIRT1 activation accelerates p53 deacetylation at K382 and stimulates cells to undergo senescence.
Some proteins have been shown to act as negative regulators of SIRT1. Hypermethylated in cancer 1 (HIC1) directly regulates SIRT1 transcription and modulates p53-dependent DNA damage responses [166]. HIC1 interacts with SIRT1 and forms a transcriptional repression complex. The HIC1-SIRT1 complex binds to the SIRT1 promoter and represses SIRT1 transcription; therefore, HIC1 suppresses SIRT1-mediated p53 deacetylation and stimulates p53-induced apoptotic responses. Since p53 up-regulates HIC1 [167,168], p53, SIRT1, and HIC1 may form a feedback loop. DBC1, initially cloned from a region homozygously deleted in breast cancers, directly binds to SIRT1 and inhibits its deacetylation activity, resulting in enhancements in acetylated p53 and its transcriptional activity [169,170]. Furthermore, the knockdown of DBC1 reduces p53 acetylation and its apoptotic activities. SET7/9, a member of the SET domain-containing methyltransferase family, negatively regulates SIRT1, which enhances p53 acetylation at K382 [171]. SET7/9 interacts with SIRT1 both in vitro and in vivo. Although SET7/9 methylates SIRT1 at multiple lysine residues, the methylation of SIRT1 does not affect its deacetylase activity. Instead, the physical association of SET7/9 with SIRT1 prevents SIRT1 binding to p53, which enhances p53 acetylation and subsequent transactivation. Insulin-like growth factor 1 (IGF-1) inhibits SIRT1 deacetylase activity and promotes p53-mediated cellular senescence [172]. IGF-1 plays a dual role by promoting cell proliferation and cellular senescence. While an acute IGF-1 treatment promoted cell proliferation, prolonged exposure to IGF-1 induced p53-dependent cellular senescence. In this case, the prolonged IGF-1 treatment attenuated SIRT1 deacetylase activity, which increased p53 acetylation at K382. Therefore, the prolonged IGF-1 treatment enhanced p53 transcriptional activity and up-regulated p21, thereby promoting cell cycle arrest and cellular senescence. Phosphofurin acidic cluster sorting protein 2 (PACS-2) modulates SIRT1-mediated p53 deacetylation at K382 to induce cell cycle arrest [173]. In the nucleus, PACS-2 associates with SIRT1. In response to DNA damage, PACS-2 translocates into the nucleus and directly interferes with the interaction between p53 and SIRT1, thereby promoting the acetylation of p53 and subsequent transactivation of the p21 gene. Brahma-related gene-1 (BRG1), a specific ATPase of the switch/sucrose non-fermentable chromatin-remodeling complex, inhibits SIRT1-mediated p53 deacetylation and regulates p53-dependent cellular senescence [174]. BRG1 interacts with SIRT1 and interferes with the deacetylation of p53 at K382, which up-regulates p21. The knockdown of BRG1 induces senescence and attenuates the proliferation of colorectal cancer cells in vivo. TSPY-Like 2 (TSPYL2), a nucleosome assembly protein, regulates p53 acetylation at K382 by inhibiting SIRT1 and enhancing the HAT activity of p300 [175]. TSPYL2 represses the function of SIRT1 by disrupting its interaction with target proteins. Under normal conditions, SIRT1 maintains p300 and p53 in a hypoacetylated status, which inhibits the HAT activity of SIRT1 and p53-mediated pro-apoptotic responses. However, under DNA damage conditions, TSPYL2 promotes the transactivation of pro-apoptotic p53 target genes by preventing the SIRT1 interaction with p300.
In addition to these findings, several kinases have been associated with the regulation of the SIRT1/p53 axis. Dual specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) and DYRK3 activate SIRT1, thereby promoting deacetylation of p53 at K382 [176]. DYRK1A and DYRK3 directly bind to SIRT1 and phosphorylate the T522 residue, which enhances SIRT1 deacetylase activity. The knockdown of DYRK1A and DYRK3 results in the hyperacetylation of p53 and sensitizes cells to DNA damage-induced cell death. Mammalian Sterile 20-like kinase 1 (MST1), a serine/threonine kinase, regulates p53 deacetylation in a SIRT1-mediated manner [177]. MST1 phosphorylates SIRT1, and this post-translational modification attenuates the deacetylation ability of SIRT1, resulting in the activation of p53. MST1 enhances p53 transcriptional activity and promotes p53-mediated apoptosis. Inhibition of Casein Kinase-2 (CK2) promotes p53-mediated cell cycle arrest through the attenuation of SIRT1 in glioma cells [178]. CK2 phosphorylates SIRT1 and increases the ability of SIRT1 to deacetylate p53 at K382 [179]. Treatments with CK2 inhibitors have been shown to down-regulate SIRT1 activity, leading to the activation of p53 and apoptosis. AMP-activated protein kinase (AMPK) inhibits the deacetylase activity of SIRT1 and antagonizes its suppressive effects towards p53 [180]. AMPK directly binds to SIRT1 through the deacetylase domain of SIRT1 and phosphorylates T344. This phosphorylation suppresses the deacetylase activity of SIRT1 towards p53 by interfering with the interaction between SIRT1 and p53, thereby inhibiting the transcription of p53 target genes, including p21 and BAX. HIPK2 suppresses the activation of SIRT1 and stimulates the transactivation of p53 [181]. In response to DNA damage, HIPK2 forms a complex with SIRT1. HIPK2 interacts via its N-terminal region containing a kinase domain with the central SIR domain of SIRT1, and it phosphorylates SIRT1 at S682. This modification suppresses the deacetylase activity of SIRT1, thereby inducing the acetylation of p53 at K382. The HIPK2-mediated inhibition of SIRT1 up-regulates proapoptotic p53 target gene expression and promotes apoptosis.
Previous studies revealed the involvement of miRNAs in the regulation of SIRT1. miR-34a was shown to down-regulate the expression of SIRT1 through a miR-34a-binding site in its 3’ UTR region [182]. miR-34a also promoted the acetylation of p53 at K382 by decreasing the expression of SIRT1, which led to the transactivation of p53 target genes, including p21 and PUMA. Since miR-34a has been identified as a direct transcriptional target of p53 [183], it may be involved in a positive feedback loop that increases p53 activity through the regulation of the SIRT1/p53 axis. miR-128 directly suppresses the expression of SIRT1 via a miR-128-binding site within the 3’ UTR of SIRT1 [184]. Furthermore, the miR-128-mediated inhibition of SIRT1 increases acetylated p53 at K382 and transactivates p53 target genes, including p21, PUMA, and NOXA.
6.6. SIRT3
The regulatory mode of SIRT3 related to the p53 deacetylation currently remains unclear. Zinc finger matrin-type 1 (ZMAT1), a member of the Cys2His2 (C2H2)-type zinc finger family, has been shown to contribute to the anti-cancer effects of SIRT3 in pancreatic ductal adenocarcinoma [185]. ZMAT1 induces the expression of SIRT3 and subsequent upregulation of p53, thereby exerting inhibitory effects on cancer. ZMAT1 binds to the promoter of SIRT3 and induces its transcription, which leads to the induction of p53. ZMAT1 regulates cancer cell proliferation and apoptosis in a p53-dependent manner. The SIRT3/p53 axis in pancreatic cancer cells need to be investigated in future studies.
7. Conclusions
Since the discovery of p53 in 1979, numerous studies have revealed its function as a tumor suppressor [6]. p53 acts as a transcriptional factor that regulates more than 200 target genes and exerts a number of biological effects. Post-translational modifications play an essential role in the precise regulation of the selectivity of different target genes and subsequent physiological outcomes. p53 undergoes various post-translational modifications, which modulate the transcriptional activity, degradation, and localization of p53. Acetylation is a key modification for the activation of p53 and its promoter specificity and, thus, contributes to cell fate determination. Therefore, further studies on the regulatory mode of p53 acetylation will provide insights into the diversity of the p53 network.
p53 plays a crucial role in tumor suppression, with its dysfunction frequently detected in p53 wild-type and p53-mutated cancers. Several therapeutic strategies have been suggested for the targeting of p53 [186]. The therapeutic strategy for p53 wild-type cancers is to inhibit p53 degradation by blocking interactions between p53 and MDM2, thereby promoting the accumulation of p53 and its anti-tumor activity. The treatment approach for p53-mutated cancers is to reactivate wild-type p53 or promote the degradation of mutant p53. Since p53 acetylation is essential for its activation, it may be an attractive therapeutic target in the p53 reactivation strategy, and various small-molecule compounds have been shown to target p53 deacetylases, including HDAC1 and SIRT1 [187,188]. However, the proteins that comprise the HDAC family are structurally similar and, thus, small-molecule approaches that target p53 deacetylase have a number of limitations, such as low specificity towards the substrate [11]. The targeting of regulators of p53 deacetylases may increase specificity and effectiveness towards p53 deacetylases. A more detailed understanding of the regulatory mode of p53 acetylation will contribute to the development of future cancer therapies.
Author Contributions
Conceptualization, M.N.; design, M.N.; data generation, M.N.; writing—original draft preparation, M.N. writing—review and editing, D.M., C.M., H.A., M.A., Y.I. and H.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Grants-in-Aid for Scientific Research (C) (No. 20K07052) and a Grant-in-Aid for Scientific Research (B) (No. 21H02650) from the Japan Society for the Promotion of Science (JSPS), a Grant-in-Aid for Young Scientists (No. 18K16081) from JSPS, and Grants-in-Aid for Research (No. 2021103, 2022005, and 2221102) from Nagoya City University. This work was also supported by the Japan Science and Technology Agency (JST) SPRING, Grant Number JPMJSP2130.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [Green Version]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M.; Feng, Z.; Hu, W. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laptenko, O.; Prives, C. Transcriptional regulation by p53: One protein, many possibilities. Cell Death Differ. 2006, 13, 951–961. [Google Scholar] [CrossRef] [Green Version]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Kon, N.; Gu, A.P.; Tavana, O.; Gu, W. Deciphering the acetylation code of p53 in transcription regulation and tumor suppression. Oncogene 2022, 41, 3039–3050. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.M.; Quelle, D.E. p53 Acetylation: Regulation and Consequences. Cancers 2014, 7, 30–69. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kon, N.; Ou, Y.; Wang, S.J.; Li, H.; Rustgi, A.K.; Gu, W. mTOR inhibition acts as an unexpected checkpoInt. in p53-mediated tumor suppression. Genes Dev. 2021, 35, 59–64. [Google Scholar] [CrossRef]
- Tang, Y.; Luo, J.; Zhang, W.; Gu, W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation is indispensable for p53 activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, C.; Wu, Z.; Mazur, S.J.; Borges, H.; Rossi, M.; Lin, T.; Wang, J.Y.; Anderson, C.W.; Appella, E.; Xu, Y. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol. Cell Biol. 2006, 26, 6859–6869. [Google Scholar] [CrossRef] [Green Version]
- Knights, C.D.; Catania, J.; Di Giovanni, S.; Muratoglu, S.; Perez, R.; Swartzbeck, A.; Quong, A.A.; Zhang, X.; Beerman, T.; Pestell, R.G.; et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell Biol. 2006, 173, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Tsay, Y.G.; Tan, B.C.; Lo, W.Y.; Lee, S.C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J. Biol. Chem. 2003, 278, 25568–25576. [Google Scholar] [CrossRef] [Green Version]
- Joubel, A.; Chalkley, R.J.; Medzihradszky, K.F.; Hondermarck, H.; Burlingame, A.L. Identification of new p53 acetylation sites in COS-1 cells. Mol. Cell Proteom. 2009, 8, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Eckner, R.; Ewen, M.E.; Newsome, D.; Gerdes, M.; DeCaprio, J.A.; Lawrence, J.B.; Livingston, D.M. Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 1994, 8, 869–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrivia, J.C.; Kwok, R.P.; Lamb, N.; Hagiwara, M.; Montminy, M.R.; Goodman, R.H. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 1993, 365, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Sellers, W.R.; Livingston, D.M.; Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 1994, 77, 799–800. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, M.; Kohno, T.; Okudela, K.; Otsuka, A.; Sasaki, H.; Tanabe, C.; Sakiyama, T.; Hirama, C.; Kitabayashi, I.; Minna, J.D.; et al. Mutations and deletions of the CBP gene in human lung cancer. Clin. Cancer Res. 2005, 11, 512–519. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, Y.; Guo, G.; Huang, Y.; Hu, X.; Tang, A.; Gao, S.; Wu, R.; Chen, C.; Li, X.; Zhou, L.; et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011, 43, 875–878. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189–195. [Google Scholar] [CrossRef]
- So, C.K.; Nie, Y.; Song, Y.; Yang, G.Y.; Chen, S.; Wei, C.; Wang, L.D.; Doggett, N.A.; Yang, C.S. Loss of heterozygosity and internal tandem duplication mutations of the CBP gene are frequent events in human esophageal squamous cell carcinoma. Clin. Cancer Res. 2004, 10, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, T.; Kawabata, M.; Ohshima, T.; Ikeda, M.A. Growth suppression of human carcinoma cells by reintroduction of the p300 coactivator. Proc. Natl. Acad. Sci. USA 2002, 99, 13073–13078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayther, S.A.; Batley, S.J.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.F.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.M.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef]
- Ohshima, T.; Suganuma, T.; Ikeda, M. A novel mutation lacking the bromodomain of the transcriptional coactivator p300 in the SiHa cervical carcinoma cell line. Biochem. Biophys. Res. Commun. 2001, 281, 569–575. [Google Scholar] [CrossRef]
- Lill, N.L.; Grossman, S.R.; Ginsberg, D.; DeCaprio, J.; Livingston, D.M. Binding and modulation of p53 by p300/CBP coactivators. Nature 1997, 387, 823–827. [Google Scholar] [CrossRef]
- Avantaggiati, M.L.; Ogryzko, V.; Gardner, K.; Giordano, A.; Levine, A.S.; Kelly, K. Recruitment of p300/CBP in p53-dependent signal pathways. Cell 1997, 89, 1175–1184. [Google Scholar] [CrossRef] [Green Version]
- Giannini, R.; Cavallini, A. Expression analysis of a subset of coregulators and three nuclear receptors in human colorectal carcinoma. Anticancer Res. 2005, 25, 4287–4292. [Google Scholar]
- Ying, M.Z.; Wang, J.J.; Li, D.W.; Yu, G.; Wang, X.; Pan, J.; Chen, Y.; He, M.X. The p300/CBP associated factor is frequently downregulated in intestinal-type gastric carcinoma and constitutes a biomarker for clinical outcome. Cancer Biol. Ther. 2010, 9, 312–320. [Google Scholar] [CrossRef]
- Bassi, C.; Li, Y.T.; Khu, K.; Mateo, F.; Baniasadi, P.S.; Elia, A.; Mason, J.; Stambolic, V.; Pujana, M.A.; Mak, T.W.; et al. The acetyltransferase Tip60 contributes to mammary tumorigenesis by modulating DNA repair. Cell Death Differ. 2016, 23, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- ME, L.L.; Vidal, F.; Gallardo, D.; Diaz-Fuertes, M.; Rojo, F.; Cuatrecasas, M.; López-Vicente, L.; Kondoh, H.; Blanco, C.; Carnero, A.; et al. New p53 related genes in human tumors: Significant downregulation in colon and lung carcinomas. Oncol. Rep. 2006, 16, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sun, J.; Chen, T.; Tao, Z.; Zhang, X.; Tian, F.; Zhou, X.; Lu, D. Tat-interactive Protein-60KDA (TIP60) Regulates the Tumorigenesis of Lung Cancer In Vitro. J. Cancer 2017, 8, 2277–2281. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, H.; Pan, H.; Yang, Y.; Huang, G.; Yang, Y.; Zhou, W.P.; Pan, Z.Y. The histone acetyltransferase hMOF suppresses hepatocellular carcinoma growth. Biochem. Biophys. Res. Commun. 2014, 452, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Bacolla, A.; Chaudhary, S.; Hunt, C.R.; Pandita, S.; Chauhan, R.; Gupta, A.; Tainer, J.A.; Pandita, T.K. Histone Acetyltransferase MOF Orchestrates Outcomes at the Crossroad of Oncogenesis, DNA Damage Response, Proliferation, and Stem Cell Development. Mol. Cell Biol. 2020, 40, e00232-20. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Ogryzko, V.V.; Nishikawa, J.; Howard, B.H.; Nakatani, Y. A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 1996, 382, 319–324. [Google Scholar] [CrossRef]
- Schiltz, R.L.; Nakatani, Y. The PCAF acetylase complex as a potential tumor suppressor. Biochim. Biophys. Acta 2000, 1470, M37–M53. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Dong, L.; Tang, M.; Zhang, P.; Zhang, C.; Cao, Z.; Zhu, Q.; Chen, Y.; Wang, H.; et al. Histone H1 acetylation at lysine 85 regulates chromatin condensation and genome stability upon DNA damage. Nucleic Acids Res. 2018, 46, 7716–7730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozdağ, H.; Batley, S.J.; Försti, A.; Iyer, N.G.; Daigo, Y.; Boutell, J.; Arends, M.J.; Ponder, B.A.; Kouzarides, T.; Caldas, C. Mutation analysis of CBP and PCAF reveals rare inactivating mutations in cancer cell lines but not in primary tumours. Br. J. Cancer 2002, 87, 1162–1165. [Google Scholar] [CrossRef]
- Ikura, T.; Ogryzko, V.V.; Grigoriev, M.; Groisman, R.; Wang, J.; Horikoshi, M.; Scully, R.; Qin, J.; Nakatani, Y. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 2000, 102, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Jiang, X.; Chen, S.; Fernandes, N.; Price, B.D. A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc. Natl. Acad. Sci. USA 2005, 102, 13182–13187. [Google Scholar] [CrossRef] [Green Version]
- Borrow, J.; Stanton, V.P., Jr.; Andresen, J.M.; Becher, R.; Behm, F.G.; Chaganti, R.S.; Civin, C.I.; Disteche, C.; Dubé, I.; Frischauf, A.M.; et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 1996, 14, 33–41. [Google Scholar] [CrossRef]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ increases p53 acetylation and premature senescence through its complex formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokudai, S.; Aikawa, Y.; Tagata, Y.; Tsuchida, N.; Taya, Y.; Kitabayashi, I. Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell-cycle arrest. J. Biol. Chem. 2009, 284, 237–244. [Google Scholar] [CrossRef] [Green Version]
- Hilfiker, A.; Hilfiker-Kleiner, D.; Pannuti, A.; Lucchesi, J.C. mof, a putative acetyl transferase gene related to the Tip60 and MOZ human genes and to the SAS genes of yeast, is required for dosage compensation in Drosophila. Embo J. 1997, 16, 2054–2060. [Google Scholar] [CrossRef]
- Taipale, M.; Rea, S.; Richter, K.; Vilar, A.; Lichter, P.; Imhof, A.; Akhtar, A. hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol. Cell Biol. 2005, 25, 6798–6810. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Corsa, C.A.; Pan, P.W.; Wu, L.; Ferguson, D.; Yu, X.; Min, J.; Dou, Y. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol. Cell Biol. 2010, 30, 5335–5347. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M.; Stanek, T.J.; Frank, A.; Murphy, M.E.; McMahon, S.B. Acetylation of the DNA binding domain regulates transcription-independent apoptosis by p53. J. Biol. Chem. 2009, 284, 20197–20205. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Zhang, L.; Hu, L.; Peng, Q.; Han, W.; Du, X.; Ke, Y. hALP, a novel transcriptional U three protein (t-UTP), activates RNA polymerase I transcription by binding and acetylating the upstream binding factor (UBF). J. Biol. Chem. 2011, 286, 7139–7148. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.; Liu, H.; Wang, Q.; Tang, Z.; Hou, L.; Zhang, B. Molecular cloning of a novel human gene encoding histone acetyltransferase-like protein involved in transcriptional activation of hTERT. Biochem. Biophys. Res. Commun. 2003, 311, 506–513. [Google Scholar] [CrossRef]
- Shen, Q.; Zheng, X.; McNutt, M.A.; Guang, L.; Sun, Y.; Wang, J.; Gong, Y.; Hou, L.; Zhang, B. NAT10, a nucleolar protein, localizes to the midbody and regulates cytokinesis and acetylation of microtubules. Exp. Cell Res. 2009, 315, 1653–1667. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, X.; Jin, K.; Lu, M.; Zhang, C.; Du, X.; Xing, B. NAT10 is upregulated in hepatocellular carcinoma and enhances mutant p53 activity. BMC Cancer 2017, 17, 605. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Tan, Y.; Zhang, C.; Zhang, Y.; Zhang, L.; Ren, P.; Deng, H.; Luo, J.; Ke, Y.; Du, X. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016, 17, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Huang, S.M.; Baglia, L.A.; McCance, D.J. The E6 protein of human papillomavirus type 16 binds to and inhibits co-activation by CBP and p300. Embo J. 1999, 18, 5061–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, H.; Degenkolbe, R.; Bernard, H.U.; O’Connor, M.J. The human papillomavirus type 16 E6 oncoprotein can down-regulate p53 activity by targeting the transcriptional coactivator CBP/p300. J. Virol. 1999, 73, 6209–6219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, D.; Ogryzko, V.; Kao, H.Y.; Nash, A.; Chen, H.; Nakatani, Y.; Evans, R.M. A viral mechanism for inhibition of p300 and PCAF acetyltransferase activity. Cell 1999, 96, 393–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, A.; Lai, C.H.; Zhao, X.; Saito, S.; Hamilton, M.H.; Appella, E.; Yao, T.P. p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. Embo J. 2001, 20, 1331–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobet, E.; Zeng, X.; Zhu, Y.; Keller, D.; Lu, H. MDM2 inhibits p300-mediated p53 acetylation and activation by forming a ternary complex with the two proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 12547–12552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbatini, P.; McCormick, F. MDMX inhibits the p300/CBP-mediated acetylation of p53. DNA Cell Biol. 2002, 21, 519–525. [Google Scholar] [CrossRef]
- Kitagawa, M.; Lee, S.H.; McCormick, F. Skp2 suppresses p53-dependent apoptosis by inhibiting p300. Mol. Cell 2008, 29, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Shiseki, M.; Nagashima, M.; Pedeux, R.M.; Kitahama-Shiseki, M.; Miura, K.; Okamura, S.; Onogi, H.; Higashimoto, Y.; Appella, E.; Yokota, J.; et al. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 2003, 63, 2373–2378. [Google Scholar] [PubMed]
- Nagashima, M.; Shiseki, M.; Miura, K.; Hagiwara, K.; Linke, S.P.; Pedeux, R.; Wang, X.W.; Yokota, J.; Riabowol, K.; Harris, C.C. DNA damage-inducible gene p33ING2 negatively regulates cell proliferation through acetylation of p53. Proc. Natl. Acad. Sci. USA 2001, 98, 9671–9676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedeux, R.; Sengupta, S.; Shen, J.C.; Demidov, O.N.; Saito, S.; Onogi, H.; Kumamoto, K.; Wincovitch, S.; Garfield, S.H.; McMenamin, M.; et al. ING2 regulates the onset of replicative senescence by induction of p300-dependent p53 acetylation. Mol. Cell Biol. 2005, 25, 6639–6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, T.; Murayama, A.; Katagiri, N.; Ohta, Y.M.; Fujita, E.; Masumoto, H.; Ema, M.; Takahashi, S.; Kimura, K.; Yanagisawa, J. RNA content in the nucleolus alters p53 acetylation via MYBBP1A. Embo J. 2011, 30, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahata, B.; Sundqvist, A.; Xirodimas, D.P. Recruitment of RPL11 at promoter sites of p53-regulated genes upon nucleolar stress through NEDD8 and in an Mdm2-dependent manner. Oncogene 2012, 31, 3060–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, D.; Li, L.; Lou, H.; Sun, H.; Ngai, S.M.; Shao, G.; Tang, J. The ribosomal protein S26 regulates p53 activity in response to DNA damage. Oncogene 2014, 33, 2225–2235. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Z.; Yang, J.E.; Luo, Y.W.; Liu, F.T.; Yuan, Y.F.; Zhuang, S.M. A p53/lnc-Ip53 Negative Feedback Loop Regulates Tumor Growth and Chemoresistance. Adv. Sci. 2020, 7, 2001364. [Google Scholar] [CrossRef]
- Aikawa, Y.; Nguyen, L.A.; Isono, K.; Takakura, N.; Tagata, Y.; Schmitz, M.L.; Koseki, H.; Kitabayashi, I. Roles of HIPK1 and HIPK2 in AML1- and p300-dependent transcription, hematopoiesis and blood vessel formation. Embo J. 2006, 25, 3955–3965. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, T.G.; Möller, A.; Sirma, H.; Zentgraf, H.; Taya, Y.; Dröge, W.; Will, H.; Schmitz, M.L. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002, 4, ncb715. [Google Scholar] [CrossRef]
- Puca, R.; Nardinocchi, L.; Sacchi, A.; Rechavi, G.; Givol, D.; D’Orazi, G. HIPK2 modulates p53 activity towards pro-apoptotic transcription. Mol. Cancer 2009, 8, 85. [Google Scholar] [CrossRef] [PubMed]
- Dornan, D.; Eckert, M.; Wallace, M.; Shimizu, H.; Ramsay, E.; Hupp, T.R.; Ball, K.L. Interferon regulatory factor 1 binding to p300 stimulates DNA-dependent acetylation of p53. Mol. Cell Biol. 2004, 24, 10083–10098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, M.; Carbone, R.; Sebastiani, C.; Cioce, M.; Fagioli, M.; Saito, S.; Higashimoto, Y.; Appella, E.; Minucci, S.; Pandolfi, P.P.; et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000, 406, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Shima, Y.; Shima, T.; Chiba, T.; Irimura, T.; Pandolfi, P.P.; Kitabayashi, I. PML activates transcription by protecting HIPK2 and p300 from SCFFbx3-mediated degradation. Mol. Cell Biol. 2008, 28, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Salomoni, P.; Pandolfi, P.P. The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2000, 2, E85–E90. [Google Scholar] [CrossRef]
- Pan, X.; Zhao, J.; Zhang, W.N.; Li, H.Y.; Mu, R.; Zhou, T.; Zhang, H.Y.; Gong, W.L.; Yu, M.; Man, J.H.; et al. Induction of SOX4 by DNA damage is critical for p53 stabilization and function. Proc. Natl. Acad. Sci. USA 2009, 106, 3788–3793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Gao, M.; Dong, W.; Hu, M.; Li, J.; Shi, X.; Hao, Y.; Li, Y.; Huang, C. p85α mediates p53 K370 acetylation by p300 and regulates its promoter-specific transactivity in the cellular UVB response. Oncogene 2011, 30, 1360–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.J.; Rivera, M.N.; Coffman, E.J.; Haber, D.A. The WTX tumor suppressor enhances p53 acetylation by CBP/p300. Mol. Cell 2012, 45, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.S.; Hubberstey, A.V.; Palissot, V.; et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 4115–4120. [Google Scholar] [CrossRef] [Green Version]
- Sinha, S.; Malonia, S.K.; Mittal, S.P.; Mathai, J.; Pal, J.K.; Chattopadhyay, S. Chromatin remodelling protein SMAR1 inhibits p53 dependent transactivation by regulating acetyl transferase p300. Int. J. Biochem. Cell Biol. 2012, 44, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Dai, C.; Qin, J.; Gu, W. Negative regulation of the p300-p53 interplay by DDX24. Oncogene 2016, 35, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, C.; Kawarada, Y.; Inoue, Y.; Suzuki, C.; Mitamura, K.; Morishita, D.; Ohoka, N.; Imamura, T.; Hayashi, H. Transcriptional Coactivator TAZ Negatively Regulates Tumor Suppressor p53 Activity and Cellular Senescence. Cells 2020, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Colosimo, A.L.; Yang, X.J.; Liao, D. Adenovirus E1B 55-kilodalton oncoprotein inhibits p53 acetylation by PCAF. Mol. Cell Biol. 2000, 20, 5540–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zeng, S.X.; Lee, H.; Lu, H. MDM2 mediates p300/CREB-binding protein-associated factor ubiquitination and degradation. J. Biol. Chem. 2004, 279, 20035–20043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zeng, S.X.; Dai, M.S.; Yang, X.J.; Lu, H. MDM2 inhibits PCAF (p300/CREB-binding protein-associated factor)-mediated p53 acetylation. J. Biol. Chem. 2002, 277, 30838–30843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grishina, I.; Debus, K.; García-Limones, C.; Schneider, C.; Shresta, A.; García, C.; Calzado, M.A.; Schmitz, M.L. SIAH-mediated ubiquitination and degradation of acetyl-transferases regulate the p53 response and protein acetylation. Biochim. Biophys. Acta 2012, 1823, 2287–2296. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, S.; Takayama, S.; Froesch, B.A.; Zapata, J.M.; Reed, J.C. p53-inducible human homologue of Drosophila seven in absentia (Siah) inhibits cell growth: Suppression by BAG-1. Embo J. 1998, 17, 2736–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Stefano, V.; Soddu, S.; Sacchi, A.; D’Orazi, G. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21Waf1 after nonapoptotic DNA damage. Oncogene 2005, 24, 5431–5442. [Google Scholar] [CrossRef] [Green Version]
- D’Orazi, G.; Cecchinelli, B.; Bruno, T.; Manni, I.; Higashimoto, Y.; Saito, S.; Gostissa, M.; Coen, S.; Marchetti, A.; Del Sal, G.; et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002, 4, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys. Acta 2011, 1813, 1245–1253. [Google Scholar] [CrossRef]
- Dai, C.; Shi, D.; Gu, W. Negative regulation of the acetyltransferase TIP60-p53 interplay by UHRF1 (ubiquitin-like with PHD and RING finger domains 1). J. Biol. Chem. 2013, 288, 19581–19592. [Google Scholar] [CrossRef]
- Dar, A.; Shibata, E.; Dutta, A. Deubiquitination of Tip60 by USP7 determines the activity of the p53-dependent apoptotic pathway. Mol. Cell Biol. 2013, 33, 3309–3320. [Google Scholar] [CrossRef] [Green Version]
- Charvet, C.; Wissler, M.; Brauns-Schubert, P.; Wang, S.J.; Tang, Y.; Sigloch, F.C.; Mellert, H.; Brandenburg, M.; Lindner, S.E.; Breit, B.; et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol. Cell 2011, 42, 584–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidu, S.R.; Lakhter, A.J.; Androphy, E.J. PIASy-mediated Tip60 sumoylation regulates p53-induced autophagy. Cell Cycle 2012, 11, 2717–2728. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Chen, Y.; Song, Q.; Xu, D.; Wang, Y.; Ma, D. PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia 2009, 11, 345–354. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.; Tang, Y.; Jung, S.Y.; Qin, J.; Aaronson, S.A.; Gu, W. Differential effects on p53-mediated cell cycle arrest vs. apoptosis by p90. Proc. Natl. Acad. Sci. USA 2011, 108, 18937–18942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Wang, J.; Wang, J.; Wang, R.; Liu, Z.; Yu, Y.; Lu, H. ING5 is a Tip60 cofactor that acetylates p53 in response to DNA damage. Cancer Res. 2013, 73, 3749–3760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, S.M.; Hagen, J.; Tompkins, V.S.; Thies, K.; Quelle, F.W.; Quelle, D.E. Nuclear interactor of ARF and Mdm2 regulates multiple pathways to activate p53. Cell Cycle 2014, 13, 1288–1298. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wu, L.; Corsa, C.A.; Kunkel, S.; Dou, Y. Two mammalian MOF complexes regulate transcription activation by distinct mechanisms. Mol. Cell 2009, 36, 290–301. [Google Scholar] [CrossRef] [Green Version]
- McCullough, C.E.; Marmorstein, R. Molecular Basis for Histone Acetyltransferase Regulation by Binding Partners, Associated Domains, and Autoacetylation. ACS Chem. Biol. 2016, 11, 632–642. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Liu, X.; Zhang, C.; Xing, B.; Du, X. Autoacetylation of NAT10 is critical for its function in rRNA transcription activation. Biochem. Biophys. Res. Commun. 2017, 483, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cai, S.; Zhang, C.; Liu, Z.; Luo, J.; Xing, B.; Du, X. Deacetylation of NAT10 by Sirt1 promotes the transition from rRNA biogenesis to autophagy upon energy stress. Nucleic Acids Res. 2018, 46, 9601–9616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W.; Röske, A.; Niesporek, S.; Noske, A.; Buckendahl, A.C.; Dietel, M.; Gekeler, V.; Boehm, M.; Beckers, T.; Denkert, C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: Specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 2008, 14, 1669–1677. [Google Scholar] [CrossRef] [Green Version]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology 2012, 143, 811–820.e815. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche, F.R.; Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Scholman, K.; Denkert, C.; Dietel, M.; et al. Class I histone deacetylases 1, 2 and 3 are highly expressed in renal cell cancer. BMC Cancer 2008, 8, 381. [Google Scholar] [CrossRef] [Green Version]
- Mithraprabhu, S.; Kalff, A.; Chow, A.; Khong, T.; Spencer, A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics 2014, 9, 1511–1520. [Google Scholar] [CrossRef] [Green Version]
- Ito, A.; Kawaguchi, Y.; Lai, C.H.; Kovacs, J.J.; Higashimoto, Y.; Appella, E.; Yao, T.P. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. Embo J. 2002, 21, 6236–6245. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Krämer, O.H. HDAC2: A critical factor in health and disease. Trends Pharm. Sci. 2009, 30, 647–655. [Google Scholar] [CrossRef]
- Zhu, P.; Martin, E.; Mengwasser, J.; Schlag, P.; Janssen, K.P.; Göttlicher, M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004, 5, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, P.; Seidler, B.; Schüler, S.; Schnieke, A.; Göttlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [Green Version]
- Niegisch, G.; Knievel, J.; Koch, A.; Hader, C.; Fischer, U.; Albers, P.; Schulz, W.A. Changes in histone deacetylase (HDAC) expression patterns and activity of HDAC inhibitors in urothelial cancers. Urol. Oncol. 2013, 31, 1770–1779. [Google Scholar] [CrossRef]
- Moreno, D.A.; Scrideli, C.A.; Cortez, M.A.; de Paula Queiroz, R.; Valera, E.T.; da Silva Silveira, V.; Yunes, J.A.; Brandalise, S.R.; Tone, L.G. Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 665–673. [Google Scholar] [CrossRef]
- Marquard, L.; Poulsen, C.B.; Gjerdrum, L.M.; de Nully Brown, P.; Christensen, I.J.; Jensen, P.B.; Sehested, M.; Johansen, P.; Ralfkiaer, E. Histone deacetylase 1, 2, 6 and acetylated histone H4 in B- and T-cell lymphomas. Histopathology 2009, 54, 688–698. [Google Scholar] [CrossRef]
- Adams, H.; Fritzsche, F.R.; Dirnhofer, S.; Kristiansen, G.; Tzankov, A. Class I histone deacetylases 1, 2 and 3 are highly expressed in classical Hodgkin’s lymphoma. Expert Opin. Targets 2010, 14, 577–584. [Google Scholar] [CrossRef]
- Brandl, A.; Wagner, T.; Uhlig, K.M.; Knauer, S.K.; Stauber, R.H.; Melchior, F.; Schneider, G.; Heinzel, T.; Krämer, O.H. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. J. Mol. Cell Biol. 2012, 4, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Qin, M.; Wu, H.P.; Khamis, M.Y.; Li, Y.H.; Ma, L.Y.; Liu, H.M. A Review of Progress in Histone Deacetylase 6 Inhibitors Research: Structural Specificity and Functional Diversity. J. Med. Chem. 2021, 64, 1362–1391. [Google Scholar] [CrossRef]
- Seidel, C.; Schnekenburger, M.; Dicato, M.; Diederich, M. Histone deacetylase 6 in health and disease. Epigenomics 2015, 7, 103–118. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Sun, X.; Zhang, L.; Yan, B.; Xie, S.; Liu, R.; Liu, M.; Zhou, J. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein Cell 2013, 5, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, C.A.; Khanim, F.L.; Hayden, R.; Bunce, C.M.; White, D.A.; Drayson, M.T.; Craddock, C.; Turner, B.M. Histone deacetylases in acute myeloid leukaemia show a distinctive pattern of expression that changes selectively in response to deacetylase inhibitors. Leukemia 2005, 19, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett 2017, 391, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., III; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Sengupta, N.; Villagra, A.; Rezai-Zadeh, N.; Seto, E. Histone deacetylase 8 safeguards the human ever-shorter telomeRes. 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell Biol. 2006, 26, 5259–5269. [Google Scholar] [CrossRef] [Green Version]
- Deardorff, M.A.; Bando, M.; Nakato, R.; Watrin, E.; Itoh, T.; Minamino, M.; Saitoh, K.; Komata, M.; Katou, Y.; Clark, D.; et al. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 2012, 489, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Stünkel, W.; Peh, B.K.; Tan, Y.C.; Nayagam, V.M.; Wang, X.; Salto-Tellez, M.; Ni, B.; Entzeroth, M.; Wood, J. Function of the SIRT1 protein deacetylase in cancer. Biotechnol. J. 2007, 2, 1360–1368. [Google Scholar] [CrossRef]
- Huffman, D.M.; Grizzle, W.E.; Bamman, M.M.; Kim, J.S.; Eltoum, I.A.; Elgavish, A.; Nagy, T.R. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007, 67, 6612–6618. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Nihal, M.; Singh, C.K.; Zhong, W.; Liu, X.; Ahmad, N. Pro-Proliferative Function of Mitochondrial Sirtuin Deacetylase SIRT3 in Human Melanoma. J. Invest. Derm. 2016, 136, 809–818. [Google Scholar] [CrossRef] [Green Version]
- Hua, W.K.; Qi, J.; Cai, Q.; Carnahan, E.; Ayala Ramirez, M.; Li, L.; Marcucci, G.; Kuo, Y.H. HDAC8 regulates long-term hematopoietic stem-cell maintenance under stress by modulating p53 activity. Blood 2017, 130, 2619–2630. [Google Scholar] [CrossRef]
- Longo, V.D.; Kennedy, B.K. Sirtuins in aging and age-related disease. Cell 2006, 126, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control. of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control. of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef] [Green Version]
- Torrens-Mas, M.; Oliver, J.; Roca, P.; Sastre-Serra, J. SIRT3: Oncogene and Tumor Suppressor in Cancer. Cancers 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B.; et al. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Patel, K.; Muldoon-Jacobs, K.; Bisht, K.S.; Aykin-Burns, N.; Pennington, J.D.; van der Meer, R.; Nguyen, P.; Savage, J.; Owens, K.M.; et al. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell 2010, 17, 41–52. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, A.; Chen, Q. SirT3 and p53 Deacetylation in Aging and Cancer. J. Cell Physiol. 2017, 232, 2308–2311. [Google Scholar] [CrossRef]
- Xiong, Y.; Wang, L.; Wang, S.; Wang, M.; Zhao, J.; Zhang, Z.; Li, X.; Jia, L.; Han, Y. SIRT3 deacetylates and promotes degradation of P53 in PTEN-defective non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2018, 144, 189–198. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Zhou, L.M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, C.; Inoue, Y.; Hayashi, H. Pseudokinase tribbles 1 (TRB1) negatively regulates tumor-suppressor activity of p53 through p53 deacetylation. Biol. Pharm. Bull. 2015, 38, 618–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, K.; Sakamoto, J.; Matsumoto, Y.; Ikuta, K.; Goto, N.; Morita, Y.; Ohno, M.; Nishi, K.; Eto, K.; Kimura, Y.; et al. Nardilysin controls intestinal tumorigenesis through HDAC1/p53-dependent transcriptional regulation. JCI Insight 2018, 3, e91316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Hao, Q.; Luo, J.; Xiong, J.; Zhang, S.; Wang, T.; Bai, L.; Wang, W.; Chen, M.; Wang, W.; et al. USP4 inhibits p53 and NF-κB through deubiquitinating and stabilizing HDAC2. Oncogene 2016, 35, 2902–2912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.; Jia, M.Y.; Fang, W.Y.; Chen, X.J.; Mu, L.L.; Wang, Z.Y.; Shen, Y.; Xiang, R.F.; Wang, L.N.; Wang, L.; et al. FLT3 inhibition upregulates HDAC8 via FOXO to inactivate p53 and promote maintenance of FLT3-ITD+ acute myeloid leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.J.; Kho, J.H.; Kang, M.R.; Um, S.J. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol. Cell 2007, 28, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazza, M.; Pelicci, P.G. Is PML a Tumor Suppressor? Front. Oncol. 2013, 3, 174. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Iemura, S.; Natsume, T.; Miyazawa, K.; Imamura, T. Suppression of p53 activity through the cooperative action of Ski and histone deacetylase SIRT1. J. Biol. Chem. 2011, 286, 6311–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Yang, H.; Kong, Q.; Li, J.; Lee, S.M.; Gao, B.; Dong, H.; Wei, J.; Song, J.; Zhang, D.D.; et al. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 2012, 46, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Liu, Y.; Zhuang, H.; Yang, B.; Hei, K.; Xiao, M.; Hou, C.; Gao, H.; Zhang, X.; Jia, C.; et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017, 45, 9947–9959. [Google Scholar] [CrossRef]
- Lee, H.; Jung, T.Y.; Lim, S.H.; Choi, E.J.; Lee, J.; Min, D.S. Phospholipase D2 is a positive regulator of sirtuin 1 and modulates p53-mediated apoptosis via sirtuin 1. Exp. Mol. Med. 2021, 53, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Savkur, R.S.; Burris, T.P. The coactivator LXXLL nuclear receptor recognition motif. J. Pept. Res. 2004, 63, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Li, Z.; Xiong, J.; Geng, Z.; Wei, W.; Zhang, Y.; Wu, G.; Zhuang, T.; Tian, X.; Liu, Z.; et al. LARP7 ameliorates cellular senescence and aging by allosterically enhancing SIRT1 deacetylase activity. Cell Rep. 2021, 37, 110038. [Google Scholar] [CrossRef]
- Chen, W.Y.; Wang, D.H.; Yen, R.C.; Luo, J.; Gu, W.; Baylin, S.B. Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 2005, 123, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Guerardel, C.; Deltour, S.; Pinte, S.; Monte, D.; Begue, A.; Godwin, A.K.; Leprince, D. Identification in the human candidate tumor suppressor gene HIC-1 of a new major alternative TATA-less promoter positively regulated by p53. J. Biol. Chem. 2001, 276, 3078–3089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wales, M.M.; Biel, M.A.; el Deiry, W.; Nelkin, B.D.; Issa, J.P.; Cavenee, W.K.; Kuerbitz, S.J.; Baylin, S.B. p53 activates expression of HIC-1, a new candidate tumour suppressor gene on 17p13.3. Nat. Med. 1995, 1, 570–577. [Google Scholar] [CrossRef]
- Kim, J.E.; Chen, J.; Lou, Z. DBC1 is a negative regulator of SIRT1. Nature 2008, 451, 583–586. [Google Scholar] [CrossRef]
- Zhao, W.; Kruse, J.P.; Tang, Y.; Jung, S.Y.; Qin, J.; Gu, W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008, 451, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, D.; Zhao, Y.; Tu, B.; Zheng, Z.; Wang, L.; Wang, H.; Gu, W.; Roeder, R.G.; Zhu, W.G. Methyltransferase Set7/9 regulates p53 activity by interacting with Sirtuin 1 (SIRT1). Proc. Natl. Acad. Sci. USA 2011, 108, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- Atkins, K.M.; Thomas, L.L.; Barroso-González, J.; Thomas, L.; Auclair, S.; Yin, J.; Kang, H.; Chung, J.H.; Dikeakos, J.D.; Thomas, G. The multifunctional sorting protein PACS-2 regulates SIRT1-mediated deacetylation of p53 to modulate p21-dependent cell-cycle arrest. Cell Rep. 2014, 8, 1545–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Fu, Y.; Hu, F.; Lan, J.; Xu, F.; Yang, X.; Luo, X.; Wang, J.; Hu, J. Loss of BRG1 induces CRC cell senescence by regulating p53/p21 pathway. Cell Death Dis. 2017, 8, e2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magni, M.; Buscemi, G.; Maita, L.; Peng, L.; Chan, S.Y.; Montecucco, A.; Delia, D.; Zannini, L. TSPYL2 is a novel regulator of SIRT1 and p300 activity in response to DNA damage. Cell Death Differ. 2019, 26, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Xie, Q.; Wu, J.; Bai, Y.; Mao, B.; Dong, Y.; Bi, W.; Ji, G.; Tao, W.; Wang, Y.; et al. MST1 promotes apoptosis through regulating Sirt1-dependent p53 deacetylation. J. Biol. Chem. 2011, 286, 6940–6945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, D.; Sharma, V.; Ghosh, S.; Mehta, V.S.; Sen, E. Inhibition of Casein kinase-2 induces p53-dependent cell cycle arrest and sensitizes glioblastoma cells to tumor necrosis factor (TNFα)-induced apoptosis through SIRT1 inhibition. Cell Death Dis. 2012, 3, e271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.; Jung, J.W.; Kim, M.K.; Chung, J.H. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS ONE 2009, 4, e6611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.W.; Wong, L.L.; Tse, E.Y.; Liu, H.F.; Leong, V.Y.; Lee, J.M.; Hardie, D.G.; Ng, I.O.; Ching, Y.P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrad, E.; Polonio-Vallon, T.; Meister, M.; Matt, S.; Bitomsky, N.; Herbel, C.; Liebl, M.; Greiner, V.; Kriznik, B.; Schumacher, S.; et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016, 23, 110–122. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Wentzel, E.A.; Kent, O.A.; Ramachandran, K.; Mullendore, M.; Lee, K.H.; Feldmann, G.; Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J.; et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol. Cell 2007, 26, 745–752. [Google Scholar] [CrossRef] [Green Version]
- Adlakha, Y.K.; Saini, N. miR-128 exerts pro-apoptotic effect in a p53 transcription-dependent and -independent manner via PUMA-Bak axis. Cell Death Dis. 2013, 4, e542. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Li, Z.; Wang, S.; Zhou, Z.; Liu, C.; Zhuang, H.; Zhou, Q.; Huang, S.; Zhang, C.; Hou, B. ZMAT1 acts as a tumor suppressor in pancreatic ductal adenocarcinoma by inducing SIRT3/p53 signaling pathway. J. Exp. Clin. Cancer Res. 2022, 41, 130. [Google Scholar] [CrossRef]
- Yang, C.; Lou, G.; Jin, W.L. The arsenal of TP53 mutants therapies: Neoantigens and bispecific antibodies. Signal Transduct. Target. 2021, 6, 219. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, W.; El-Deiry, W.S. Current strategies to target p53 in cancer. Biochem. Pharm. 2010, 80, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic of p53 domains and acetylation sites. The domain structure of p53 and major p53 acetylation sites are shown above. They are: TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA-binding domain; TD, tetramerization domain; CTD, C-terminal domain. The corresponding modifying enzymes are also indicated.
Figure 1.
Schematic of p53 domains and acetylation sites. The domain structure of p53 and major p53 acetylation sites are shown above. They are: TAD, transactivation domain; PRD, proline-rich domain; DBD, DNA-binding domain; TD, tetramerization domain; CTD, C-terminal domain. The corresponding modifying enzymes are also indicated.

Figure 2.
Regulators of p53 acetyltransferase. The acetyltransferase activity towards p53 is affected by various factors, including viral oncoproteins, E3 ubiquitin ligases, phosphatases, lncRNAs, and other proteins. Arrows and perpendicular bars indicate enhancing and suppressive effects towards p53 acetyltransferases, respectively.
Figure 2.
Regulators of p53 acetyltransferase. The acetyltransferase activity towards p53 is affected by various factors, including viral oncoproteins, E3 ubiquitin ligases, phosphatases, lncRNAs, and other proteins. Arrows and perpendicular bars indicate enhancing and suppressive effects towards p53 acetyltransferases, respectively.

Figure 3.
Regulators of p53 deacetylase. The deacetylation activity towards p53 is influenced by multiple factors, including E3 ubiquitin ligases, kinases, miRNAs, and other proteins. Arrows and perpendicular bars indicate enhancing and suppressive effects towards p53 deacetylases, respectively.
Figure 3.
Regulators of p53 deacetylase. The deacetylation activity towards p53 is influenced by multiple factors, including E3 ubiquitin ligases, kinases, miRNAs, and other proteins. Arrows and perpendicular bars indicate enhancing and suppressive effects towards p53 deacetylases, respectively.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nagasaka, M.; Miyajima, C.; Aoki, H.; Aoyama, M.; Morishita, D.; Inoue, Y.; Hayashi, H. Insights into Regulators of p53 Acetylation. Cells 2022, 11, 3825. https://doi.org/10.3390/cells11233825
AMA Style
Nagasaka M, Miyajima C, Aoki H, Aoyama M, Morishita D, Inoue Y, Hayashi H. Insights into Regulators of p53 Acetylation. Cells. 2022; 11(23):3825. https://doi.org/10.3390/cells11233825
Chicago/Turabian StyleNagasaka, Mai, Chiharu Miyajima, Hiromasa Aoki, Mineyoshi Aoyama, Daisuke Morishita, Yasumichi Inoue, and Hidetoshi Hayashi. 2022. "Insights into Regulators of p53 Acetylation" Cells 11, no. 23: 3825. https://doi.org/10.3390/cells11233825
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.