
Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease

Institute of Clinical Medicine, University of Eastern Finland (UEF), 70211 Kuopio, Finland
Cells 2022, 11(6), 978; https://doi.org/10.3390/cells11060978
Submission received: 21 January 2022
/
Revised: 1 March 2022
/
Accepted: 10 March 2022
/
Published: 12 March 2022
(This article belongs to the Special Issue Focus on Cellular Parkinson’s Disease—from Gut to Brain)
{kind=link}
Abstract
:Several bacterial species can generate hydrogen sulfide (H2S). Study evidence favors the view that the microbiome of the colon harbors increased amounts of H2S producing bacteria in Parkinson’s disease. Additionally, H2S can easily penetrate cell membranes and enter the cell interior. In the cells, excessive amounts of H2S can potentially release cytochrome c protein from the mitochondria, increase the iron content of the cytosolic iron pool, and increase the amount of reactive oxygen species. These events can lead to the formation of alpha-synuclein oligomers and fibrils in cells containing the alpha-synuclein protein. In addition, bacterially produced H2S can interfere with the body urate metabolism and affect the blood erythrocytes and lymphocytes. Gut bacteria responsible for increased H2S production, especially the mucus-associated species of the bacterial genera belonging to the Desulfovibrionaceae and Enterobacteriaceae families, are likely play a role in the pathogenesis of Parkinson’s disease. Special attention should be devoted to changes not only in the colonic but also in the duodenal microbiome composition with regard to the pathogenesis of Parkinson’s disease. Influenza infections may increase the risk of Parkinson’s disease by causing the overgrowth of H2S-producing bacteria both in the colon and duodenum.
1. Introduction
In humans, hydrogen sulfide (H2S) plays various roles in a myriad of physiologic processes relating to inflammatory, immune, endocrine, respiratory, vascular and neuromodulatory actions [1]. Furthermore, H2S is endogenously produced in the human cells by enzymes including cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), which both use L-cysteine as a substrate, and via 3-mercapto-sulfurtransferase (3-MST) pathway that uses 3-mercaptopyruvate as a substrate. Both the brain and colonic tissue generate and modulate H2S production via the activity of CSE and CBS [2]. In healthy humans, the plasma baseline levels of H2S have been reported to lie in the range of 34 μM–274 μM [3,4]. In the gut lumen, the sulfate-reducing bacteria (SRB) and the bacteria of the desulfhydrase enzyme are notable H2S producers. In addition, various bacteria which are homologs of the mammalian CBS, CSE, and 3-MST enzymes are capable of producing H2S. At low concentrations, bacterially produced H2S displays cytoprotective properties by maintaining gut mucus integrity but is toxic to the host at high concentrations [5]. When administered to the human body, H2S diffuses rapidly to blood and, after administration, only a small part of H2S remains in a soluble form [6]. In the blood, H2S combines easily with hemoglobin (Hb) and methemoglobin (metHb) [7,8]. MetHb and H2S form relatively stable metHb-H2S complexes which have a slow reduction rate. Thus, metHb keeps Hb in an oxygen-binding form and acts as a scavenger and regulator of sulfide in the blood [8,9]. In the cells, the main route of H2S elimination is mitochondria, by a chain of several oxidative enzymes [10]. Overall, the toxicity-dose relationship in H2S exposition has been documented to be extremely steep and toxic symptoms such as hypotension and apnea occur when the concentration of free H2S reaches only a few moles per litre in the blood [6]. High H2S concentrations are toxic to cells, causing the inhibition of the cytochrome oxidase, a hemeprotein which is the last enzyme of the electron transport chain in the mitochondria [11]. In addition, H2S induces a release of cytochrome c protein from the mitochondrial membrane, an event though to be associated with the etiopathogenesis of Parkinson’s disease (PD) [12,13].
2. H2S Releases Cytochrome c from the Mitochondria—Start for Alpha-Synuclein Aggregation
Cytochrome c is (Cyt c) is a small heme protein responsible for the electron shuttle between Complex III and Complex IV of the respiratory chain in the mitochondria. It binds to the mitochondrial inner membrane partly by weak readily mobilized electrostatic interactions [14]. In an experiment where cultured human lung fibroblasts were treated with increased H2S concentrations, a release of Cyt c from the mitochondria was observed [15]. In a study where human gingival epithelial cells were incubated for up to three days with air containing a low concentration (50 ng/mL) of H2S, a remarkable release of Cyt c from the mitochondria was noted [16]. Accordingly, when human dental pulp stem cells were exposed to air somposed of a similar low concentration of H2S, significant increases in the apoptotic enzyme levels (caspase-9 and -3), accompanied by increases in the Cyt c release from the mitochondria were observed [17]. Special information on the dynamics of Cyt c release from the mitochondria provides information on the effects of rotenone, a poisonous isoflavone which has been used in animal studies to induce experimental PD. In a proportional rotenone titration, a proportional increase in the release of Cyt c from the mitochondria was documented in this study [18]. Of importance, neurons can tolerate Cyt c release into the cytosol without inducing apoptosis. Although Cyt c concentrations were already modestly increased in the cytosol, the activity of the apoptosis indicator caspase 9 was not increased [18]. In its compact state, Cyt c possesses peroxidase activity [19]. In the preapoptotic phase when Cyt c relase has already started, Cyt c can interact with anionic lipids and due to these interactions, Cyt c can develop remarkable peroxidase activity [20,21]. The anionic lipids containing Cyt c-peroxidase can utilize cytoplasmic alpha-synuclein (aSyn) as a peroxidase substrate which leads to an aggregation of aSyn and Cyt c, a reaction reflecting mostly a protective role of aSyn against apoptosis [21]. The native Cyt c, which leads to peroxidase activity, can potentially take part in the aSyn oligomerization and aggregation in the cytosol. It has been shown that aSyn aggregation and aSyn radical formation occurs in the interaction between Cyt c, aSyn and hydrogen peroxide, a member of the reactive oxygen species (ROS) [22,23]. Alterations in the iron metabolism induced by H2S probably take part in the aSyn aggregation. It has been reported that sulfide releases iron from the mammalian ferritin and rises the ferrous iron levels of the cytosolic labile iron pool (LIP) and some evidence favors the view that H2S can reduce intracellular bound ferric iron to form unbound ferrous iron [24,25,26]. Furthermore, it has been demonstrated that ferrous iron promotes both aSyn aggregation and transmission by inhibiting the autophagosome-lysosome fusion [27]. In case the iron content of the LIP reaches abnormally high levels, ROS formation in the cell eventually increases [26,28]. The possible presence of magnetite nanoparticles produced by some Desulfovibrio gut bacteria may be an additional factor in the increase in cytosolic ROS levels in the gut cells [13,29,30]. The increased ROS formation likely favors the emergence of aSyn oligomers and fibrils in the presence of Cyt c and aSyn (Figure 1)
3. Overgrowth of H2S Producing Gut Bacteria—Putative Consequences
Several bacterial species generate H2S in the human gastrointestinal canal. A prominent overgrowth of these species may lead to a spectrum of undesired consequences. Notably, H2S is capable of penetrating cell membranes easily without any help of a facilitator for this transport [31]. To counteract the toxic effects of H2S the colonic mucosal cells are subject to an efficient mechanism to dispose H2S by oxidizing it to thiosulfates [32]. In mitochondria, a chain of enzymatic reactions starting with the actions of sulfide quinone oxidoreductase (SQR) catalyze the oxidation of sulfide to thiosulfate, which is further oxidized to sulfate and excreted by the kidney [10]. In case H2S is produced in abnormally high amounts by the gut bacteria, the concentration of H2S may exceed the capacity of the gut cells to detoxify all H2S and part of it may end up in the blood. Experimentally, it has been shown that free H2S levels are reduced in the plasma and gastrointestinal tissue of germ-free mice, indicating that the gut microbiota activity can increase plasma H2S concentrations [33]. A large study where plasma metabolome data were integrated with the gut microbiome data of PD patients and healthy controls revealed changes in sulfur metabolism driven by Akkermansia muciniphila and Bilophila wadsworthia bacteria [34]. Notably, the secretion potential of H2S was detected to increase in the PD microbiome in that study. Further support for the view that increased H2S production occurs in PD is a study where blood metHb content was reported to be increased in PD patients [35]. In addition, it has been shown that PD patients have significantly higher concentrations of H2S in the cerebrospinal fluid when compared to control subjects [36]. Studies providing information on the correlations between blood concentrations of H2S and quantities of gut bacteria producing H2S are not availabe. If the production of H2S by gut bacteria leads to unphysiologically high blood concentrations, the brain tissue may be especially sensitive to the toxic actions of H2S as the activity of SQR enzyme activity is apparently very low in the mammalian brain cells [37]. As to changes in the blood constituents, CD8+ cytotoxic T-lymphocytes have been especially reported to be decreased in the blood of PD patients and this decrease seems to be associated with the severity of PD [38]. Furthermore, H2S effects may provide an explanation for the finding as it has been found that exogenous H2S induces the cell death of peripheral blood lymphocytes with relevant subset specifity for the CD8+ T lymphocytes and natural killer cells [39]. As an additional observation, increased H2S levels likely modify plasma uric acid (UA) concentrations which reflect the intestinal metabolism of dietary purines such as xanthine [40]. Additionally, H2S has been reported to decrease UA formation from xanthine, giving one explanation for the finding that plasma UA levels are lowered in patients with PD [41,42].
4. H2S and aSyn Containing Gut Cells
Of special concern is the part of the gut epithelial cells which faces the gut lumen without vascular covering. Consequently, those epithelial cells such as the enteroendocrine cells (EECs) which lack the protective role of blood Hb and metHb to scavenge H2S are at an increased risk of H2S toxicity. An excess production of H2S by bacterial species belonging to the genus Desulfovibrio (gDSV) has been suggested to induce Cyt c release from the mitochondria and aSyn oligomerization in the EECs [12,13]. It has been reported that aSyn is expressed in EECs both of the small and large intestine and that EECs are connected to enteric nerves [43,44]. In addition, EECs express both pre- and postsynaptic proteins, implying that EECs both send and receive neural signals [44]. Similarly to EECs, enteric neurons carry a potential risk to produce aSyn oligomers when exposed to high levels of H2S as enteric neurons contain aSyn [45]. Hypothetically, if aSyn oligomers are produced in EECs and enteric neurons, part of them are possibly secreted in the blood. Significantly elevated levels of oligomeric forms of aSyn have been observed in the blood plasma of PD patients [46]. However, the origin of these oligomeric aggregates has remained an open issue. As a potential mechanism, aSyn oligomers and fibrils propagate, in the same way as prions, from the EECs and enteric neurons by a cell-to-cell mechanism to the lower brain stem via the vagal nerve (Figure 1). Experimentally, it has been shown that different forms of aSyn, after being injected into the intestinal wall, are transported to the brainstem via the vagal nerve [47]. In another study on transgenic rats expressing an excess of human aSyn, injections of aSyn fibrils into the duodenum wall induced aSyn pathology both in the parasympathetic and sympathetic pathways [48].
5. H2S Producing Colonic Bacteria and PD
With regard to humans, the excess production of H2S by gut bacteria may induce the emergence of PD. Sulfate-reducing bacteria (SRB) form the main group of H2S-producing bacteria in the feces of healthy people [49]. In this group, the most prevalent genera are the hydrogen- and lactate utilizing gDSV, acetate utilizing genus Desulfobacter, and hydrogen and propionate utilizing genus Desulfobulbus [50,51]. Notably, SRB are the only gut microbes that rely on inorganic sulfate for energy conservation [51]. Numerous other bacteria that do not belong to the SRB group produce H2S as well. Among others, bacterial genera such as Alistipes, Bacteroides, Escherichia, Enterobacter, Clostridium, Collinsella, Fusobacterium, Klebsiella, Oscillibacter, Prevotella, Proteus, Porphyromonas, Streptococcus, Veillonella, and Yearsinia include species that have the capacity for primary H2S production via cysteine degradation [51,52]. Of special interest, Bilophila wadsworthia bacteria produces H2S via the taurine degradation pathway [51]. As to specific bacterial species known to produce H2S, Helicobacter pylori, Clostridium difficile, and Desulfovibrio desufuricans have been reported to be associated with the occurrence of PD [13,53,54].
Numerous studies have been conducted on the microbiome changes in PD with variable results. A large meta-analysis of ten case–control studies on the gut microbiome in PD showed that the fecal samples of PD patients were most consistently enriched by the genera Lactobacillus, Bifidobacterium and Akkermansia [55]. In contrast, the butyrate-producing genera Roseburia, Blautia, Faecalibacterium, Moryella and Anaerostipes were found to be less enriched. In a large microbiome-wide association study (MWAS) the genera Prevotella, Corynebacterium_1, and Porphyromonas, as a cluster of co-occurring opportunistic pathogens, were reported to be enriched in PD [56]. As noted, the genera Prevotella and Porphyromonas are H2S producers. In addition, sulfite reductase, an iron flavoprotein enzyme which produces H2S, has been suggested to be present in the species of the genus Corynebacterium [57]. However, concerning the enrichment of the genus Prevotella in the fecal samples of PD patients in the MWAS study, contrasting results have been reported in some other studies [58,59,60]. In the MWAS study, the relative abundances of the genera Butyricoccus, Lachnospira, Fusikatenibacter, Roseburia, Blautia, Agathobacter and Faecalibacterium, all of which are butyrate producers, were found to be significantly decreased in the PD gut microbiome. An overgrowth of H2S-producing bacteria provides a reasonable explanation for the reduced population of butyrate-reducing bacteria. Furthermore, H2S inhibits acetyl-CoA synthesis and produces CoA-persulfide when reacting with CoA, an essential molecule in the butyrate synthesis pathways [61,62]. In an innovative study on a synthetic microbiome community, composed of several butyrate-producing bacteria, it was observed that H2S, in concentrations similar to those produced by Desulfovibrio piger, inhibited butyrate production across the designed bacterial community, including the members Faecalibacterium prausnitzii and and Roseburia intestinalis [63]. Decreased butyrate production most probably reflects the amount of butyrate-producing gut bacteria. Advocating this view, it has been reported that the amount of butyrate correlate with the abundancies of bacteria of the genus Blautia and bacterial species such as Faecalibacterium prausnitzii and Roseburia faecis [64]. An enrichment of the genera Lactobacillus and Bifidobacterium in the fecal samples of PD patients has been reported in several studies [55,65]. The finding can be explained, at least partly, by the availability of these probiotics as commercial products.
Concerning SRB such as gDSV, their role in the gut microbiota of PD patients requires particular attention. In an Italian study, many bacterial genera, including gDSV, were observed to be enriched in the fecal samples of PD patients. However, after adjusting for several covariates, only the genus Veillonella remained as the enriched genus [66]. The finding is of interest as Veillonella species produce H2S from L-cysteine and an enrichment of this genus has been found to be enriched in the fecal microbiome of the PD patients in an earlier study [52,58,67]. In two Chinese studies, the relative abundance of the family Desulfovibrionaceae was observed to be significantly increased in the fecal samples of PD patients [58,68]. In addition, in a study from southern China, the relative abundance of gDSV was increased in PD patients [68]. Further confirmation for the overgrowth of the family Desulfovibrionaceae in the gut microbiota of PD patients is provided in a Canadian study where the Desulfovibrionaceae and Christensenellaceae families showed overgrowth in the fecal samples of PD patients [69]. The family Christensenellaceae has been reported to be significantly increased in the PD microbiota [55]. One explanation for its increased abundance is linked to body mass index (BMI). Patients with PD have significantly lower BMIs when compared to healthy controls and on the other hand, an inverse correlation exists between BMI and the relative abundance of the Christensenellaceae family [70,71].
In contrast to a focus on the relative abundances of a multitude of gut bacteria representing various taxonomic levels, one study provided data on the absolute concentrations of the gDSV species Desulfovibrio desulfuricans, D. fairfieldensis, D. piger and D. vulgaris in the fecal samples of PD patients and healthy controls [13]. Excluding D. vulgaris, not found in the PD samples, the gDSV species were present at significantly higher concentrations in the PD samples than in the control samples. In addition, the presence of the periplasmic [FeFe]-hydrogenase, which was used as a common denominator of different gDSV species, correlated with the presence of PD [13]. Apparent cross-feeding takes place between gDSV and genus Akkermansia. In studies on fecal samples, the relative overgrowth of the genus Akkermansia is a common finding in PD patients [55,56,65]. Akkermansia muciniphila, a bacterium residing abundantly in the mucus layer of the large intestine releases sulfate in mucin fermentation, thus offering sulfate to the mucosa-associated SRB [72,73]. A study on sequence analyses of functional gene clones of colonic biopsy samples revealed that SRB populations associate with the mucosa throughout the colon and are phylogenetically related to hydrogenotrophic Desulfovibrio piger, Desulfovibrio desulfuricans and Bilophila wadsworthia bacteria [74]. In colonic mucus samples of healthy people, Bilophila wadsworthia was reported to have a high colonisation rate [75]. Of apparent importance, the relative abundance of the genus Bilophila has been reported to be significantly increased in the fecal samples of PD patients when compared to controls, and to correlate with the clinical stage of PD by the Hoehn and Yahr clinical criteria [76]. As an apparent weakness, the gut microbiome studies on PD have focused mainly on the relative abundancies of various bacterial taxa occurring in the fecal samples. The composition of the mucosal-associated microbiome evidently differs significantly from that of the fecal microbiome, as demonstrated in an analysis comprising both sigmoidoscopy and fecal samples [77]. Genus Escherichia resides especially in the mucosal area [77,78]. Notably, a significantly more intense staining for Eschericia coli has been reported in the sigmoid mucosal samples of PD patients than in the samples of the control subjects [79]. In agreement with this study, the relative abundance of the genus Escherichia-Shigella has been reported to be significantly increased in the fecal samples of PD patient and to correlate with the severity and duration of PD [80].
6. H2S Producing Small Intestinal Bacteria and PD
Apart from the changes in the colonic microbiota composition, changes in the quantities of small intestinal bacteria likely relate to the pathogenesis of PD as it suggests the high prevalence of small intestinal bacterial overgrowth (SIBO) in the PD population. According to a large meta-analysis, based on lactulose-hydrogen breath test results, a strong association was reported to exist between SIBO and PD [81]. Although increased hydrogen production from the small intestinal microbiome seems to take place in several PD subjects, it is not yet known which bacterial genera or species in the small intestine explain this increase. Hydrogen-utilizing bacteria such as Desulfovibrio species and Bilophila wadsworthia likely benefit from these circumstances. At a general level, the small intestinal microbiome composition has been found to differ markedly from the fecal microbiome composition in humans [82]. A study performed on SIBO and non-SIBO subjects provides interesting information about the duodenal microbiome changes in the SIBO subjects at a general level [82]. In that study, over four-fold higher relative abundance of the class Gammaproteobacteria and three-fold higher relative abundance of the class Deltaproteobacteria was reported to be present in the duodenal aspirates of SIBO subjects when compared to non-SIBO subjects. Of further interest, the family Enterobacteriaceae represented 89 % of the total relative abundance of the Gammaproteobacteria in the duodenum of the SIBO subjects [83]. Notably, hydrogen sulfide-producing genera such as Escherichia, Klebsiella, and Proteus belong to the family Enterobacteriaceae. As to the family members of the class Deltaproteobacteria, especially concerning the family Desulfovibrionaceae, they are best known for their ability to produce H2S. Swallowed bacteria from the oral cavity, emerging, e.g., from the areas of periodontal infections, may possibly play a role in modulating the small intestinal microbiome composition. In a large-scale cohort study, periodontitis was shown to increase the risk of PD [84]. Subgingival deposits occurring with periodontal disease, including cases with aggressive periodontitis, have been reported to contain SRB (which mainly represent the class Deltaproteobacteria) and, among others, bacterial genera such as Porphyromonas and Prevotella [85,86]. Of interest, in a Finnish study on oral mucosal biofilm, significantly increased abundances of the Prevotella and Veillonella genera were observed in PD patients [87].
7. Viral Infections, PD, and H2S Producing Gut Bacteria
Influenza infection evidently increases the risk of PD. In a population-based case–control study in Canada, a significant association was reported to exist between a history of severe influenza and PD [88]. Recently, a case–control study based on the data of over 60 000 individuals from the Danish National Patient Registry showed that a history of influenza was significantly associated with a later occurrence of PD [89]. In addition, the Spanish flu (influenza A subtype H1N1) has been considered to be a risk factor for a later PD occurrence. As to animal studies, it has been shown that influenza A virus (H5N1) can induce a disruption of the mucus layer integrity of the small intestine and cause enteric dysbiosis by increasing the relative amounts of Gammaproteobacteria and Bacilli [90]. In a mice study, the influenza A (PR8) virus infection was demonstrated to induce a significant increase in the intestinal Escherichia coli species [91]. As to another study, influenza A (PR8) infection caused a significant increase in the Enterobacteriaceae population in the stool samples of the wild-type mice [92]. In addition, influenza A virus H3N2 infection has been reported to increase the fecal content of the genus Escherichia in mice [93]. As to human studies, an interesting cross-sectional study on alterations in the gut microbiota of 30 patients with COVID-19 infection, 24 patients with influenza (H1N1), and 30 matched healthy controls showed that the relative abundance of the genus Escherichia-Shigella was significantly higher in the fecal samples of H1N1 patients than in controls [94].
Hepatitis C virus (HCV) infection has been reported to be a significant risk factor for Parkinson’s disease [95]. HCV infection can be classified into three types, which consist of the persistently normal serum alanine transferase type, chronic hepatitis, and liver cirrhosis. In all these stages, an enrichment of the family Enterobacteriaceae present in the gut microbiome has been a consistent finding [96]. Evidently, influenza and hepatitis C virus infections may be inductors for the development of PD by inducing gut dysbacteriosis, whereby an overgrowth of the H2S-producing members of the family Enterobacteriaceae, especially the genus Escherichia, play a crucial role. Clinical studies on fecal microbiome content in PD provide support for the view that the Enterobacteriaceae family is an important player in the pathogenesis of PD. Notably, this family has been reported to be significantly more abundant in the fecal samples of PD patients when compared to healthy controls [80,97,98]. Postural instability and gait difficulty have been reported to correlate with the relative abundance of the Enterobacteriaceae in the fecal samples of PD patients [97]. In addition, an increase in Enterobacteriaceae in the fecal samples has been reported to be associated with the non-tremor dominant subtype of PD [99].
8. Bacterially Produced H2S and Risk Factors for PD
Advancing age has been established as the largest risk factor for developing PD. Globally, the prevalence of PD begins rise steeply in the age range of 60–70 years and peaks in the age range of 80–90 years [100]. As to the colonic microbiome in advancing age, a community study on changes in the microbiome composition during the aging process showed that in older people, a sharp and continuous increase takes place in the relative abundance of the H2S-producing Desulfovibrio, Bilophila and Corynebacterium gut bacteria [101]. In case additional and substantial overgrowth of the H2S producing bacterial genera such as Escherichia or gDSV takes place, the risk for PD development will likely increase as a function of age.
In addition to aging, male gender is an established risk factor for PD. Furthemore, with regard to the incidence rates, male to female ratios have been reported to vary from 1.37 to 3.7 [102]. Estrogen, especially 17beta-estradiol which has been reported to display neuroprotective properties may provide an explanation for this difference [103,104]. It has been stated that long-term exposure to estrogen may be important in the PD risk reduction [104]. Experimental studies on ischemia models have shown that 17beta-estradiol prevents the release of Cyt c from mitochondria and by this action, estradiol displays cytoprotective properties [105,106]. Ultimately, given that H2S levels increase in the gut cells and perhaps even at the brain level, estadiol can probably counteract H2S actions by preventing the release of Cyt c from the mitochondrial membrane.
9. Conclusions
Considerable amounts of evidence support the view that an overgrowth of H2S-producing bacteria takes place in the gut microbiome of PD patients. It is plausible that by inhibiting acetyl-CoA synthesis, H2S decreases the amount of butyrate-producing gut bacteria. Furthemore, H2S can diffuse easily to gut cells and the vascular compartment. In blood circulation, some part of the bacterially produced H2S may end up at the brain level. When the gut cells are exposed to unphysiologically high concentrations of H2S, the release of Cyt c from the mitochondria will likely begin. In addition, H2S increases the iron content in the labile iron pool of the cell cytosol and the ROS content. In case the cell expresses aSyn, in the way that EECs and enteric neurons do, a development of aSyn oligomers and fibrils may start in the presence of Cyt c and increased ROS. The toxic oligomers and fibrils may spread to the lower brainstem via the vagal nerve and some oligomers will likely end up in the blood circulation. As to H2S producers, species of the genera Desulfovibrio, Escherichia, Bilophila, Porhyromonas, Prevotella, Corynebacterium, Veillonella, Helicobacter, and Clostridium invite particular attention when identifying the role of different bacterial genera in the etiology of PD. The bacterial composition of the microbiome in duodenum require special attention in PD as it is likely that the quantities and characteristics of the small intestinal bacteria, including the H2S-producing bacteria, differ significantly from those of the colon. Notably, studies on the bacterial composition of duodenal microbiome in PD are lacking. In future, microbiological and metabolomics-based studies on duodenal aspirates combined with the corresponding analyses of fecal samples may offer key information on the pathogenesis of PD. In case H2S plays a substantial role in the pathogenesis of PD, several strategies are already available to counteract its actions. In 2003, Braak and his colleagues proposed that PD is caused by an intestinal pathogen [107]. H2S could be that ”pathogen”.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Ulla Sederlöf, Senior Lecturer, Information Technology, Metropolia UAS, is acknowledged as the designer of Figure 1.
Conflicts of Interest
The author (K.M.) declares no conflict of interest.
References
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linden, D.R.; Sha, L.; Mazzone, A.; Stoltz, G.J.; Bernard, C.E.; Furne, J.K.; Levitt, M.D.; Farrugia, G.; Szurszewski, J.H. Production of the gaseous signal molecule hydrogen sulfide in mouse tissue. J. Neurochem. 2008, 106, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R479–R1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karunya, R.; Jayaprakash, K.S.; Gaikwad, R.; Sajeesh, P.; Ramshad, K.; Muraleedharan, K.M.; Dixit, M.; Thangaraj, P.R.; Sen, A.K. Rapid measurement of hydrogen sulfide in human blood plasma using a microfluid method. Sci. Rep. 2019, 9, 3258. [Google Scholar] [CrossRef] [PubMed]
- Buret, A.G.; Allain, T.; Motta, J.-P.; Wallace, J.L. Effects of hydrogen sulfide on the microbiome: From toxicity to therapy. Antioxid. Redox Sign. 2022, 36, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Haouzi, P.; Sonobe, T.; Judenherc-Haouzi, A. Hydrogen sulfide intoxication induced brain injury and methylene blue. Neurobiol. Dis. 2020, 133, 104474. [Google Scholar] [CrossRef] [PubMed]
- Klingerman, C.M.; Trushin, N.; Prokopczyk, B.; Haouzi, P. H2S concentration in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R630–R638. [Google Scholar] [CrossRef]
- Bianco, C.L.; Savitsky, A.; Feelisch, M.; Cortese-Krott, M.M. Investigations on the role of hemoglobin in sulfide metabolism by intact human red blood cells. Biochem. Pharmacol. 2018, 149, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Fago, A. Reactions of ferric hemoglobin and myoglobin with hydrogen sulfide under physiologic conditions. J. Inorg. Biochem. 2018, 182, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Zaorska, E.; Tomasova, L.; Loszelewski, D.; Ostaszewski, R.; Ufnal, M. Hydrogen sulfide in pharmacotherapy, beyond the hydrogen sulfide-donors. Biomolecules 2020, 10, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorman, D.C.; Moulin, F.J.-M.; McManus, B.E.; Mahle, K.C.; James, A.; Struve, M.F. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: Correlation with tissue sulfide concentrations in the rat brin, liver, lung, and nasal epithelium. Toxicol. Sci. 2002, 65, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Murros, K.E. Sulfate reducing gut bacteria and Parkinson’s disease. Eur. J. Neurol. 2021, 28, e21. [Google Scholar] [CrossRef] [PubMed]
- Murros, K.E.; Huynh, V.A.; Takala, T.M.; Saris, P.E.J. Desulfovibrio bacteria are associated with Parkinson’s disease. Front. Cell. Infect. Microbiol. 2021, 11, 652617. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanism of cytochrome c release from mitochondria. Cell Death Diff. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baskar, R.; Li, L.; Moore, P.K. Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. Faseb J. 2007, 21, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Calenic, B.; Yaegaki, K.; Murata, T.; Imai, T.; Aoyama, I.; Sato, T.; Ii, H. Oral malodorous compound triggers mitochondrial-dependent apoptosis and causes genomic DNA damage in human gingival epithelial cells. J. Periodontal Res. 2010, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, C.; Yaegaki, K.; Calenic, B.; Ishkitiev, N.; Imai, T.; Ii, H.; Aoyama, I.; Kobayashi, H.; Izumi, Y.; Haapasalo, M. Hydrogen sulfide causes apoptosis in human pulp stem cells. J. Endod. 2011, 37, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Clayton, R.; Clark, J.B.; Sharpe, M. Cytochrome c release from rat brain mitocondria is proportional to the mitochondrial funtional deficit: Implications for apoptosis and neurodegenerative disease. J. Neurochem. 2005, 92, 840–849. [Google Scholar] [CrossRef]
- Tomasková, N.; Varhac, T.; Lysáková, V.; Musatov, A.; Sedlák, E. Peroxidase activity of cytochrome c in its compact state dependes on dynamics of the heme region. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Belikova, N.A.; Vladimirov, Y.A.; Osipov, A.N.; Kapralov, A.A.; Tyurin, V.A.; Potapovich, M.V.; Basova, L.V.; Peterson, J.; Kurnikov, I.V.; Kagan, V.E. Peroxidase activity and structural transitions of cytochrome c bound to cardiolipin-contiainig membranes. Biochemistry 2006, 45, 4998–5009. [Google Scholar] [CrossRef] [Green Version]
- Bayir, H.; Kapralov, A.A.; Jiang, J.; Huang, Z.; Tyurina, Y.Y.; Tyurin, V.A.; Zhao, Q.; Belikova, N.A.; Vlasova, I.I.; Maeda, A.; et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome c. J. Biol. Chem. 2008, 284, 15951–15969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, M.; Takeda, A.; Hsu, L.J.; Takenouchi, T.; Masliah, E. Role of cytochrome c as astimulator of α-synuclein aggregation in Lewy-body disease. J. Biol. Chem. 1999, 274, 28849–28852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Ganini, D.; Mason, R.P. Role of cytochrome c in α-synuclein radical formation: Implications of α-synuclein in neuronal death in Maneb- and paraquat-induced model of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassanelli, S.; Moulis, J. Sulfide is an efficient iron releasing agent for mammalian ferritins. Biochim. Biophys. Acta 2001, 1547, 174–182. [Google Scholar] [CrossRef]
- Hälldin, J.; Land, T. Sulfide increases labile iron pool in RD4 cells. Biometals 2008, 21, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Truong, D.H.; Eghbal, M.A.; Hindmarsh, W.; Roth, S.R.; O’Brien, P.J. Molecular mechanisms of hydrogen sulfide toxicity. Drug Metab. Rev. 2006, 38, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, X.; Huang, S.; Li, G.; Mo, M.; Zhang, L.; Chen, C.; Guo, W.; Zhou, M.; Wu, Z.; et al. Iron promotes α-synuclein aggregation and transmission by inhibiting TFEB-mediated autophagosome-lysosome fusion. J. Neurochem. 2018, 145, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I. Labile iron in cells and body fluids: Physiology, pathology, and pharmacology. Front. Pharmacol. 2014, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murros, K.; Wasiljeff, J.; Macias-Sánchez, E.; Faivre, D.; Soinne, L.; Valtonen, J.; Pohja, M.; Saari, P.; Pesonen, L.J.; Salminen, J.M. Magnetic nanoparticles in human cervical skin. Front. Med. 2019, 6, 123. [Google Scholar] [CrossRef] [PubMed]
- Könczöl, M.; Ebeling, S.; Goldenberg, E.; Treude, F.; Gminski, R.; Gieré, R.; Grobéty, B.; Rothen-Rutishauser, B.; Merfort, I.; Merch-Sundermann, V. Cytotoxicity and genotoxicity of size-fractionde iron oxide (magnetite) in A549 human lung epithelial cells: Role of ROS, JNK, and NF-κB. Chem. Res. Toxicol. 2011, 24, 1460–1475. [Google Scholar] [CrossRef] [PubMed]
- Mathai, J.C.; Missner, A.; Kügler, P.; Saparov, S.M.; Zeidel, M.L.; Lee, J.K.; Pohl, P. No facilitator required fro membrane transport of hydrogen sulfide. PNAS 2009, 106, 16633–16638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.B.; Lin, H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganism 2015, 3, 866–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Carlström, M.; Borniquel, S.; Jädert, C.; Kevill, C.G.; Lundberg, J. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic. Biol. Med. 2013, 60, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertel, J.; Harms, A.C.; Heinken, A.; Baldini, F.; Thinnes, C.C.; Glaab, E.; Vasco, D.A.; Pietzner, M.; Stewart, I.D.; Wareham, N.J.; et al. Integrated analyses on microbiome and longitudinal metabolome data reveal microbial-host interactions on sulfur metabolism in Parkinson’s disease. Cell Rep. 2019, 29, 1767–1777.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makletsova, M.G.; Rikhireva, G.T.; Poleshuk, V.V.; Grjakalov, K.V.; Timerbaeva, S.L.; Fedorova, T.N. The effects of antioxidants on in vivo and in vitro methemoglobin formation in erythorcytes of patients with Parkinson’s disease. Biomed. Khim. 2016, 62, 193–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, V.; Neri, C.; Pieragostino, D.; Spalloni, A.; Persichilli, S.; Gastaldi, M.; Mercuri, N.B.; Longone, P.; Urbani, A. Investigating different forms of hydrogen sulfide in cerebrospinal fulid of various neurological disorders. Metabolites 2021, 11, 152. [Google Scholar] [CrossRef] [PubMed]
- Lagouette, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, D.; Grozdanov, V.; Ruf, W.P.; Kassubek, J.; Ludolph, A.C.; Weishaupt, J.H.; Danzer, K.M. T-cell dysregulation is associated with disease severity in Parkinson’s disease. J. Neuroinflamm. 2021, 18, 250. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, P.; Gobbi, G.; Sponzilli, I.; Pambianco, M.; Malinverno, C.; Cacchioli, A.; De Panfilis, G.; Vitale, M. Exogenous hydrogen sulfide induces functional inhibition and cell desth of cytotoxic lymphocytes subsets. J. Cellul. Physiol. 2007, 213, 826–833. [Google Scholar] [CrossRef]
- Hyndman, D.; Liu, S.; Miner, J.N. Urate handling in the human body. Curr. Rheumatol. Rep. 2016, 18, 34. [Google Scholar] [CrossRef] [Green Version]
- Annanmäki, T.; Muuronen, A.; Murros, K. Low plasma uric acid in Parkinson’s disease. Mov. Disord. 2007, 22, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Pardue, S.; Kolluru, G.K.K.; Shen, X.; Lewis, S.; Saffle, C.; Kelley, E.; Kevil, C. Hydrogen sulfide stimulates xanthine oxidoreductase conversion to nitrite reductase and formation of NO. Redox Biol. 2020, 34, 101447. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Hiniker, A.; Kuo, Y.-M.; Nussbaum, R.L.; Liddle, R.A. α-Synuclein in gut endocrine cells and its implications for Parkinsons’s disease. JCI Insight 2017, 2, e92295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddle, R.A. Interactions of gut endocrine cells with epithelium and neurons. Compr. Physiol. 2019, 8, 1019–1030. [Google Scholar] [CrossRef]
- Bu, L.-L.; Huang, K.-X.; Zheng, D.-Z.; Lin, D.-Y.; Chen, Y.; Jing, X.-N.; Liang, Y.-R.; Tao, E.-X. Alpha-synuclein accumulation and its phosphorylation in the enteric nervous system of patients without neurodegeneration: An explorative study. Front. Aging Neurosci. 2020, 12, 575481. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.L.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synucelin protein in human plasma as a potential biomarker for Parkinson’s disease. Faseb J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Nyengaard, J.R.; Tamgüney, G.; Jensen, P.H.; et al. Evidence of bidirectional and trans-synaptic parasymphathetic and symphathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Dordević, D.; Janciková, S.; Vitezová, M.; Kushkevych, I. Hydrogen sulfide toxicity in the gut environment: Meta-analysis of sulfate-reducing and lactic acid bacteria in inflammatory processes. J. Adv. Res. 2021, 27, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Macfarlane, S.; Macfarlane, G.T. Metabolic interactions involving sulphate-reducing and methanogenic bacteria in the human large intestine. FEMS Microbiol. 1993, 12, 117–125. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braccia, J.; Jiang, X.; Pop, M.; Hall, A.B. The capacity to produce hydrogen sulfide (H2S) via cysteine degradation is ubiquitos in the human gut microbiome. Front. Microbiol. 2021, 12, 3193. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-K.; Wang, J.-H.; Lei, W.-Y.; Chen, C.-L.; Chang, C.-Y.; Liou, L.-S. Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Park. Rel. Dis. 2018, 47, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Ploner, A.; Ludvigsson, J.F.; Williams, D.M.; Larsson, H.; Pedersen, N.L.; Wirdefeldt, P.K. Clostridium difficile infection and risk of Parkinson’s disaease: A Swedish population-based cohort study. Eur. J. Neurol. 2020, 27, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiota suggest alterations linked to intestinal inflammation. Npj. Park. Dis. 2021, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Wallen, Z.D.; Appah, M.; Dean, M.N.; Sesler, C.L.; Factor, S.A.; Molho, E.; Zabetian, C.P.; Standaert, D.G.; Payami, H. Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. Npj. Park. Dis. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Blachier, F.; Davila, A.-M.; Mimoun, S.; Benetti, P.-H.; Atanasiu, C.; Andriamihaja, M.; Benamouzig, R.; Boillaud, F.; Tomé, D. Luminal sulfide and large intestine mucosa: Friend or foe? Amino Acids 2009, 39, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, C.-C.; Chiang, H.-L.; Liou, J.-M.; Chang, C.-M.; Lu, T.-P.; Chuang, E.Y.; Tai, Y.-C.; Cheng, C.; Lin, H.-Y.; et al. Altered microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J. Neuroinflamm. 2019, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Petrov, V.A.; Saltykova, I.V.; Zhukova, I.A.; Alifirova, V.M.; Zhukova, N.G.; Dorofeeva, Y.B.; Tyakht, A.V.; Kovarsky, B.A.; Alekseev, D.G.; Kostryukova, E.S.; et al. Analysis of gut microbiota in patients with Parkinson’s disease. Bull. Exp. Biol. Med. 2017, 162, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Liu, F.; Wang, K.; Wang, L.; Liang, S.; Tao, H.; Zhu, B.; Alkasir, R. Analysis of the gut microflora in patients with Parkinson’s disease. Front. Neurosci. 2019, 13, 1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babidge, W.; Millard, S.; Roediger, W. Sulfides impair short chain fatty acid β-oxidation at acyl-CoA dehydrogenase level in colonocytes: Implications for ulcerative colitis. Mol. Cell. Biochem. 1998, 181, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Moon, S.; Kim, H.; Yadav, P.K.; Guha, A.; Cho, U.-S.; Banerjee, R. A catalytic trisulfide in human sulfide quinone oxidoreductase catalyzes Coenzyme A persulfide synthesis and inhibits butyrate oxidation. Cell. Chem. Biol. 2019, 26, 1515–1525.e4. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.L.; Connors, B.M.; Stevenson, D.M.; Hromada, S.E.; Hamilton, J.J.; Amador-Noguez, D.; Venturelli, O.S. Design of synthetic human gut microbiome assembly and butyrate production. Nature Comm. 2021, 12, 3254. [Google Scholar] [CrossRef] [PubMed]
- de la Cuesta-Zuluaga, J.; Mueller, N.T.; Alvarez-Quintero, R.; Velásguez-Mejia, E.; Sierra, J.A.; Corrales-Agudelo, V.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Higher fecal short-chain fatty acid levels are associated with gut microbiome dysbiosis, obesity, hypertension and cardiometbolic disease risk factors. Nutritients 2019, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.-J.; Liang, C.-Y.; Yang, L.-Q.; Ren, S.-M.; Xia, Y.-M.; Cui, L.; Li, X.-F.; Gao, B.-L. Association of Parkinson’s disease with microbes and microbiological therapy. Front. Cell. Infect. Microbiol. 2021, 11, 619354. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Palmas, V.; Melis, M.; Pisanu, S.; Cusano, R.; Uva, P.; Perra, D.; Madau, V.; Sarchioto, M.; Oppo, V.; et al. Gut microbiota and metabolome alterations associated with Parkinson’s disease. Msystems 2020, 5, e00561-20. [Google Scholar] [CrossRef] [PubMed]
- Washio, J.; Sakuma, Y.; Shimada, Y.; Takahashi, N. Hydrogen-sulfide production from various substrates by oral Veillonella and effects of lactate on the production. J. Med. Microbiol. 2009, 54, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Zheng, W.; He, Y.; Tang, W.; Wei, X.; He, R.; Huang, W.; Su, Y.; Huang, Y.; Zhou, H.; et al. Gut microbiota in patients with Parkinson’s disease in southern China. Park. Rel. Dis. 2018, 53, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, M.S.; Yu, A.C.; Golz, E.; Sundvick, K.; Kliger, D.; Radisavljevic, N.; Foulger, L.M.; Mackenzie, M.; Huan, T.; Finlay, B.; et al. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov. Disord. 2020, 35, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- van der Marck, M.; Dicke, H.C.; Uc, E.Y.; Kentin, Z.H.A.; Borm, G.F.; Bloem, B.R.; Overeem, S.; Munneke, M. Body mass index in Parkinson’s disease: A meta-analysis. Park. Rel. Dis. 2012, 18, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.L.; Ley, R. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. MMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Geerlings, S.Y.; Kostopoulos, I.; de Vos, W.M.; Belzer, C. Akkermansia muciniphila in he human gastrointrestinal tract: When, where, and how? Microorganisms 2018, 6, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; de Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nava, G.M.; Carbonero, F.; Croix, J.A.; Greenberg, E.; Gaskins, H.R. Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 2012, 6, 57–80. [Google Scholar] [CrossRef] [PubMed]
- Earley, H.; Lennon, G.; Balfe, A.; Coffey, J.C.; Winter, D.C.; O’Connell, P.R. The abundance of Akkermansia muciniphila and its relationship in health and ulceratice colitis. Sci. Rep. 2019, 9, 15683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldini, F.; Hertel, J.; Sandt, E.; Thinnes, C.C.; Neuberger-Castillo, L.; Pavelka, L.; Betsou, F.; Krüger, R.; Thiele, I.; on behalf of the NCER-PD Consortium. Parkinson’s disease-associated alterations of the gut microbiome predict disease-relevant changes in metabolic functions. BMC Biol. 2020, 18, 62. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sicard, J.F.; Le Bihan, G.; Vogeleer, P.; Jacques, M.; Harel, J. Interactions of intestinal bacteria with components of the intestinal mucus. Front. Cell Infect. Microbiol. 2017, 7, 387. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Shannon, K.M.; Kordower, J.H.; Voigt, R.M.; Shaikh, M.; Jaglin, J.A.; Estes, J.D.; Dodiya, H.B.; Keshavarzian, A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinosn’s disease. PLoS ONE 2011, 6, e28032. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural changes of gut microbiota in Parkinson’s disease and its correlations with clinical features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef]
- Li, X.; Feng, X.; Jiang, Z.; Jiang, Z. Association of small intestinal bacterial overgrowth with Parkinson’s disease: A systematic review and meta-analysis. Gut Pathog. 2021, 13, 25. [Google Scholar] [CrossRef]
- Leite, G.G.S.; Weitsman, S.; Parodi, G.; Celly, S.; Sedighi, R.; Sanchez, M.; Morales, W.; Villanueva-Millan, M.J.; Barlow, G.M.; Mathur, R.; et al. Mapping the segmental microbiomes in the human small bowel in comparison with stool: A REIMAGINE study. Digest. Dis. Sci. 2020, 65, 2595–2604. [Google Scholar] [CrossRef] [Green Version]
- Leite, G.; Morales, W.; Weitsman, S.; Celly, S.; Parodi, G.; Mathur, R.; Barlow, G.M.; Sedighi, R.; Millan, M.J.V.; Rezaie, A.; et al. The duodenal microbiome is altered in small intestinal bacterial overgrowth. PloS ONE 2020, 15, e02344906. [Google Scholar] [CrossRef]
- Chen, C.-K.; Wu, Y.-T.; Chang, Y.-C. Periodontal inflammatory disease is associated with the risk of Parkinson’s disease: A population-based retrospective matched-cohort study. PeerJ 2017, 10, e3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The subgingival micrbiome of periodontal pockets with different probing depths in chronic and aggressive periodontitis: A pilot study. Front. Cell. Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Kushkevych, I.; Coufalová, M.; Vitezová, M.; Ritmann, S.K.-M.R. Sulfate-reducing bacteria of the oral cavity and their relation with periodontitis-recent advances. J. Clin. Med. 2020, 9, 2347. [Google Scholar] [CrossRef]
- Pereira, P.A.B.; Aho, V.T.E.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. Oral and nasal microbiota in Parkinson’s disease. Park. Rel. Dis. 2017, 38, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.A.; Tsui, J.K.; Marion, S.A.; Shen, H.; Teschke, K. Asoociation of Parkinson’s disease with infections and occupational exposure to possible vectors. Mov. Disord 2012, 27, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Cocoros, N.M.; Svensson, E.; Szépligeti, S.K.; Vestergaard, S.V.; Szentkúti, P.; Thomsen, R.W.; Borghammer, P.; Sorensen, H.T.; Henderson, V.W. Long-term risk of Parkinson’s disease following influenza and other infections. JAMA Neurol. 2021, 78, 1461–1470. [Google Scholar] [CrossRef]
- Yildiz, S.; Mazel-Sanchez, B.; Kandasamy, M.; Manicassamy, B.; Schmolke, M. Influenza A virus infection impacts systemic microbiota dynamics and causes quantitative enteric dysbiosis. Microbiome 2018, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, F.; Wei, H.; Lian, Z.-X.; Sun, R.; Tian, Z. Respiratory influenza virus infection induces intestinal immune injury via microbiota-mediated Th17 cell-dependent inflammation. J. Exp. Med. 2014, 211, 2397–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deriu, E.; Boxx, G.M.; He, X.; Pan, C.; Benavidez, S.D.; Cen, L.; Rozengurt, N.; Shi, W.; Cheng, G. Influenza virus affects interstinal microbiota and secondary Salmonella infection in the gut through type 1 interferons. PloS Pathog. 2016, 15, e1005572. [Google Scholar] [CrossRef]
- Sencio, V.; Barthelemy, A.; Tavares, L.P.; Machado, M.G.; Soulard, D.; Cuinat, C.; Queiroz-Junior, C.M.; Noordine, M.-L.; Salomé-Desnoulez, S.; Deryuter, L.; et al. Gut dysbiosis during influenza contributes to pulmonary pneumococcal superinfection through altered short-chain fatty acid production. Cell Rep. 2020, 30, 2934–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.; Chen, Y.; Wu, Z.; Chen, Y.; Gao, H.; Lv, L.; Guo, F.; Zhang, X.; Luo, R.; Huang, C.; et al. Alterations of the gut microbiota in patients with Coronavirus disease 2019 or H1N1 influenza. Clin. Infect. Dis. 2020, 71, 2669–2678. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.Y.-Y.; Kang, K.-H.; Chen, S.L.-S.; Chiu, S.Y.-H.; Yen, A.M.-F.; Fann, J.C.-T.; Su, C.-W.; Liu, H.-C.; Lee, C.-Z.; Fu, W.-M.; et al. Hepatitis C virus infection: A risk for Parkinson’s disease. J. Viral Hepat. 2015, 22, 784–791. [Google Scholar] [CrossRef] [PubMed]
- El-Mowafy, M.; Elgaml, A.; El-Mesery, M.; Sultan, S.; Ahmed, T.A.E.; Gomaa, A.I.; Aly, M.; Mottawea, W. Changes of gut-microbiota-liver axis in hepatitis C virus infections. Biology 2021, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinicla phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Unger, M.M.; Spiegel, J.; Dillmann, K.-U.; Grundmann, D.; Philippelt, H.; Bürmann, J.; Fassbender, K.; Scwiertz, A.; Schäfer, K.-H. Short chain fatty acids and gut microbiota differ between patients witn Parkinson’n disease and age-matched controls. Park. Rel. Dis. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Vascellari, S.; Melis, M.; Palmas, V.; Serra, A.; Perra, D.; Santoru, M.L.; Oppo, V.; Uva, P.; Atzori, L.; Morelli, M.; et al. Clinical phenotypes of Parkinson’s disease associate with distinct gut microbiota and metabolome enterotypes. Biomolecules 2021, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- GBD 2016 Parkinson’s Disease Collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhu, H.; Qiu, P. Aging progression of human microbiota. BMC Microbiol. 2019, 19, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillies, G.; Pienaar, I.S.; Vohra, S.; Qamhawi, Z. Sex differences in Parkinson’s disease. Front. Neuroendocrin. 2014, 35, 370–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourque, M.; Dluzen, D.E.; Di Paolo, T. Neuroprotective actions of sex steroids in Parkinson’s disease. Front Neuroendocrinol. 2009, 30, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Gatto, N.M.; Deapen, D.; Stoyanoff, S.; Pinder, R.; Bordelon, T.; Ritz, B. Lifetime exposure to estrogens and Parkinson’s disease in California teachers. Park. Rel. Dis. 2014, 20, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Bagetta, C.; Chiappetta, O.; Amantea, D.; Iannone, M.; Rotiroti, D.; Costa, A.; Nappi, G.; Corasaniti, M.T. Estradiol reduces cytochrome c translocation and minimizes hippocampal damage caused by transient global ischemia in rat. Neurosci. Lett. 2004, 368, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Morkuniene, R.; Arandarcikaite, O.; Borutaite, V. Estradiol prevents release of cytochrome c form mitochondria and inhibits ischemia-induced apoptosis in perfused heart. Exp. Gerontol. 2006, 41, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural. Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
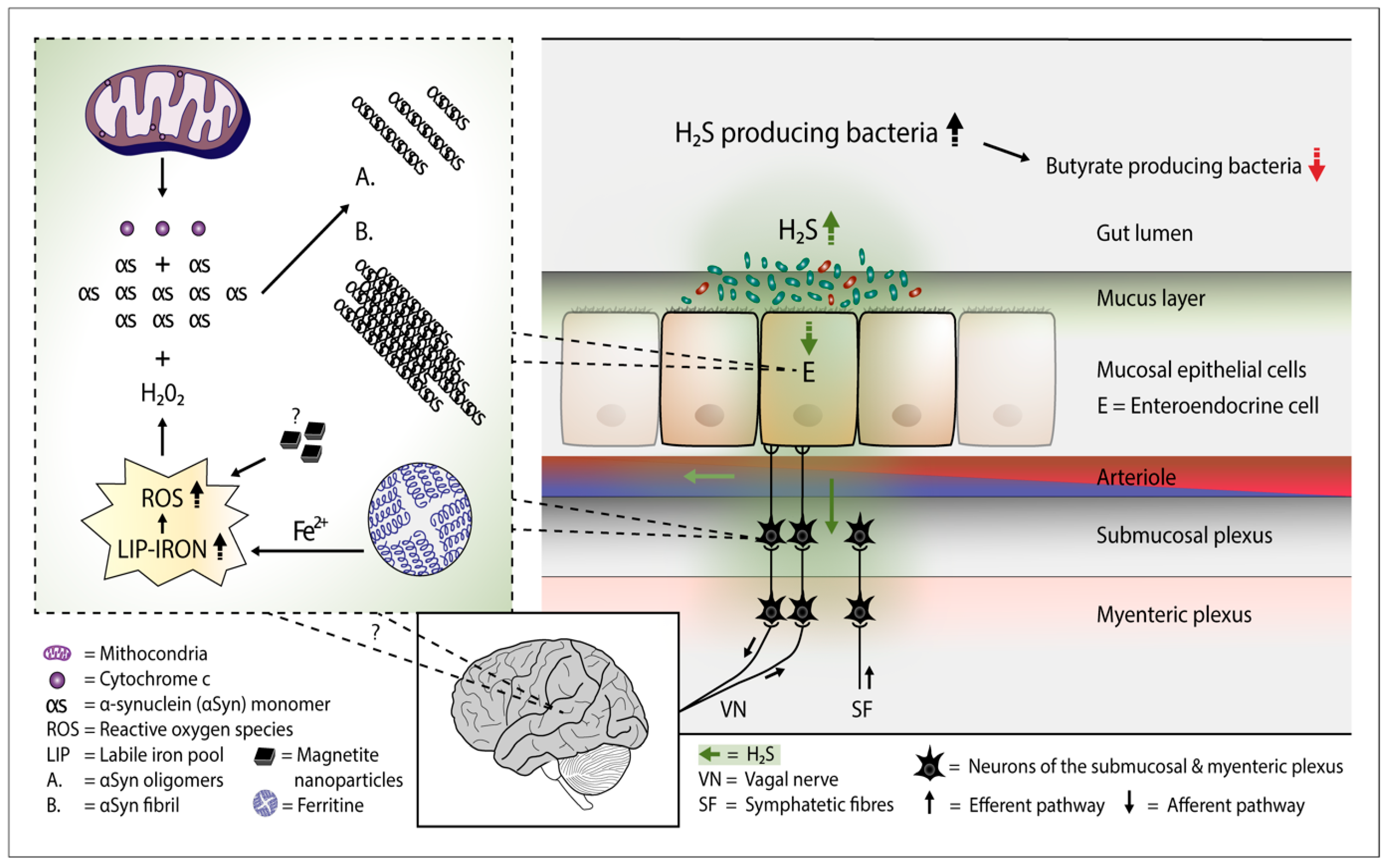
Figure 1.
Plausible pathophysiological mechanism of Parkinson’s disease. Overgrowth of H2S producing gut bacteria raises H2S concentrations in the gut cells and blood. In the gut cells, excessively increased H2S releases Cyt c from the mitochondria and increases cytosolic iron (Fe2+) levels. Consequently, the amount of reactive oxygen species (ROS) increases. The presence of magnetite nanoparticles originating from the Desulfovibrio species can further increase the emergence of ROS. The co-occurence of aSyn, Cyt c, and ROS (especially hydrogen peroxide) leads to aSyn aggregation. Emerged aSyn aggregates (oligomers and fibrils) may spread in a prion-like manner to the lower brain stem via the vagal nerve. In the blood H2S combines with hemoglobin. Part of H2S may remain in a free form and possibly induce aSyn aggregation even in the brain neurons.
Figure 1.
Plausible pathophysiological mechanism of Parkinson’s disease. Overgrowth of H2S producing gut bacteria raises H2S concentrations in the gut cells and blood. In the gut cells, excessively increased H2S releases Cyt c from the mitochondria and increases cytosolic iron (Fe2+) levels. Consequently, the amount of reactive oxygen species (ROS) increases. The presence of magnetite nanoparticles originating from the Desulfovibrio species can further increase the emergence of ROS. The co-occurence of aSyn, Cyt c, and ROS (especially hydrogen peroxide) leads to aSyn aggregation. Emerged aSyn aggregates (oligomers and fibrils) may spread in a prion-like manner to the lower brain stem via the vagal nerve. In the blood H2S combines with hemoglobin. Part of H2S may remain in a free form and possibly induce aSyn aggregation even in the brain neurons.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Murros, K.E. Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells 2022, 11, 978. https://doi.org/10.3390/cells11060978
AMA Style
Murros KE. Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease. Cells. 2022; 11(6):978. https://doi.org/10.3390/cells11060978
Chicago/Turabian StyleMurros, Kari Erik. 2022. "Hydrogen Sulfide Produced by Gut Bacteria May Induce Parkinson’s Disease" Cells 11, no. 6: 978. https://doi.org/10.3390/cells11060978
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.