
Nuclear Localization Sequence of FGF1 Is Not Required for Its Intracellular Anti-Apoptotic Activity in Differentiated Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Cell Lines
2.3. Expression and Purification of Recombinant Proteins
2.4. Transfections and Silencing
2.5. Subcellular Fractionation
2.6. Viability and Apoptosis Measurments
2.7. Thermal Stability Measurments
2.8. FGF1-Induced Signaling and FGF1 Degradation in Cell-Conditioned Medium
2.9. Statistical Analyses
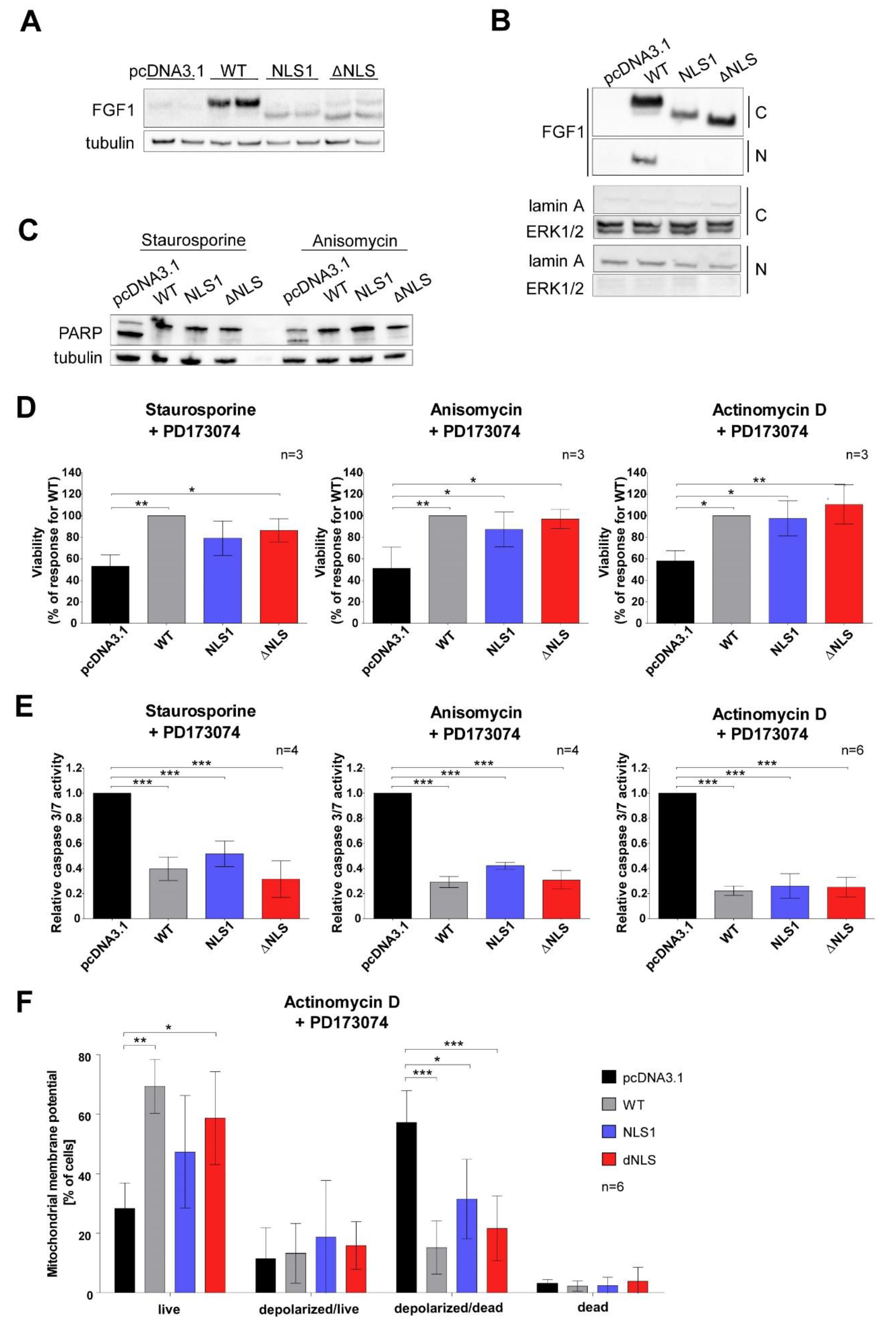
3. Results
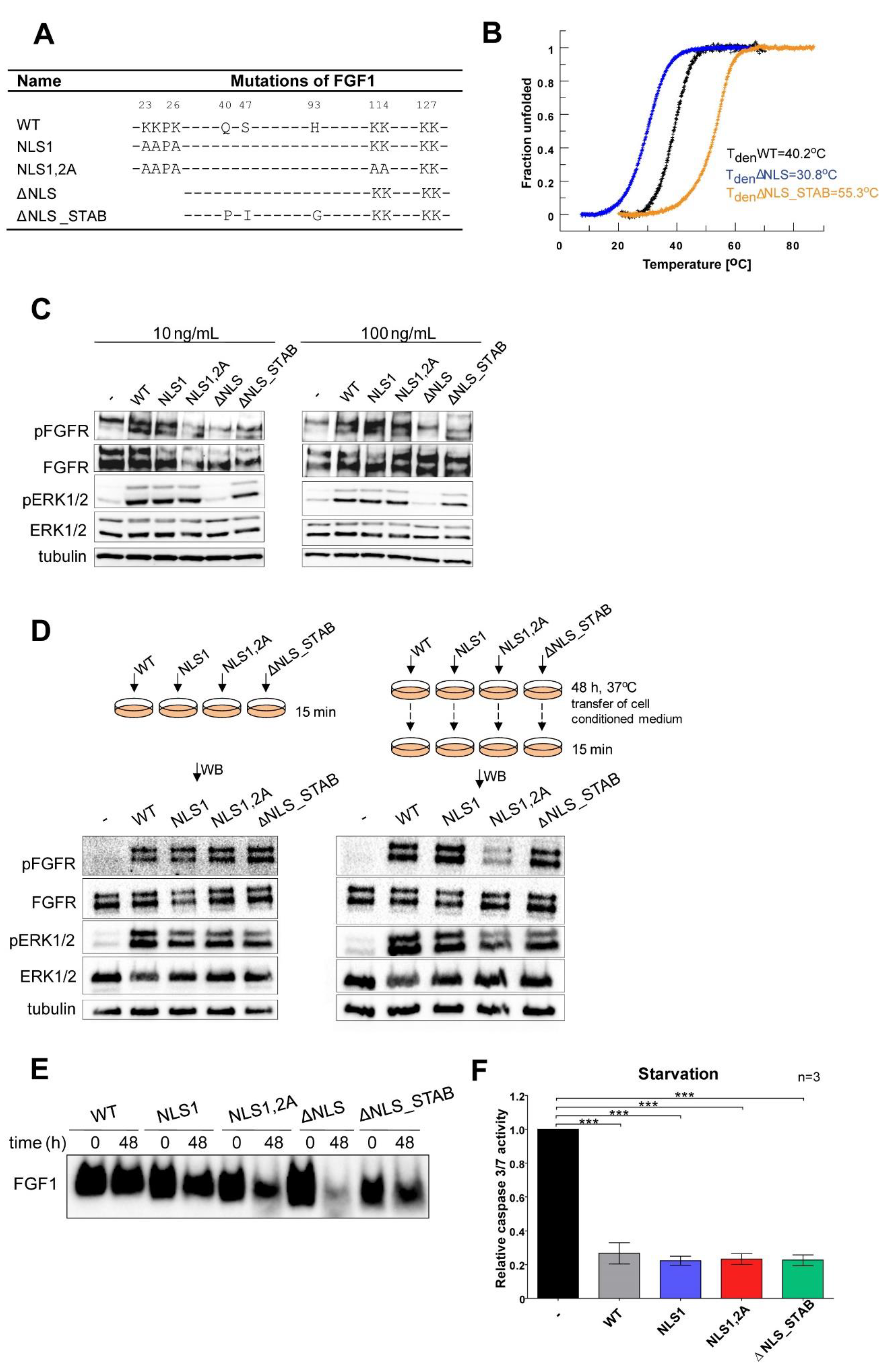
3.1. Characterisation of Recombinant NLS Variants of the FGF1 Protein
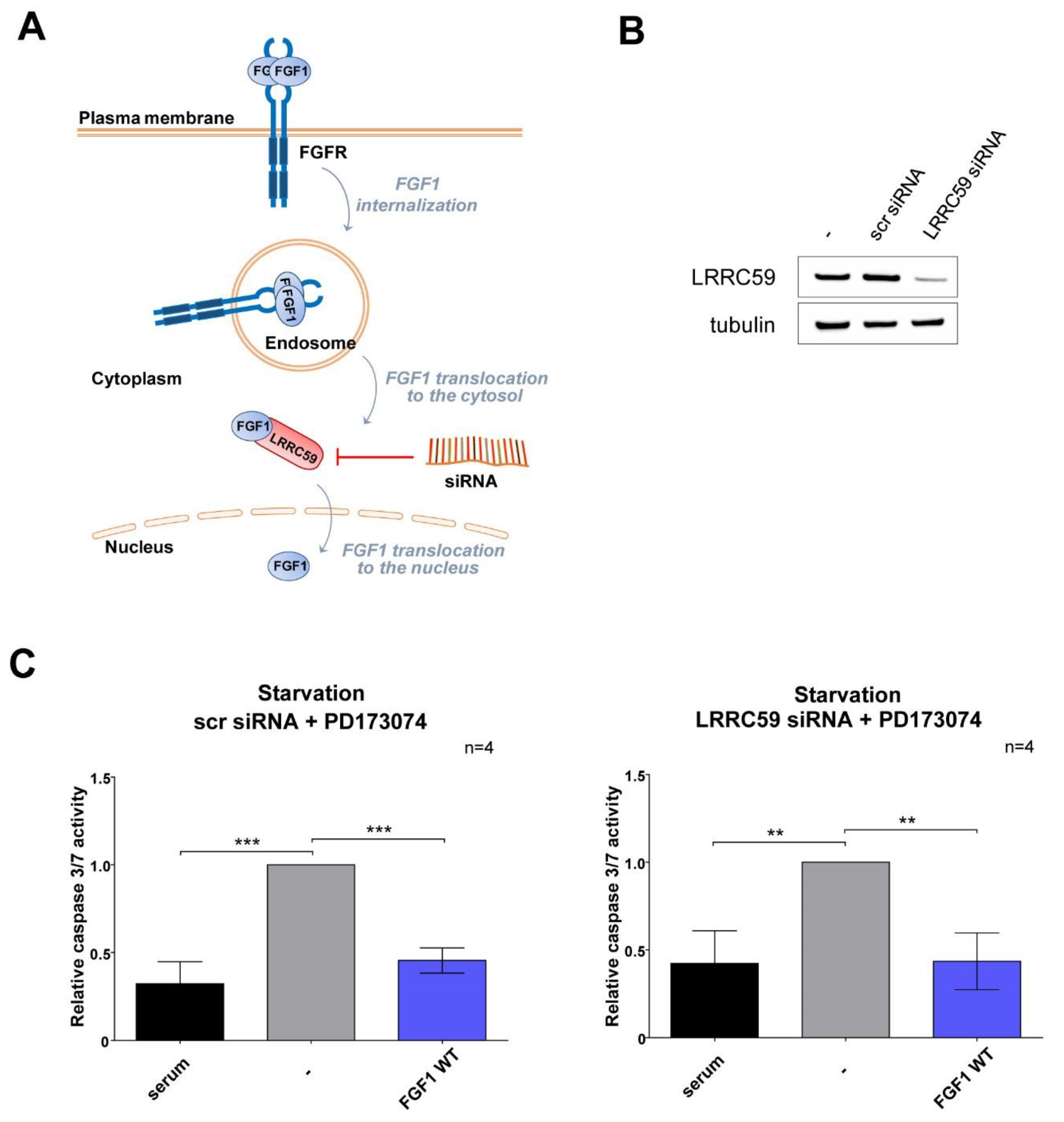
3.2. Exogenously Added NLS Variants of FGF1 Deficient in Nuclear Translocation Retain Intracellular Anti-Apoptotic Properties
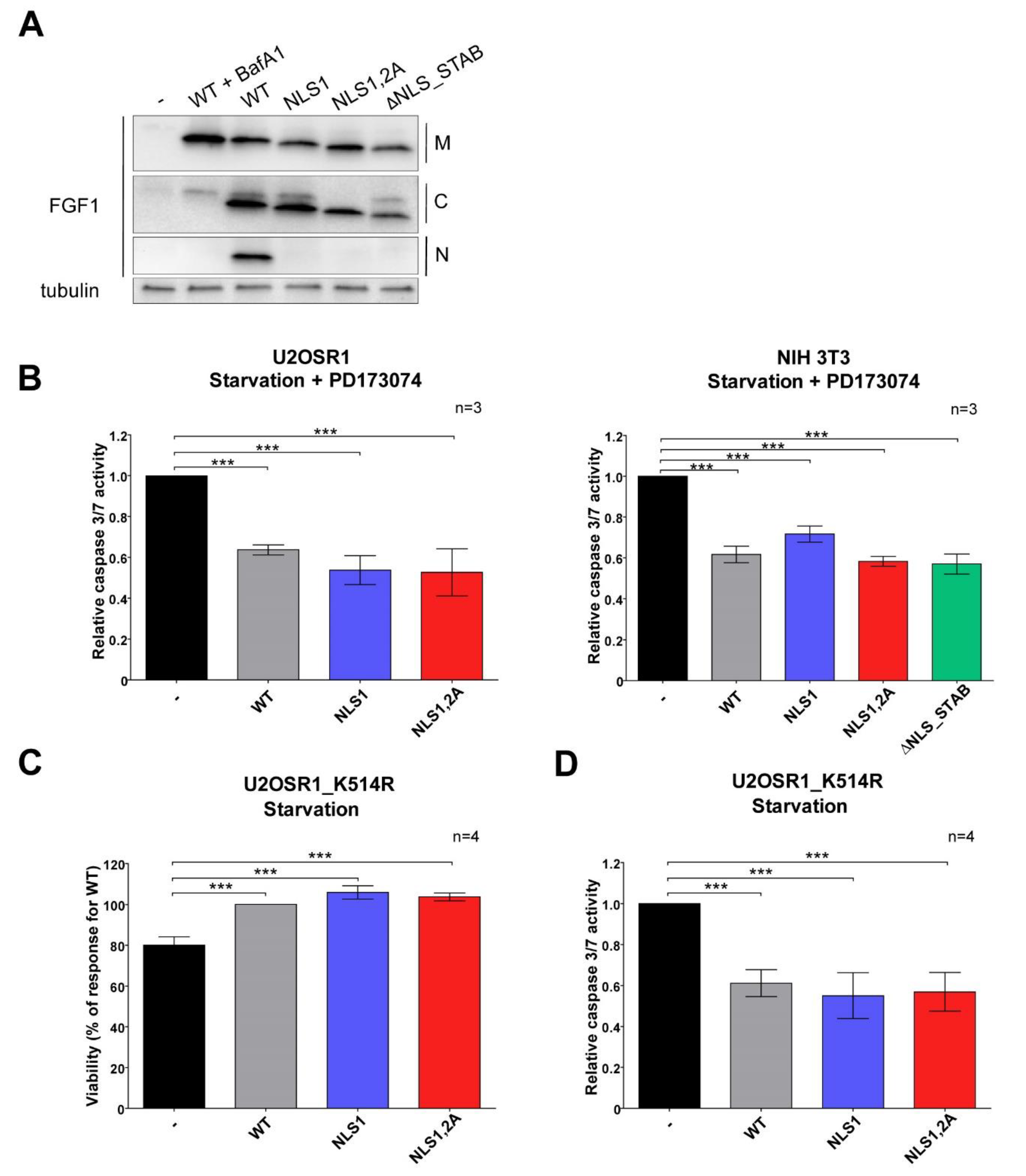
3.3. Wild-Type FGF1 Retains Its Anti-Apoptotic Potential despite Being Prevented from Nuclear Translocation
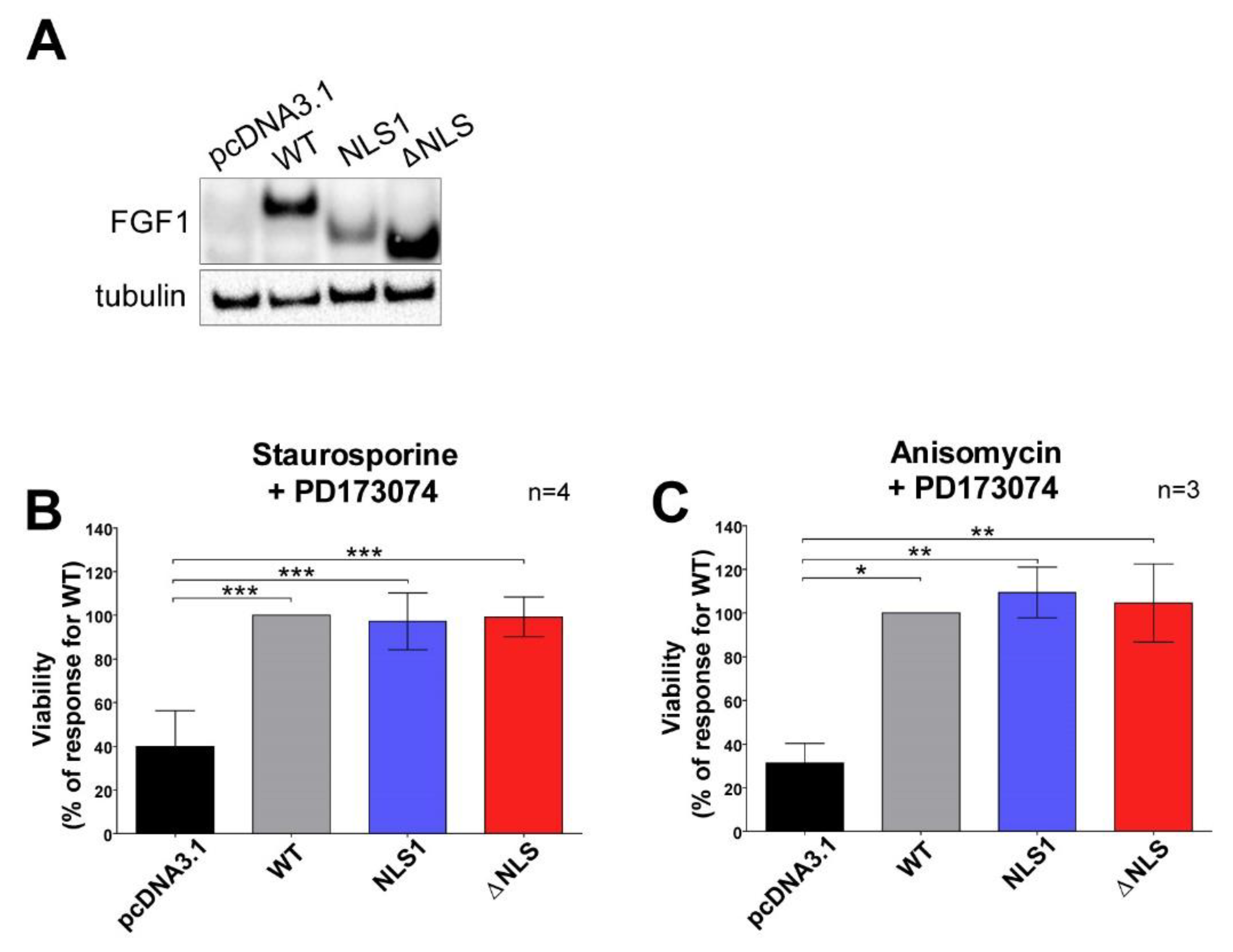
3.4. Mutations in NLS Sequence of Transiently Expressed FGF1 Do Not Affect Its Pro-Survival Response in Differentiated Cells
3.5. Stably Expressed NLS Mutants Exhibit Anti-Apoptotic Activity in U2OS Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BafA1 | Bafilomycin A1 |
| FGF1 | fibroblast growth factor 1 |
| FGFR | fibroblast growth factor receptor |
| NLS | nuclear localization sequence |
| NES | nuclear export sequence |
| PARP | Poly (ADP-ribose) polymerase |
| WT | wild type |
References
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Imamura, T.; Oka, S.; Tanahashi, T.; Okita, Y. Cell cycle-dependent nuclear localization of exogenously added fibroblast growth factor-1 in BALB/c 3T3 and human vascular endothelial cells. Exp. Cell Res. 1994, 215, 363–372. [Google Scholar] [CrossRef]
- Wiedlocha, A.; Falnes, P.O.; Madshus, I.H.; Sandvig, K.; Olsnes, S. Dual mode of signal transduction by externally added acidic fibroblast growth factor. Cell 1994, 76, 1039–1051. [Google Scholar] [CrossRef]
- Olsnes, S.; Klingenberg, O.; Wiedlocha, A. Transport of exogenous growth factors and cytokines to the cytosol and to the nucleus. Physiol. Rev. 2003, 83, 163–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imamura, T.; Engleka, K.; Zhan, X.; Tokita, Y.; Forough, R.; Roeder, D.; Jackson, A.; Maier, J.A.; Hla, T.; Maciag, T. Recovery of mitogenic activity of a growth factor mutant with a nuclear translocation sequence. Science 1990, 249, 1567–1570. [Google Scholar] [CrossRef]
- Imamura, T.; Tokita, Y.; Mitsui, Y. Identification of a heparin-binding growth factor-1 nuclear translocation sequence by deletion mutation analysis. J. Biol. Chem. 1992, 267, 5676–5679. [Google Scholar] [CrossRef]
- Dang, C.V.; Lee, W.M. Nuclear and nucleolar targeting sequences of c-erb-A, c-myb, N-myc, p53, HSP70, and HIV tat proteins. J. Biol. Chem. 1989, 264, 18019–18023. [Google Scholar] [CrossRef]
- Silver, P.; Goodson, H. Nuclear protein transport. Crit. Rev. Biochem. Mol. Biol. 1989, 24, 419–435. [Google Scholar] [CrossRef]
- Luo, Y.; Gabriel, J.L.; Wang, F.; Zhan, X.; Maciag, T.; Kan, M.; McKeehan, W.L. Molecular modeling and deletion mutagenesis implicate the nuclear translocation sequence in structural integrity of fibroblast growth factor-1. J. Biol. Chem. 1996, 271, 26876–26883. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.; Zhan, X.; Maciag, T. Mutagenesis of the nuclear localization sequence in EGF-1 alters protein stability but not mitogenic activity. Biochem. Biophys. Res. Commun. 1994, 198, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Malecki, J.; Wiedlocha, A.; Ehsani, M.; Marcinkowska, E.; Nilsen, T.; Olsnes, S. Two nuclear localization signals required for transport from the cytosol to the nucleus of externally added FGF-1 translocated into cells. Biochemistry 2005, 44, 6071–6080. [Google Scholar] [CrossRef]
- Malecki, J.; Wesche, J.; Skjerpen, C.S.; Wiedlocha, A.; Olsnes, S. Translocation of FGF-1 and FGF-2 across vesicular membranes occurs during G1-phase by a common mechanism. Mol. Biol. Cell 2004, 15, 801–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, V.; Wiedlocha, A.; Haugsten, E.M.; Khnykin, D.; Wesche, J.; Olsnes, S. Different abilities of the four FGFRs to mediate FGF-1 translocation are linked to differences in the receptor C-terminal tail. J. Cell Sci. 2006, 119, 4332–4341. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, V.; Zhen, Y.; Zakrzewska, M.; Haugsten, E.M.; Walchli, S.; Nilsen, T.; Olsnes, S.; Wiedlocha, A. Phosphorylation of fibroblast growth factor (FGF) receptor 1 at Ser777 by p38 mitogen-activated protein kinase regulates translocation of exogenous FGF1 to the cytosol and nucleus. Mol. Cell. Biol. 2008, 28, 4129–4141. [Google Scholar] [CrossRef] [Green Version]
- Klingenberg, O.; Wiedocha, A.; Citores, L.; Olsnes, S. Requirement of phosphatidylinositol 3-kinase activity for translocation of exogenous aFGF to the cytosol and nucleus. J. Biol. Chem. 2000, 275, 11972–11980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewska, M.; Sorensen, V.; Jin, Y.; Wiedlocha, A.; Olsnes, S. Translocation of exogenous FGF1 into cytosol and nucleus is a periodic event independent of receptor kinase activity. Exp. Cell Res. 2011, 317, 1005–1015. [Google Scholar] [CrossRef]
- Malecki, J.; Wiedlocha, A.; Wesche, J.; Olsnes, S. Vesicle transmembrane potential is required for translocation to the cytosol of externally added FGF-1. EMBO J. 2002, 21, 4480–4490. [Google Scholar] [CrossRef] [Green Version]
- Belkowski, S.M.; Levine, J.E.; Prystowsky, M.B. Requirement of PI3-kinase activity for the nuclear transport of prolactin in cloned murine T lymphocytes. J. Neuroimmunol. 1999, 94, 40–47. [Google Scholar] [CrossRef]
- Wesche, J.; Malecki, J.; Wiedlocha, A.; Skjerpen, C.S.; Claus, P.; Olsnes, S. FGF-1 and FGF-2 require the cytosolic chaperone Hsp90 for translocation into the cytosol and the cell nucleus. J. Biol. Chem. 2006, 281, 11405–11412. [Google Scholar] [CrossRef] [Green Version]
- Skjerpen, C.S.; Wesche, J.; Olsnes, S. Identification of ribosome-binding protein p34 as an intracellular protein that binds acidic fibroblast growth factor. J. Biol. Chem. 2002, 277, 23864–23871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Yang, L.; Shi, L.; Li, Q.; Zhang, G.; Wu, J.; Zheng, J.; Jiao, B. Nuclear translocation of fibroblast growth factor-2 (FGF2) is regulated by Karyopherin-beta2 and Ran GTPase in human glioblastoma cells. Oncotarget 2015, 6, 21468–21478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Miyamoto, Y.; Tsujii, A.; Moriyama, T.; Ikuno, Y.; Shiromizu, T.; Serada, S.; Fujimoto, M.; Tomonaga, T.; Naka, T.; et al. Cell surface localization of importin alpha1/KPNA2 affects cancer cell proliferation by regulating FGF1 signalling. Sci. Rep. 2016, 6, 21410. [Google Scholar] [CrossRef]
- Nilsen, T.; Rosendal, K.R.; Sorensen, V.; Wesche, J.; Olsnes, S.; Wiedlocha, A. A nuclear export sequence located on a beta-strand in fibroblast growth factor-1. J. Biol. Chem. 2007, 282, 26245–26256. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Sorensen, V.; Skjerpen, C.S.; Haugsten, E.M.; Jin, Y.; Walchli, S.; Olsnes, S.; Wiedlocha, A. Nuclear import of exogenous FGF1 requires the ER-protein LRRC59 and the importins Kpnalpha1 and Kpnbeta1. Traffic 2012, 13, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Wiedlocha, A.; Nilsen, T.; Wesche, J.; Sorensen, V.; Malecki, J.; Marcinkowska, E.; Olsnes, S. Phosphorylation-regulated nucleocytoplasmic trafficking of internalized fibroblast growth factor-1. Mol. Biol. Cell 2005, 16, 794–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostas, M.; Lampart, A.; Bober, J.; Wiedlocha, A.; Tomala, J.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Translocation of Exogenous FGF1 and FGF2 Protects the Cell against Apoptosis Independently of Receptor Activation. J. Mol. Biol. 2018, 430, 4087–4101. [Google Scholar] [CrossRef] [PubMed]
- Renaud, F.; Desset, S.; Oliver, L.; Gimenez-Gallego, G.; Van Obberghen, E.; Courtois, Y.; Laurent, M. The neurotrophic activity of fibroblast growth factor 1 (FGF1) depends on endogenous FGF1 expression and is independent of the mitogen-activated protein kinase cascade pathway. J. Biol. Chem. 1996, 271, 2801–2811. [Google Scholar] [CrossRef] [Green Version]
- Bouleau, S.; Grimal, H.; Rincheval, V.; Godefroy, N.; Mignotte, B.; Vayssiere, J.L.; Renaud, F. FGF1 inhibits p53-dependent apoptosis and cell cycle arrest via an intracrine pathway. Oncogene 2005, 24, 7839–7849. [Google Scholar] [CrossRef]
- Bouleau, S.; Parvu-Ferecatu, I.; Rodriguez-Enfedaque, A.; Rincheval, V.; Grimal, H.; Mignotte, B.; Vayssiere, J.L.; Renaud, F. Fibroblast Growth Factor 1 inhibits p53-dependent apoptosis in PC12 cells. Apoptosis Int. J. Program. Cell Death 2007, 12, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Delmas, E.; Jah, N.; Pirou, C.; Bouleau, S.; Le Floch, N.; Vayssiere, J.L.; Mignotte, B.; Renaud, F. FGF1 C-terminal domain and phosphorylation regulate intracrine FGF1 signaling for its neurotrophic and anti-apoptotic activities. Cell Death Dis. 2016, 7, e2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Enfedaque, A.; Bouleau, S.; Laurent, M.; Courtois, Y.; Mignotte, B.; Vayssiere, J.L.; Renaud, F. FGF1 nuclear translocation is required for both its neurotrophic activity and its p53-dependent apoptosis protection. Biochim. Biophys. Acta 2009, 1793, 1719–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewska, M.; Wiedlocha, A.; Szlachcic, A.; Krowarsch, D.; Otlewski, J.; Olsnes, S. Increased protein stability of FGF1 can compensate for its reduced affinity for heparin. J. Biol. Chem. 2009, 284, 25388–25403. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewska, M.; Krowarsch, D.; Wiedlocha, A.; Olsnes, S.; Otlewski, J. Highly stable mutants of human fibroblast growth factor-1 exhibit prolonged biological action. J. Mol. Biol. 2005, 352, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, A.; Sochacka, M.; Czyrek, A.; Opalinski, L.; Krowarsch, D.; Otlewski, J.; Zakrzewska, M. Low Stability of Integrin-Binding Deficient Mutant of FGF1 Restricts Its Biological Activity. Cells 2019, 8, 899. [Google Scholar] [CrossRef] [Green Version]
- Suh, J.M.; Jonker, J.W.; Ahmadian, M.; Goetz, R.; Lackey, D.; Osborn, O.; Huang, Z.; Liu, W.; Yoshihara, E.; van Dijk, T.H.; et al. Endocrinization of FGF1 produces a neomorphic and potent insulin sensitizer. Nature 2014, 513, 436–439. [Google Scholar] [CrossRef]
- Low, B.C.; Lim, Y.P.; Lim, J.; Wong, E.S.; Guy, G.R. Tyrosine phosphorylation of the Bcl-2-associated protein BNIP-2 by fibroblast growth factor receptor-1 prevents its binding to Cdc42GAP and Cdc42. J. Biol. Chem. 1999, 274, 33123–33130. [Google Scholar] [CrossRef] [Green Version]
- Roumiantsev, S.; Krause, D.S.; Neumann, C.A.; Dimitri, C.A.; Asiedu, F.; Cross, N.C.; Van Etten, R.A. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell 2004, 5, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lampart, A.; Sluzalska, K.D.; Czyrek, A.; Szerszen, A.; Otlewski, J.; Wiedlocha, A.; Zakrzewska, M. Nuclear Localization Sequence of FGF1 Is Not Required for Its Intracellular Anti-Apoptotic Activity in Differentiated Cells. Cells 2022, 11, 522. https://doi.org/10.3390/cells11030522
Lampart A, Sluzalska KD, Czyrek A, Szerszen A, Otlewski J, Wiedlocha A, Zakrzewska M. Nuclear Localization Sequence of FGF1 Is Not Required for Its Intracellular Anti-Apoptotic Activity in Differentiated Cells. Cells. 2022; 11(3):522. https://doi.org/10.3390/cells11030522
Chicago/Turabian StyleLampart, Agata, Katarzyna Dominika Sluzalska, Aleksandra Czyrek, Aleksandra Szerszen, Jacek Otlewski, Antoni Wiedlocha, and Malgorzata Zakrzewska. 2022. "Nuclear Localization Sequence of FGF1 Is Not Required for Its Intracellular Anti-Apoptotic Activity in Differentiated Cells" Cells 11, no. 3: 522. https://doi.org/10.3390/cells11030522