
Pediatric Atypical Hemolytic Uremic Syndrome Advances

 , and
, and
Abstract
:1. Introduction
2. The Complement System
3. Genetics
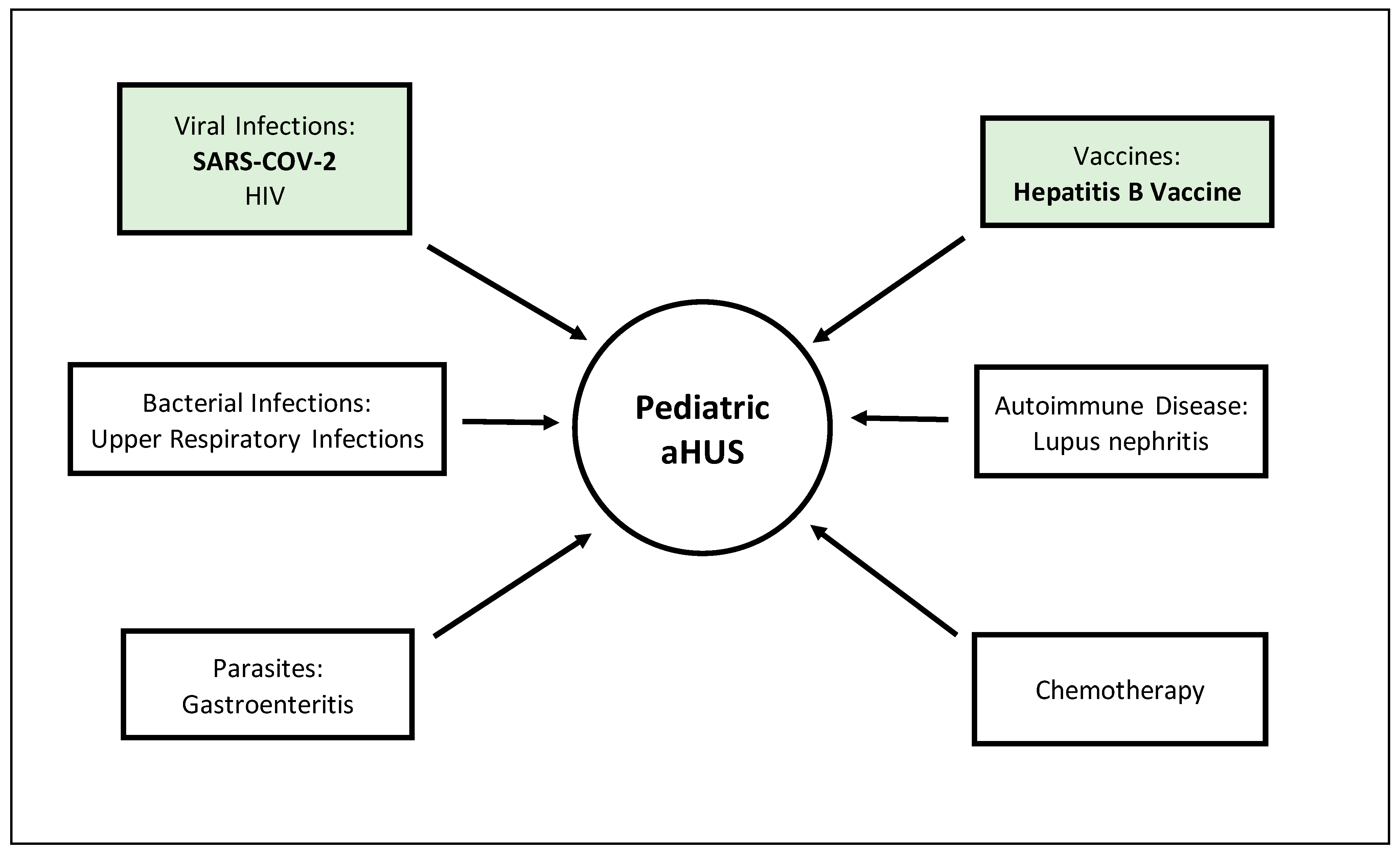
4. Triggers
4.1. COVID-19
4.2. Hepatitis B Vaccine
5. Manifestations
5.1. Renal Manifestations
5.2. Neurological Manifestations
5.3. Ocular Manifestations
5.4. Cardiovascular Manifestations
5.5. Pulmonary Manifestations
5.6. Gastrointestinal and Dermatologic Manifestations
6. Selected Mutations and Anti-Complement Factor Antibodies
6.1. Complement Factor H and Anti-Complement Factor H Antibodies
6.2. Complement Factor I and Anti-Complement Factor I Autoantibodies
6.3. Diacylglycerol Kinase Epsilon
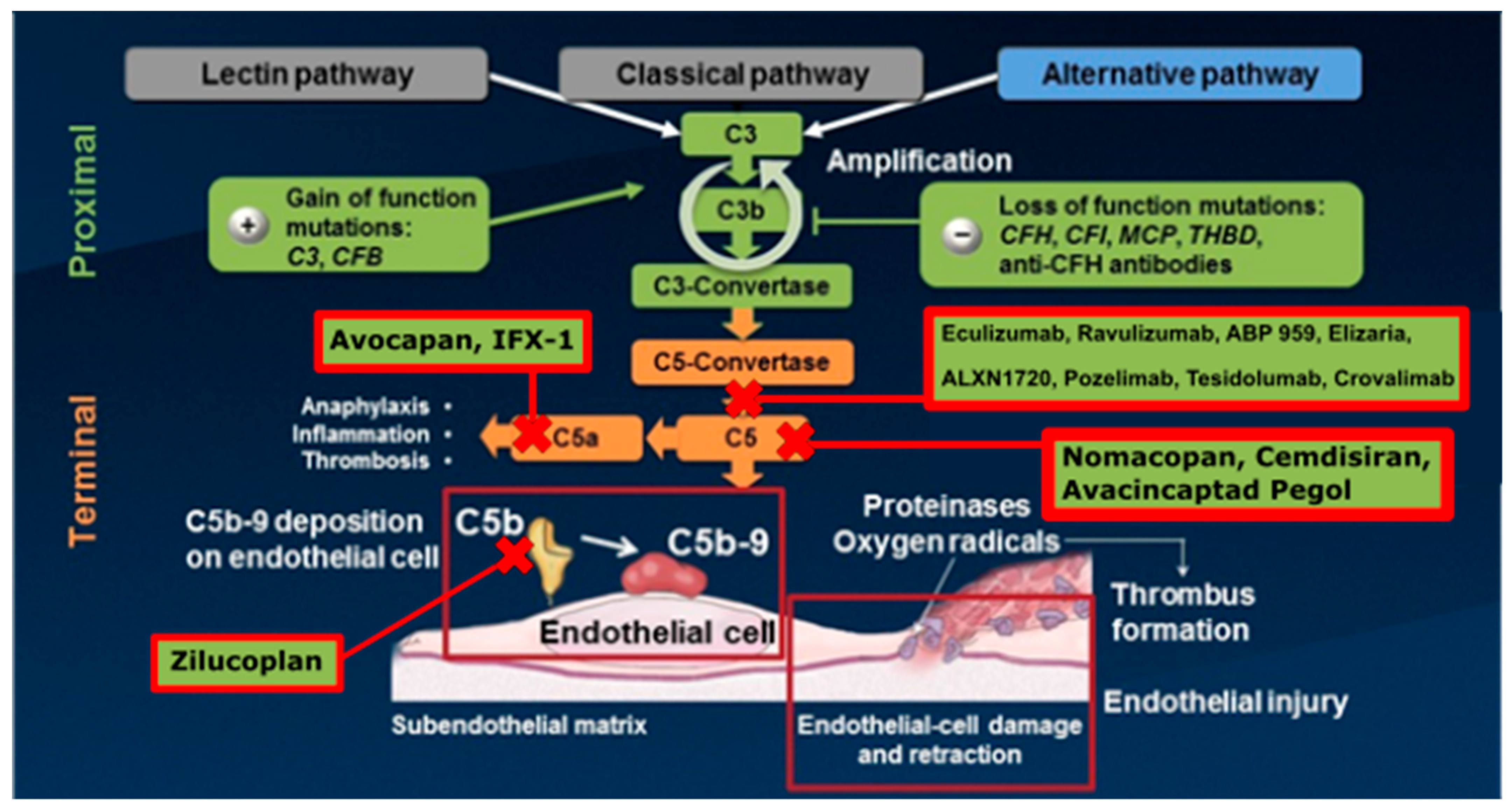
7. Current Therapeutics
7.1. Immunotherapy
7.2. Eculizumab
7.3. Ravulizumab
7.4. Kidney Transplant Setting
7.5. Biosimilars
8. Future Therapeutic Options
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, K.; Desai, K.; Gullapalli, L.; Druyts, E.; Balijepalli, C. Epidemiology of Atypical Hemolytic Uremic Syndrome: A Systematic Literature Review. Clin. Epidemiol. 2020, 12, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Lapeyraque, A.L.; Bitzan, M.; Al-Dakkak, I.; Francis, M.; Huang, S.S.; Kaprielian, R.; Larratt, L.; Pavenski, K.; Ribic, C.; Tosikyan, A.; et al. Clinical Characteristics and Outcome of Canadian Patients Diagnosed With Atypical Hemolytic Uremic Syndrome. Can. J. Kidney Health Dis. 2020, 7, 2054358119897229. [Google Scholar] [CrossRef] [Green Version]
- Noris, M.; Bresin, E.; Mele, C.; Remuzzi, G. Genetic Atypical Hemolytic-Uremic Syndrome. 2007 Nov 16 [Updated 2016 Jun 9]. In Gene Reviews® [Internet]; University of Washington: Seattle, WA, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1367/ (accessed on 15 November 2021).
- Sawai, T.; Nangaku, M.; Ashida, A.; Fujimaru, R.; Hataya, H.; Hidaka, Y.; Kaname, S.; Okada, H.; Sato, W.; Yasuda, T.; et al. Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Pediatr. Int. 2014, 56, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragon-Durey, M.A.; Sethi, S.K.; Bagga, A.; Blanc, C.; Blouin, J.; Ranchin, B.; Andre, J.L.; Takagi, N.; Cheong, H.I.; Hari, P.; et al. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2010, 21, 2180–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, M.; Go, R.S.; Abraham, R.S.; Fervenza, F.C.; Sethi, S.; Bryant, S.C.; Spears, G.M.; Murray, D.L.; Willrich, M.A.V. Diagnostic Utility of Complement Serology for Atypical Hemolytic Uremic Syndrome. Mayo Clin. Proc. 2018, 93, 1351–1362. [Google Scholar] [CrossRef] [PubMed]
- Tomazos, I.; Garlo, K.; Wang, Y.; Chen, P.; Laurence, J. Triggers in Patients with Atypical Hemolytic Uremic Syndrome: An Observational Cohort Study Using a US Claims Database. Blood 2020, 136, 30–31. [Google Scholar] [CrossRef]
- McFarlane, P.A.; Bitzan, M.; Broome, C.; Baran, D.; Garland, J.; Girard, L.P.; Grewal, K.; Lapeyraque, A.L.; Patriquin, C.J.; Pavenski, K.; et al. Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Can. J. Kidney Health Dis. 2021, 8, 20543581211008707. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Pryzdial, E.L.G. Is the COVID-19 thrombotic catastrophe complement-connected? J. Thromb. Haemost. 2020, 18, 2812–2822. [Google Scholar] [CrossRef]
- Trimarchi, H.; Coppo, R. COVID-19 and acute kidney injury in pediatric subjects: Is there a place for eculizumab treatment? J. Nephrol. 2020, 33, 1119–1120. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Quincy Brown, J.; Vander Heide, R.S. Pulmonary and cardiac pathology in African American patients with COVID-19: An autopsy series from New Orleans. Lancet Respir. Med. 2020, 8, 681–686. [Google Scholar] [CrossRef]
- Mastellos, D.C.; Reis, E.S.; Lambris, J.D. Editorial: Therapeutic Modulation of the Complement System: Clinical Indications and Emerging Drug Leads. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Mastellos, D.C.; da Silva, B.G.P.; Fonseca, B.A.; Fonseca, N.P.; Auxiliadora-Martins, M.; Mastaglio, S.; Ruggeri, A.; Sironi, M.; Radermacher, P.; Chrysanthopoulou, A.; et al. Complement C3 vs C5 inhibition in severe COVID-19: Early clinical findings reveal differential biological efficacy. Clin. Immunol. 2020, 220, 108598. [Google Scholar] [CrossRef] [PubMed]
- Mastaglio, S.; Ruggeri, A.; Risitano, A.M.; Angelillo, P.; Yancopoulou, D.; Mastellos, D.C.; Huber-Lang, M.; Piemontese, S.; Assanelli, A.; Garlanda, C.; et al. The first case of COVID-19 treated with the complement C3 inhibitor AMY-101. Clin. Immunol. 2020, 215, 108450. [Google Scholar] [CrossRef] [PubMed]
- Diurno, F.; Numis, F.G.; Porta, G.; Cirillo, F.; Maddaluno, S.; Ragozzino, A.; De Negri, P.; Di Gennaro, C.; Pagano, A.; Allegorico, E.; et al. Eculizumab treatment in patients with COVID-19: Preliminary results from real life ASL Napoli 2 Nord experience. Eur. Rev. Med Pharmacol. Sci. 2020, 24, 4040–4047. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Lipton, M.; Broglie, L.; Jain, N.G.; Uy, N.S. Eculizumab treatment for renal failure in a pediatric patient with COVID-19. J. Nephrol. 2020, 33, 1373–1376. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, F.; O’Halloran, A.; Alghamdi, A.; Chen, C.; Trissal, M.; Traum, A.; DeCourcey, D. Toddler With New Onset Diabetes and Atypical Hemolytic-Uremic Syndrome in the Setting of COVID-19. Pediatrics 2021, 147, e2020016774. [Google Scholar] [CrossRef]
- Kaufeld, K.; Reinhardt, M.; Schröder, C.; Bräsen, J.H.; Wiech, T.; Brylka, P.; Khaled, A.; Bergmann, C.; Haller, H.; Gäckler, A.; et al. Atypical HUS triggered by infection with SARS-CoV2. Kidney Int. Rep. 2021, 6, 2709–2712. [Google Scholar] [CrossRef]
- Avcı, Z.; Bayram, C.; Malbora, B. Hepatitis B vaccine-associated atypical hemolytic uremic syndrome. Turk. J. Haematol. 2013, 30, 418–419. [Google Scholar] [CrossRef]
- Geerdink, L.M.; Westra, D.; Wijk, J.A.; van Dorresteijn, E.M.; Lilien, M.R.; Davin, J.C.; Kömhoff, M.; van Hoeck, K. Atypical hemolytic uremic syndrome in children: Complement mutations and clinical characteristics. Pediatr. Nephrol. 2012, 27, 1283–1291. [Google Scholar]
- Bernabeu, A.I.A.; Escribano, T.C.; Vilarino, M.C. Atypical Hemolytic Uremic Syndrome: New Challenges in the Complement Blockage Era. Nephron 2020, 144, 537–549. [Google Scholar] [CrossRef]
- Yenerel, M.N. Atypical Hemolytic Uremic Syndrome: Differential Diagnosis from TTP/HUS and Management. Atipik Hemolitik Üremik Sendrom: Ttp/Hüs ile Ayırıcı Tanısı ve Tedavisi. Turk. J. Haematol. 2014, 31, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Loirat, C.; Fremeaux-Bacchi, V. Atypical hemolytic uremic syndrome. Orphanet. J. Rare Dis. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loirat, C.; Macher, M.A.; Elmaleh-Berges, M.; Kwon, T.; Deschenes, G.; Goodship, T.H.; Majoie, C.; Davin, J.C.; Blanc, R.; Savatovsky, J.; et al. Non-atheromatous arterial stenoses in atypical haemolytic uraemic syndrome associated with complement dysregulation. Nephrol. Dial. Transplant. 2010, 25, 3421–3425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Formeck, C.; Swiatecka-Urban, A. Extra-renal manifestations of atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2019, 34, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Fidan, K.; Goknar, N.; Gulhan, B.; Melek, E.; Yildirim, Z.Y.; Baskin, E.; Hayran, M.; Gulleroglu, K.; Ozcakar, Z.B.; Ozaltin, F.; et al. Extra-Renal manifestations of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2018, 33, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Gulleroglu, K.; Fidan, K.; Hancer, V.S.; Bayrakci, U.; Baskin, E.; Soylemezoglu, O. Neurologic involvement in atypical hemolytic uremic syndrome and successful treatment with eculizumab. Pediatr. Nephrol. 2013, 28, 827–830. [Google Scholar] [CrossRef]
- Koehl, B.; Boyer, O.; Biebuyck-Gouge, N.; Kossorotoff, M.; Fremeaux Bacchi, V.; Boddaert, N.; Niaudet, P. Neurological involvement in a child with atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2010, 25, 2539–2542. [Google Scholar] [CrossRef]
- Diamante Chiodini, B.; Davin, J.C.; Corazza, F.; Khaldi, K.; Dahan, K.; Ismaili, K.; Adams, B. Eculizumab in anti-factor h antibodies associated with atypical hemolytic uremic syndrome. Pediatrics 2014, 133, e1764–e1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Gorovoy, I.R.; Mao, J.; Jin, J.; Chen, X.; Cui, Q.N. Recurrent ocular involvement in pediatric atypical hemolytic uremic syndrome. J. Pediatr. Ophthalmol. Strabismus 2014, 51, e62–e65. [Google Scholar] [CrossRef]
- Vilalta, R.; Lara, E.; Madrid, A.; Chocron, S.; Munoz, M.; Casquero, A.; Nieto, J. Long-term eculizumab improves clinical outcomes in atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2012, 27, 2323–2326. [Google Scholar] [CrossRef] [Green Version]
- Neuhaus, T.J.; Calonder, S.; Leumann, E.P. Heterogeneity of atypical haemolytic uraemic syndromes. Arch. Dis. Child 1997, 76, 518–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Nagra, A.; Haq, M.R.; Gilbert, R.D. Eculizumab in atypical haemolytic uraemic syndrome with severe cardiac and neurological involvement. Pediatr. Nephrol. 2014, 29, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Davin, J.C.; Gracchi, V.; Bouts, A.; Groothoff, J.; Strain, L.; Goodship, T. Maintenance of kidney function following treatment with eculizumab and discontinuation of plasma exchange after a third kidney transplant for atypical hemolytic uremic syndrome associated with a CFH mutation. Am. J. Kidney Dis 2010, 55, 708–711. [Google Scholar] [CrossRef]
- Vergouwen, M.D.; Adriani, K.S.; Roos, Y.B.; Groothoff, J.W.; Majoie, C.B. Proximal cerebral artery stenosis in a patient with hemolytic uremic syndrome. AJNR. Am. J. Neuroradiol. 2008, 29, e34. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.; Stojanovic, J.; Ariceta, G.; Bitzan, M.; Besbas, N.; Frieling, M.; Karpman, D.; Landau, D.; Langman, C.; Licht, C.; et al. An audit analysis of a guideline for the investigation and initial therapy of diarrhea negative (atypical) hemolytic uremic syndrome. Pediatr. Nephrol. 2014, 29, 1967–1978. [Google Scholar] [CrossRef] [PubMed]
- Besbas, N.; Gulhan, B.; Soylemezoglu, O.; Ozcakar, Z.B.; Korkmaz, E.; Hayran, M.; Ozaltin, F. Turkish pediatric atypical hemolytic 1346. Pediatr. Nephrol. 2019, 34, 1337–1348. [Google Scholar]
- Roman-Ortiz, E.; Mendizabal Oteiza, S.; Pinto, S.; Lopez-Trascasa, M.; Sanchez-Corral, P.; Rodriguez de Cordoba, S. Eculizumab long-term therapy for pediatric renal transplant in aHUS with CFH/CFHR1 hybrid gene. Pediatr. Nephrol. 2014, 29, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Webb, T.N.; Griffiths, H.; Miyashita, Y.; Bhatt, R.; Jaffe, R.; Moritz, M.; Hofer, J.; Swiatecka-Urban, A. Atypical Hemolytic Uremic Syndrome and Chronic Ulcerative Colitis Treated with eculizumab. Int. J. Med. Pharm. Case Rep. 2015, 4, 105–112. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. CJASN 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Matrat, L.; Bacchetta, J.; Ranchin, B.; Tanné, C.; Sellier-Leclerc, A.L. Pediatric atypical hemolytic-uremic syndrome due to auto-antibodies against factor H: Is there an interest to combine eculizumab and mycophenolate mofetil? Pediatr. Nephrol. 2021, 36, 1647–1650. [Google Scholar] [CrossRef]
- Fujisawa, M.; Yasumoto, A.; Kato, H.; Sugawara, Y.; Yoshida, Y.; Yatomi, Y.; Nangaku, M. The role of anti-complement factor H antibodies in the development of atypical haemolytic uremic syndrome: A possible contribution to abnormality of platelet function. Br. J. Haematol. 2020, 189, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fan, M.N.; Yang, M.; Lu, C.; Zhang, M.; Liu, X.H.; Ma, L. Association among Complement Factor H Autoantibodies, Deletions of CFHR, and the Risk of Atypical Hemolytic Uremic Syndrome. Int. J. Environ. Res. Public Health 2016, 13, 1209. [Google Scholar] [CrossRef] [Green Version]
- Puraswani, M.; Khandelwal, P.; Saini, H.; Saini, S.; Gurjar, B.S.; Sinha, A.; Shende, R.P.; Maiti, T.K.; Singh, A.K.; Kanga, U.; et al. Clinical and Immunological Profile of Anti-factor H Antibody Associated Atypical Hemolytic Uremic Syndrome: A Nationwide Database. Front. Immunol. 2019, 10, 1282. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.Y.; Song, D.; Liu, X.R.; Chen, Z.; Xiao, H.J.; Ding, J.; Sun, S.Z.; Liu, H.Y.; Wang, S.X.; Yu, F.; et al. Immunological features and functional analysis of anti-CFH autoantibodies in patients with atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2019, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, S.; Rawat, A.; Ramachandran, R.; Hans, R.; Dawman, L.; Tiewsoh, K. Anti-complement factor I antibody associated atypical hemolytic uremic syndrome—A new insight for future perspective! Immunobiology 2020, 225, 152000. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, M.; Frémeaux-Bacchi, V.; Schaefer, F.; Choi, M.; Tang, W.H.; Le Quintrec, M.; Fakhouri, F.; Taque, S.; Nobili, F.; Martinez, F.; et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat. Genet. 2013, 45, 531–536. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Chaki, M.; Lu, D.; Ren, C.; Wang, S.S.; Rauhauser, A.; Li, B.; Zimmerman, S.; Jun, B.; Du, Y.; et al. Loss of diacylglycerol kinase epsilon in mice causes endothelial distress and impairs glomerular Cox-2 and PGE2 production. Am. J. Physiol. Ren. Physiol. 2016, 310, F895–F908. [Google Scholar] [CrossRef] [Green Version]
- Brocklebank, V.; Kumar, G.; Howie, A.J. Long-term outcomes and response to treatment in diacylglycerol kinase epsilon nephropathy. Kidney Int. 2020, 97, 1260–1274. [Google Scholar] [CrossRef] [Green Version]
- Fremeaux-Bacchi, V.; Fakhouri, F.; Garnier, A.; Bienaimé, F.; Dragon-Durey, M.A.; Ngo, S.; Moulin, B.; Servais, A.; Provot, F.; Rostaing, L.; et al. Genetics and outcome of atypical hemolytic uremic syndrome: A nationwide French series comparing children and adults. Clin. J. Am. Soc. Nephrol. 2013, 8, 554–562. [Google Scholar] [CrossRef] [Green Version]
- Raina, R.; Krishnappa, V.; Blaha, T.; Kann, T.; Hein, W.; Burke, L.; Bagga, A. Atypical Hemolytic-Uremic Syndrome: An Update on Pathophysiology, Diagnosis, and Treatment. Ther. Apher. Dial. 2019, 23, 4–21. [Google Scholar] [CrossRef] [Green Version]
- Loirat, C.; Fakhouri, F.; Ariceta, G.; Besbas, N.; Bitzan, M.; Bjerre, A.; Coppo, R.; Emma, F.; Johnson, S.; Karpman, D.; et al. HUS International. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr. Nephrol. 2016, 31, 15–39. [Google Scholar] [CrossRef]
- Dundas, S.; Murphy, J.; Soutar, R.L.; Jones, G.A.; Hutchinson, S.J.; Todd, W.T. Effectiveness of therapeutic plasma exchange in the 1996 Lanarkshire Escherichia coli O157:H7 outbreak. Lancet 1999, 354, 1327–1330. [Google Scholar] [CrossRef]
- Colic, E.; Dieperink, H.; Titlestad, K.; Tepel, M. Management of an acute outbreak of diarrhoea-associated haemolytic uraemic syndrome with early plasma exchange in adults from southern Denmark: An observational study. Lancet 2011, 378, 1089–1093. [Google Scholar] [CrossRef]
- Nakatani, T.; Tsuchida, K.; Yoshimura, R.; Sugimura, K.; Take-moto, Y. Plasma exchange therapy for the treatment of Escherichia coli O-157 associated hemolytic uremic syn-drome. Int. J. Mol. Med. 2002, 10, 585–588. [Google Scholar] [PubMed]
- Barbour, T.D.; Pickering, M.C.; Terence Cook, H. Recent insights into C3 glomerulopathy. Nephrol. Dial. Transplant. 2013, 28, 1685–1693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ort, M.; Dingemanse, J.; van den Anker, J.; Kaufmann, P. Treatment of Rare Inflammatory Kidney Diseases: Drugs Targeting the Terminal Complement Pathway. Front. Immunol. 2020, 11, 599417. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, L.A.; Fila, M.; Ardissino, G.; Al-Akash, S.I.; Evans, J.; Henning, P.; Lieberman, K.V.; Maringhini, S.; Pape, L.; Rees, L.; et al. Eculizumab is a safe and effective treatment in pediatric patients with atypical hemolytic uremic syndrome. Kidney Int. 2016, 89, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariceta, G.; Dixon, B.P.; Kim, S.H.; Kapur, G.; Mauch, T.; Ortiz, S.; Vallee, M.; Denker, A.E.; Kang, H.G.; Greenbaum, L.A. The long-acting C5 inhibitor, ravulizumab, is effective and safe in pediatric patients with atypical hemolytic uremic syndrome naïve to complement inhibitor treatment. Kidney Int. 2021, 100, 225–237. [Google Scholar] [CrossRef]
- Socié, G.; Caby-Tosi, M.P.; Marantz, J.L.; Cole, A.; Bedrosian, C.L.; Gasteyger, C.; Mujeebuddin, A.; Hillmen, P.; Vande Walle, J.; Haller, H. Eculizumab in paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome: 10-year pharmacovigilance analysis. Br. J. Haematol. 2019, 185, 297–310. [Google Scholar] [CrossRef] [Green Version]
- McKeage, K. Ravulizumab: First Global Approval. Drugs 2019, 79, 347–352. [Google Scholar] [CrossRef]
- Tanaka, K.; Adams, B.; Aris, A.M.; Fujita, N.; Ogawa, M.; Ortiz, S.; Vallee, M.; Greenbaum, L.A. The long-acting C5 inhibitor, ravulizumab, is efficacious and safe in pediatric patients with atypical hemolytic uremic syndrome previously treated with eculizumab. Pediatr. Nephrol. 2021, 36, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Ehren, R.; Habbig, S. Real-world data of six patients with atypical hemolytic uremic syndrome switched to ravulizumab. Pediatr. Nephrol. 2021, 36, 3281–3282. [Google Scholar] [CrossRef] [PubMed]
- Turkmen, K.; Baloglu, I.; Ozer, H. C3 glomerulopathy and atypical hemolytic uremic syndrome: An updated review of the literature on alternative complement pathway disorders. Int. Urol. Nephrol. 2021, 53, 2067–2080. [Google Scholar] [CrossRef]
- Miles, N.; Skerra, A. Therapeutic Development of Complement C5 Inhibitor CoversinTM with Extended Half-Life Via PASylation®. Blood 2016, 128, 5900. [Google Scholar] [CrossRef]
- Kusner, L.L.; Yucius, K.; Sengupta, M.; Sprague, A.G.; Desai, D.; Nguyen, T.; Charisse, K.; Kuchimanchi, S.; Kallanthottathil, R.; Fitzgerald, K.; et al. Investigational RNAi Therapeutic Targeting C5 Is Efficacious in Pre-clinical Models of Myasthenia Gravis. Mol. Ther. Methods Clin. Dev. 2019, 13, 484–492. [Google Scholar] [CrossRef] [Green Version]
- Bresin, E.; Daina, E.; Noris, M.; Castelletti, F.; Stefanov, R.; Hill, P.; Goodship, T.H.; Remuzzi, G. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: Prognostic significance of genetic background. Clin. J. Am. Soc. Nephrol. 2006, 1, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Managing and preventing atypical hemolytic uremic syndrome recurrence after kidney transplantation. Curr. Opin. Nephrol. Hypertens. 2013, 22, 704–712. [Google Scholar] [CrossRef]
- Gruppo, R.A.; Rother, R.P. Eculizumab for congenital atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2009, 360, 544–546. [Google Scholar] [CrossRef] [PubMed]
- Giordano, M.; Castellano, G.; Messina, G.; Divella, C.; Bellantuono, R.; Puteo, F.; Colella, V.; Depalo, T.; Gesualdo, L. Preservation of renal function in atypical hemolytic uremic syndrome by eculizumab: A case report. Pediatrics 2012, 130, e1385–e1388. [Google Scholar] [CrossRef] [Green Version]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [Green Version]
- Zuber, J.; Le Quintrec, M.; Krid, S.; Bertoye, C.; Gueutin, V.; Lahoche, A.; Heyne, N.; Ardissino, G.; Chatelet, V.; Noel, L.H.; et al. Eculizumab for atypical hemolytic uremic syndrome recurrence in renal transplantation. Am. J. Transplant. 2012, 12, 3337–3354. [Google Scholar] [CrossRef]
- Stojanovic, J.; Adamusiak, A.; Waters, A.; Sebire, N.J.; Kessaris, N.; Mamode, N.; Marks, S.D. Early relapse of atypical hemolytic uremic syndrome following ABO-incompatible living–related pediatric kidney re-transplant successfully treated with eculizumab. Pediatr. Nephrol. 2021, 36, 3271–3275. [Google Scholar] [CrossRef]
- Kant, S.; Bhalla, A.; Alasfar, S.; Alachkar, N. Ten-year outcome of Eculizumab in kidney transplant recipients with atypical hemolytic uremic syndrome- a single center experience. BMC Nephrol. 2020, 21, 189. [Google Scholar] [CrossRef] [PubMed]
- Nester, C.; Stewart, Z.; Myers, D.; Jetton, J.; Nair, R.; Reed, A.; Thomas, C.; Smith, R.; Brophy, P. Renal transplantation underprophylactic eculizumab in atypical hemolytic uremic syndromewith CFH/CFHR1 hybrid protein. Am. J. Transplant. 2012, 12, 1938–1944. [Google Scholar]
- Weitz, M.; Amon, O.; Bassler, D.; Koenigsrainer, A.; Nadalin, S. Prophylactic eculizumab prior to kidney transplantation for atypical hemolytic uremic syndrome. Pediatr. Nephrol. 2011, 26, 1325–1329. [Google Scholar] [CrossRef]
- Zimmerhackl, L.B.; Hofer, J.; Cortina, G.; Mark, W.; Würzner, R.; Jungraithmayr, T.C.; Khursigara, G.; Kliche, K.O.; Radauer, W. Prophylactic eculizumab after renal transplantation in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2010, 362, 1746–1748. [Google Scholar] [CrossRef] [Green Version]
- Krid, S.; Roumenina, L.T.; Beury, D.; Charbit, M.; Boyer, O.; Frémeaux-Bacchi, V.; Niaudet, P. Renal transplantation under prophylactic eculizumab in atypical hemolytic uremic syndrome with CFH/CFHR1 hybrid protein. Am. J. Transplant. 2012, 12, 1938–1944. [Google Scholar] [CrossRef]
- Ardissino, G.; Cresseri, D.; Tel, F.; Giussani, A.; Salardi, S.; Sgarbanti, M.; Strumbo, B.; Testa, S.; Capone, V.; Griffini, S.; et al. Kidney transplant in patients with atypical hemolytic uremic syndrome in the anti-C5 era: Single-center experience with tailored Eculizumab. J. Nephrol. 2021, 34, 2027–2036. [Google Scholar] [CrossRef]
- Siedlecki, A.M.; Isbel, N.; Vande Walle, J.; James Eggleston, J.; Cohen, D.J.; Global aHUS Registry. Eculizumab use for kidney transplantation in patients with a diagnosis of atypical hemolytic uremic syndrome. Kidney Int. Rep. 2018, 4, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, V.; Pan, J.; Chien, D.; Mytych, D.T.; Hanes, V. A randomized, double-blind, single-dose, three-arm, parallel group study to determine pharmacokinetic similarity of ABP 959 and eculizumab (Soliris®) in healthy male subjects. Eur. J. Haematol. 2020, 105, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Lavrishcheva, I.V.; Jakovenko, A.A.; Kudlay, D.A. A case report of atypical hemolytic-uremic syndrome treatment with the first Russian eculizumab in adult patient. Urol. Nephrol. Open Access J. 2020, 8, 37–40. [Google Scholar] [CrossRef]
- Devalaraja-Narashimha, K.; Ni, Y.G.; Huang, C.; Wang, M.-D.; Chaudhari, U.; Prasad, S.; Harari, O.; Rankin, A.J.; Morton, L.; Weyne, J. Pozelimab, a Human Antibody Against Complement Factor C5, Demonstrates Robust Inhibition of Alternative Complement Activity Both in Normal Human Serum and in Phase I Normal Healthy Volunteers. Blood 2019, 134, 2278. [Google Scholar] [CrossRef]
- Kassa, E.; Ciulla, T.A.; Hussain, R.M.; Dugel, P.U. Complement inhibition as a therapeutic strategy in retinal disorders. Expert Opin. Biol. Ther. 2019, 19, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, T.; Nezu, J. SKY59, A Novel Recycling Antibody for Complement-mediated Diseases. Curr. Med. Chem. 2020, 27, 4157–4164. [Google Scholar] [CrossRef] [PubMed]
- Fukuzawa, T.; Sampei, Z.; Haraya, K.; Ruike, Y.; Shida-Kawazoe, M.; Shimizu, Y.; Gan, S.W.; Irie, M.; Tsuboi, Y.; Tai, H.; et al. Long lasting neutralization of C5 by SKY59, a novel recycling antibody, is a potential therapy for complement-mediated diseases. Sci. Rep. 2017, 7, 1080. [Google Scholar] [CrossRef]
- Röth, A.; Nishimura, J.I.; Nagy, Z.; Gaàl-Weisinger, J.; Panse, J.; Yoon, S.S.; Egyed, M.; Ichikawa, S.; Ito, Y.; Kim, J.S.; et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood 2020, 135, 912–920. [Google Scholar] [CrossRef] [Green Version]
- Riedemann, N.C.; Habel, M.; Ziereisen, J.; Hermann, M.; Schneider, C.; Wehling, C.; Kirschfink, M.; Kentouche, K.; Guo, R. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin. Immunol. 2017, 180, 25–32. [Google Scholar] [CrossRef]
- NCT04115293 Safety, Tolerability, and Efficacy of Zilucoplan in Subjects with Generalized Myasthenia Gravis (RAISE). ClinicalTrials.gov. A Service of the U.S. National Institutes of Health. Available online: https://clinicaltrials.gov/ct2/show/NCT04115293?term=zilucoplan&draw=2&rank=4. (accessed on 15 November 2021).
- Drolet, D.W.; Green, L.S.; Gold, L.; Janjic, N. Fit for the Eye: Aptamers in Ocular Disorders. Nucleic Acid Ther. 2016, 26, 127–146. [Google Scholar] [CrossRef] [Green Version]
- Brandstätter, H.; Schulz, P.; Polunic, I.; Kannicht, C.; Kohla, G.; Römisch, J. Purification and biochemical characterization of functional complement factor H from human plasma fractions. Vox Sang. 2012, 103, 201–212. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Mutation | No. of Studies | Total aHUS Patients | Pooled Estimate | I2 (95% CI); p Value | Egger’s Test |
|---|---|---|---|---|---|
| (95% CI) | |||||
| CFH | 12 | 2295 | 21.41% | 85.84% (76.97–91.29%); | p = 0.5189 |
| (16.60–26.64%) | p < 0.0001 | ||||
| CD46 | 11 | 2177 | 9.98% | 76.84% (58.58–87.05%); | p = 0.2614 |
| (7.15–13.22%) | p < 0.0001 | ||||
| CFI | 12 | 2295 | 6.89% | 64.69% (34.6–80.93%); | p = 0.2206 |
| (5.01–9.05%) | p = 0.0010 | ||||
| DGKE | 4 | 558 | 6.57% | 90.06% (77.48–95.61%); | p = 0.1619 |
| (0.93–16.76%) | p < 0.0001 | ||||
| C3 | 9 | 2193 | 5.29% | 61.37% (20.05–81.34%); | p = 0.8866 |
| (3.74–7.09%) | p = 0.0080 | ||||
| THBD | 6 | 1176 | 1.74% | 77.9% (51.11–90.01%); | p = 0.6401 |
| (0.47–3.8%) | p = 0.0004 | ||||
| CFB | 5 | 1469 | 1.55% | 26.03% (0.00–70.5%); | p = 0.7374 |
| (0.99–2.32%) * | p = 0.2480 | ||||
| Others | 4 | 691 | 19.29% | 98.7% (97.98–99.16%); | p = 0.7916 |
| (1.34–50.78%) | p < 0.0001 | ||||
| Combined | 7 | 1922 | 3.06% | 84.36% (69.48–91.98%); | p = 0.0566 |
| (1.26–5.61%) | p < 0.0001 |
| Novel Triggers to aHUS | ||||||
|---|---|---|---|---|---|---|
| COVID-19 | Hepatitis B Vaccine | |||||
| Mahajan et al. | Alizadeh et al. | Kaufeld et al. | Avci et al. | Geerdink et al. | ||
| Year | 2020 | 2021 | 2021 | 2013 | 2012 | |
| Sample Size | 1 | 1 | 1 | 1 | 1 | 1 |
| Age (Months) | 14 | 1.3 | 22 | 52 | 0.15 | - |
| Symptoms at Onset | Abdominal pain Diarrhea Vomiting Myalgias Chest pain | Fever Tachycardia Tachypnea Metabolic Acidosis | Diarrhea Vomiting Loss of taste Hypertension | Loss of taste Fever Abdominal pain Hypertension | Jaundice Pulse rate: 140/min Respiratory rate: 48/min BP: 70/40 mmHg | x |
| Lab Results | ↑Ferritin >100,000 ng/mL, ↑CRP > 300 mg/dL, WBC 33,000/µL SCr 0.7 mg/dL ↓Platelets 126,000/mL ↓Hgb 6.8 g/dL ↑LDH 4087 U/L ↑Bilirubin 6.2 mg/dL Schistocytes ↓C3 33 mg/dL ↓C4 1.0 mg/dL | ↓Bicarbonate 4 mmol/L ↓PC02 17 mmHg ↓Anion gap 40 ↑Glucose 805 mg/dL ↑WBC 33,000 ↑Procalcitonin 3 ng/mL | ↑LDH 2066 U/L ↓Hgb 5.5 g/dL | ↑LDH 88,560 U/L ↓Hgb 9.4 g/dl | ↓Hgb 51 g/L Leukocyte count 10 × 109/L Platelet count 28 × 109/L ↑Reticulocyte level 3.9% ↑Urea 88 mg/dL SCr 1.1 mg/dL ↑Total bilirubin 13.7 mg/dL ↑Direct bilirubin 2.6 mg/dL Uric acid 8.1 mg/dL ↑Asp aminotransferase 96 U/L ↑LDH 4641 U/L | x |
| Progressing Lab Results | ↑SCr 8.97 mg/dL ↑BUN 170 mg/dL | ↓Platelets <100 K/uL ↑Reticulosytosis 13% ↑LDH 3190 U/L ↑Bilirubmin 1.5 mg/dL ↑Fibrinogen 557 mg/dL ↑Ferritin 1493 ng/mL ↑BUN 39 mg/dL ↑Creatinine 0.39 mg/dL C3 142 mg/dL C4 19 mg/dL | ↑SCr 6.33 mg/dL ↓Platelets 28 k cells/uL | ↑SCr 2.88 mg/dL ↓Platelets 128 k/uL | x | x |
| Organ System | Clinical Manifestations | Reported Efficacy of Eculizumab | |
|---|---|---|---|
| Renal | Glomerular thrombotic microangiopathy, Arterial TMA, and Cortical necrosis | Yes | |
| Neurological | Seizures, Headache, Altered consciousness, Hemiparesis, Vision loss, Hallucinations, Encephalopathy | Agitation, Confusion, Reduced reflexes, Hemiplegia, Nystagmus, Diplopia, Focal neurologic deficits, Coma | Yes |
| Pulmonary | Pulmonary embolism, Hemorrhage, Edema, Respiratory failure | N/A | |
| Dermatologic | Peripheral gangrene, Ischemia, Cutaneous rashes | Yes | |
| Cardiovascular | Hypertrophic cardiomyopathy, Left ventricular hypertrophy, Elevated CK-MB level, Dilated cardiomyopathy, Valve insufficiency | Tachycardia, Intracardiac thrombus, Steno-occlusive arterial disease in large arterial vessels (i.e., middle and anterior cerebral artery stenosis) | Yes |
| Ocular | Reduced visual acuity, Ocular pain, Visual scotomas, Diplopia, Blurred vision | Optic disc edema, Bilateral flame-shaped intraretinal hemorrhage, Tortuosity, Venous stasis retinopathy | Yes |
| Gastrointestinal | Vomiting, Cholelithiasis, Transaminitis, Pancreatitis, | Hepatitis, Gastrointestinal bleeding, Abdominal pain | Yes |
| Organ System Complications Due to aHUS | |||||
|---|---|---|---|---|---|
| Organ System | Authors | Year | Sample Size | Age | Outcome |
| Neurological | Gulleroglu et al. | 2013 | 2 | 14 | Neurologic symptoms and irregular cerebral MRI results |
| Diamante et al. | 2014 | 1 | <18 | Multifocal hyperintensities and altered consciousness | |
| Cardiovascular | Hu et al. | 2015 | 1 | 0.75 | Cardiomyopathy and altered cardiac function |
| Vilalta et al. | 2012 | 1 | 1 | ||
| Neuhaus et al. | 1997 | 23 | |||
| Davin et al. | 2010 | 1 | 15 | Middle and anterior cerebral artery stenosis | |
| Pulmonary | Johnson et al. | 2014 | 71 | - | 21% of aHUS patients developed respiratory failure |
| Ocular | Zheng et al. | 2014 | 1 | 11 | Decreased visual acuity (20/100 in the right eye, 20/200 in the left eye) Intraretinal hemorrhages, Venous stasis retinopathy, and Vein occlusions |
| Gastrointestinal | Besbas et al. | 2017 | 146 | - | 10% displayed vomiting, cholelithiasis, transaminitis, pancreatitis, hepatitis, and GI bleeding |
| Dragon-Durey et al. | 2010 | 45 | - | <80% of patients with anti-CFH antibodies had GI symptoms | |
| Roman-Ortiz et al. | 2014 | 1 | 9 | Abdominal pain | |
| Eculizumab Treatment | |||||
|---|---|---|---|---|---|
| Legendre et al. | Licht et al. | Cofiell et al. | Greenbaum et al. | ||
| Year | 2013 | 2014 | 2014 | 2015 | |
| Sample Size | 37 | 37 | 41 | 22 | |
| Age (Years) | ≥12 | ≥18 + 1 adolescent | ≥18 | 0.4–17 | |
| Primary Endpoints | Platelet normalization (≥150 × 109/L) LDH normalization (<upper limit of normal) eGFR via SCr improved (≥25% reduction from baseline) | ||||
| Secondary Endpoints | 26 weeks = 80% | 26 weeks = 82% | - | 27 weeks = 95% | TMA free outcomes |
| 26 weeks = 100% | 26 weeks = 76% | - | 55 days (median) = 82% | Hematologic normalization | |
| 26 weeks = 100% | - | 6 weeks = sig. ↓ | - | Complement pathway inhibition | |
| 26 weeks = 100% | 26 weeks = 80% | 6 weeks = sig. ↓ | 48 days (median) = 73% | Renal function measure normalization | |
| Current Therapeutics | Drug Class | Pathophysiology/ Mechanism of Action | Complement Pathway Proteins Affected | |
|---|---|---|---|---|
| Current Therapeutics. | Eculizumab | Monoclonal Antibody, terminal complement inhibitor | Binds to C5 and prevents cleavage to C5a and C5b | C5a and C5b levels |
| Ravulizumab | Prevents the cleavage of C5 into C5a and C5b | |||
| Nomacopan | C5aR1 antagonist | Inhibits C3a, C4a, and C5a protein function | C3a, C4a, and C5a levels | |
| Avocapan | Recombinant protein derived from a tick C5 inhibitor | Inhibits C5 and leukotriene B4 | C5 and leukotriene B4 levels | |
| Cemdisiran | Short sequences of interfering RNA | Match mRNA for the C5 protein, with N-acetylgalactosamine | C5 levels | |
| Biosimilars | ABP 959 | Biosimilar to FDA-licensed Eculizumab | Binds to C5 and prevents cleavage to C5a and C5b | C5a and C5b levels |
| Elizaria | Russian biosimilar to Eculizumab | Binds to C5 and prevents cleavage to C5a and C5b | ||
| Future Therapeutics | ALXN1720 | Anti-C5 mini body | Binds to C5 protein and blocks its activation | C5 levels |
| Pozelimab | C5 antibody | Decrease hemolysis and C5 level | ||
| Tesidolumab | C5 monoclonal IgG1 antibody | Binds to C5 preventing its cleavage | C5a and C5b levels | |
| Crovalimab | Binds to a C5 epitope | Binds to C5b and prevents the formation of the MAC complex | C5a, C5b, and MAC complex proteins | |
| IFX-1 | Targets C5a protein directly | Binds to C5a | C5a levels | |
| Zilucoplan | Binds to the C5b protein and the C5b part of C5 | Inhibits C5b binding on C5 by binding to its C5 domain | C5b levels | |
| Avacincaptad Pegol | Binds to and inhibits the C5 protein | Prevents cleavage of C5 | C5 levels | |
| Avdoralimab | Anti-C5aR1 antibody | Blocks T-cell and natural killer cell activity through C5aR1 suppression | C5aR1 levels | |
| MAC Inhbitor HMR59 | Promotes CD59 production | Enhances synthesis of CD59, which blocks C5b-9 formation | C5b-9 formation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raina, R.; Vijayvargiya, N.; Khooblall, A.; Melachuri, M.; Deshpande, S.; Sharma, D.; Mathur, K.; Arora, M.; Sethi, S.K.; Sandhu, S. Pediatric Atypical Hemolytic Uremic Syndrome Advances. Cells 2021, 10, 3580. https://doi.org/10.3390/cells10123580
Raina R, Vijayvargiya N, Khooblall A, Melachuri M, Deshpande S, Sharma D, Mathur K, Arora M, Sethi SK, Sandhu S. Pediatric Atypical Hemolytic Uremic Syndrome Advances. Cells. 2021; 10(12):3580. https://doi.org/10.3390/cells10123580
Chicago/Turabian StyleRaina, Rupesh, Nina Vijayvargiya, Amrit Khooblall, Manasa Melachuri, Shweta Deshpande, Divya Sharma, Kashin Mathur, Manav Arora, Sidharth Kumar Sethi, and Sonia Sandhu. 2021. "Pediatric Atypical Hemolytic Uremic Syndrome Advances" Cells 10, no. 12: 3580. https://doi.org/10.3390/cells10123580