Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock


Department of Science and Environmental Studies, The Education University of Hong Kong, 10 Lo Ping Road, Tai Po, N. T., Hong Kong, China
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(9), 760; https://doi.org/10.3390/catal9090760
Submission received: 25 August 2019
/
Accepted: 3 September 2019
/
Published: 10 September 2019
(This article belongs to the Special Issue Catalysis and Catalytic Processes for CO2 Conversion)
Abstract
:Processes for the conversion of CO2 to valuable chemicals are highly desired as a result of the increasing CO2 levels in the atmosphere and the subsequent elevating global temperature. However, CO2 is thermodynamically and kinetically inert to transformation and, therefore, many efforts were made in the last few decades. Reformation/hydrogenation of CO2 is widely used as a means to access valuable products such as acetic acids, CH4, CH3OH, and CO. The electrochemical reduction of CO2 using hetero- and homogeneous catalysts recently attracted much attention. In particular, molecular CO2 reduction catalysts were widely studied using transition-metal complexes modified with various ligands to understand the relationship between various catalytic properties and the coordination spheres above the metal centers. Concurrently, the coupling of CO2 with various electrophiles under homogeneous conditions is also considered an important approach for recycling CO2 as a renewable C-1 substrate in the chemical industry. This review summarizes some recent advances in the conversion of CO2 into valuable chemicals with particular focus on the metal-catalyzed reductive conversion and functionalization of CO2.
1. Introduction
Many efforts were devoted in the recent decades to utilizing CO2 as a source for renewable energy and materials. Direct gas-phase reactions of CO2 with other readily accessible small molecules, e.g., via the reformation with CH4 to produce syngas (CO/H2) and acetic acid (Equations (1) and (2)), as well as hydrogenation to produce CH4, CH3OH, and CO (Equations (3)–(5)), were widely studied using mainly heterogeneous or supported metal catalysts [1,2,3,4,5,6,7]. Although these gaseous reactions convert CO2 into useful products, the binary processes, which involve the reactions of CO2 with explosive gases such as H2 and CH4, are usually pressure-sensitive and, thus, require careful control over the reaction conditions. More handy chemical approaches via which diversified products can be derived directly from CO2 are, therefore, highly desirable.
CO2 + CH4 → 2CO + 2H2; ΔH°298K = +247.30 kJ/mol.
CO2 + CH4 → CH3COOH; ΔH°298K = +8.62 kJ/mol.
CO2 + 4H2 → CH4 + 2H2O; ΔH°298K = −165.00 kJ/mol.
CO2 + 3H2 → CH3OH + H2O; ΔH°298K = −90.70 kJ/mol.
CO2 + H2 → CO + H2O; ΔH°298K = +41.19 kJ/mol.
The catalytic reduction of CO2 to CO, CH3OH, CH4, or higher hydrocarbons is considered an attractive approach for recycling CO2, whereby the energy from intermittent renewable sources is stored at high density in the form of chemical bonds, mainly C–H. As one of the widely studied approaches, direct photochemical reduction of CO2 using nano-sized hybrid heterogeneous photocatalysts, on which light is absorbed and charge separation takes place, occurs at the solid–liquid or solid–gas interface. The photogenerated charges are then transported to the surface, where the active sites mediate the catalytic reactions [8,9,10,11,12,13,14].
The electrochemical reduction of CO2 on solid-state electrodes modified with nanocatalysts were recently shown to have good potential in yielding valuable products, e.g., acetate and ethylene, attracting much attention [15,16,17]; moreover, a large number of molecular metal catalysts were recently studied with the aim to reductively convert CO2 into fuels using solar energy and electricity as the driving force [17,18,19]. The multiple accessible oxidation states of transition-metal complexes renders them suitable candidates for mediating multielectron redox processes such as CO2 reduction, and the reactions are readily studied under homogeneous conditions using various conventional electrochemical and spectroscopic techniques. With the exception of the Lehn-type rhenium(I) tricarbonyl catalysts, i.e., Re(bpy)(CO)3Cl (bpy = 2,2′-bipyridine, see Section 2.1), which are capable of acting as both electro- and photocatalysts of CO2 reduction, the majority of the catalysts discussed herein work as stand-alone CO2 reduction electrocatalysts, although the photochemical CO2 reduction mediated by some of these catalysts was also reported in the presence of suitable photosensitizers and sacrificial donors [17,18,19,20]. On the other hand, chemical technologies or processes that utilize CO2 as a renewable, non-toxic, and cost-efficient C-1 feedstock via coupling with various nucleophiles to produce more valuable chemicals, particularly more reduced and energetic products, also gained much attention [21,22,23]. However, given the wide variety of related processes, the discussion below focuses on transition-metal catalysts for the electroreduction and functionalization of CO.
2. Electrochemical Reduction of CO2 on Molecular Transition-Metal Catalysts
The substantial energy barrier in activating CO2 is reflected by the highly negative formal reduction potential of −2.14 V vs. saturated calomel electrode (SCE) for the one-electron reduction of CO2 to CO2− [18,19,24], where a large overpotential is required for the rapid reduction of CO2 to occur, largely as a result of the structural difference between the linear CO2 and bent CO2– [19,20,24,25,26]. Alternatively, CO2 can be reduced more easily via proton-assisted multiple electron transfer, which is more favorable thermodynamically [19,20,24,25,26]. Equations (6)–(11) show the typical processes for the reduction of CO2 to different products and the corresponding reduction potentials vs. SCE [24]. These alternative pathways in turn affect the nature of products.
CO2 + 2H+ + 2e− → HCO2H; −0.85 V (vs. SCE).
CO2 + 2H+ + 2e− → CO + H2O; −0.77 V.
CO2 + 4H+ + 4e− → C + 2H2O; −0.44 V.
CO2 + 4H+ + 4e− → HCHO + H2O; −0.72 V.
CO2 + 6H+ + 6e− → CH3OH + H2O; −0.62 V.
CO2 + 8H+ + 8e− → CH4 + 2H2O; −0.48 V.
2.1. 4d and 5d Metal CO2 Reduction Electrocatalysts
A number of heavier transition-metal complexes were studied for their activity on CO2 electroreduction. In particular, 4d and 5d transition-metal complexes bearing mainly bipyridyl and bidentate phosphine ligands were widely examined for their activity toward the two-electron reduction of CO2. As these complexes are usually substitution labile and the derivatization of the related ligands are well reported, these heavier transition-metal CO2 reduction catalysts are generally well characterized, and their catalytic properties are more readily tunable. Therefore, the corresponding reaction mechanisms and intermediates are more widely reported. Examples of 4d and 5d metal catalysts are summarized in Table 1 and Table 2.
A number of 4d and 5d 2,2′-bipyridine complexes (Re, Rh, Ru, and Os) catalyze the two-electron electroreduction of CO2 to CO or formic acid/formate. For example, Re(bpy)(CO)3Cl (−1.49 V vs. SCE, 98% Faradaic efficiency (FE) in dimethylformamide (DMF)/H2O) catalyzes CO2 reduction to produce CO [27]. A series of related Re complexes with different substituted bpy and labile ligands, fac-Re(4,4′-R-bpy)(CO)3X (bpy = 2,2′-bipyridine, R = OCH3, CH3, tBu, H, CN, CF3; X = Cl, Br, py(OTf), or CH3CN(OTf)) were studied for their electrocatalytic efficiency with and without addition of a proton source (PhOH, CH3COOH, CF3CH2OH) [28]. The results showed that the catalytic activity and overpotential increase with the electron-donating ability of the bpy substituents and the addition of acid positively shifted the catalytic current response of Re(tBu-bpy)(CO)3Cl (~170 mV) [28]. Additionally, cis-[Rh(bpy)2×2]+ (X = Cl or OTf) reduces CO2 to formate (−1.55 V vs. SCE, 64% FE, turnover number (TON) = 6.8–12.3) [29]. A series of functionalized Re(I) tricarbonyl catalysts, {Re[bpyMe(ImMe)](CO)3Cl}PF6 (Scheme 1), bearing redox-active imidazolium groups in the secondary coordination sphere, were reported to demonstrate improved catalytic activities as compared to the reference Re(bpy)(CO)3Cl catalyst such that the potentials of the reductive catalytic current was approximately 170 mV less negative, and the Faradaic efficiency for CO generation was increased by 19% [30]. Recently, a water-soluble rhenium(I) tricarbonyl electrocatalyst with two hydroxymethyl moieties, [Re(CH2OH)–OH2]+ (Scheme 1), was studied for CO2 reduction in aqueous solutions. Controlled-potential electrolysis using the catalyst at −1.1 V vs. normal hydrogen electrode (NHE) in a pH 6.9 CO2-saturated aqueous solution yielded CO and HCOOH at selectivities of 95% and 4%, respectively [31].
Ru(bpy)2(CO)X (X = CO or Cl) and [M(bpy)2(CO)H]+ (M = Os and Ru) reduce CO2 to CO/H2/HCOO− and CO, respectively [32,33]. Recently, trans-Cl-Ru(mesbpy)(CO)2Cl2 (mesbpy = 6,6′-dimesityl-2,2′-bipyridine) was reported to reduce CO2 to CO and formate with turnover frequencies (TOF) = 1300 s−1 [34]. Phosphine complexes of 4d and 5d transition metals such as Rh(dppe)2Cl (dppe = 1,2-bis(diphenylphosphino)ethane) (−1.55 V vs. SCE, 42% FE) and {m-(triphos)2-[Pd(CH3CN)]2}(BF4)4} (triphos = bis(diphenylphosphinoethyl)phenylphosphine) reduce CO2 to formate and CO (Table 2) [35,36,37].
2.2. 3d Metal CO2 Reduction Electrocatalysts
Although the 4d and 5d CO2 reduction catalysts of bidentate ligands, especially bipyridyl ligands, attracted much attention, the relatively low abundancy of these metals prompted the development of cost-efficient catalysts and, subsequently, the study of 3d transition-metal catalysts for CO2 reduction. Probably as a result of their better stability in the reduced state, a number of 3d metal complexes bearing macrocyclic and tetradentate chelating ligands were investigated for activity toward CO2 electroreduction. Cobalt, iron, and nickel tetraaza macrocyclic complexes were reported to catalyze the electrochemical reduction of CO2. Eisenberg first reported the use of cobalt and nickel tetraaza macrocyclic complexes as electrocatalysts for CO2 reduction (at −1.3 to −1.6 V vs. SCE) to CO or CO/H2 mixture in aqueous acetonitrile at nearly quantitative FE (Scheme 2) [38]. Mechanistic studies for catalytic CO2 reduction were also reported using Co complexes of similar ligands [39,40,41,42].
[Ni(cyclam)]2+ and other structurally related Ni complexes (Scheme 3) exhibit remarkable efficiency and selectivity for reduction of CO2 to CO on a mercury electrode (at −0.86 V vs. SCE) in purely aqueous solution (pH 4–5) [43,44,45]. Product selectivity depends on the pH and potential at which the controlled potential electrolysis was performed [46,47]. Electrolysis of a CO2 saturated solution (pH 4.1) containing [Ni(cyclam)]2+ at −1.00 V vs. SHE (overpotential = 640 mV) on an Hg electrode yields CO almost quantitatively at 96% FE and a TON of 116 [43,44]. N-substituted Ni cyclams exhibit similar activity and produce CO at high FE of 84–92% at pH 5 and 550 mV overpotential. Formic acid is obtained similarly, with CO as the byproduct, using the dimeric {[Ni(cyclam)]2}4+ and related derivative (Scheme 3) at 75% and 68% FE, respectively, in DMF on an Hg electrode at −1.4 V vs. SCE (460 mV overpotential) [48].
The CO2 reduction activity of CoII tetraazamacrocycle [Co(CR)]2+ (CR = 2,12-dimethyl-3,7,11,17- tetra-azabicyclo [11.3.1]-heptadeca-1(17),2,11,13,15-pentaene) was studied by Tinnemans et al. and Che et al. (Scheme 4) [49,50]. Electrocatalysis using the complex on a carbon electrode yields CO selectively at 20–30% FE in acetonitrile (overpotential = 940 mV). Peters et al. later reported an enhanced FE of 45% in the presence of 10 M H2O. A related Fe pentaazamacrocycle [FeIII(LN5)Cl]+ (Scheme 4) was recently reported by Lau and co-workers to catalyze the electroreduction of CO2 to HCOOH in DMF at −1.25 V vs. SCE (310 mV overpotential) on a glassy carbon electrode, producing formic acid at 75% FE with a TOF of 0.12 s−1 [51].
Iron(0) porphyrin complexes were widely studied for electrocatalytic reduction of CO2 to CO by Savéant et al. and are among the most efficient homogeneous molecular catalysts in aprotic solvent (DMF and acetonitrile (can)) [52,53,54,55]. Fe(TPP) was found to selectively catalyze the reduction of CO2 to CO at −1.8 V vs. SCE in DMF (Scheme 5, R = H) [56,57,58,59]. The catalysis is significantly enhanced by the presence of weak Brönsted acids (water, trifluoroethanol, and phenol), as well as Lewis acids. For example, electrolysis using Fe(TPP) at −1.46 V vs. SHE on a mercury pool electrode in DMF, with phenol as the added acid, yields CO between 100% and 94% at varied phenol concentrations (0.1 to 1 M). When 1-propanol (6.7 M) is added instead, formic acid (35% yield) and CO (60% yield) were obtained on a mercury pool at −1.7 V vs. SCE, along with minor byproducts such as H2 and oxalate [59]. Fujita et al. later reported that iron(IV) and cobalt(III) corrole complexes similarly catalyze the reduction of CO2 to CO at −1.7 V vs. SCE under homogeneous conditions (Scheme 5) [60,61].
Cobalt porphyrin (CoTPP) and phtalocyanine (CoPc) complexes were also found to be catalytically active in water on carbon electrodes film-coated with the complexes [24,62,63,64,65]. By using the Co(TPP) (Scheme 6) deposited on a carbon black gas-diffusion electrode, CO is produced at −0.76 V vs. SHE (overpotential = 230 mV) at 97% FE in a 0.5 M KHCO3 solution under a high CO2 pressure of 20 atm. Similarly, CO is produced in 80% FE at pH 4.6 with TOF = 140 h−1 and TON = 1100 using cobalt chlorin (CoCh) deposited on carbon nanotubes (700 mV overpotential) [66]. Cobalt protoporphyrin (CoPP) deposited on pyrolytic graphite also catalyzes the CO2-to-CO conversion at a low pH of 3 at 60% FE and 500 mV overpotential under 10 atm of CO2, while 2.5% CH4 was obtained when the pH was lowered to 1 [24].
The introduction of intramolecular phenolic protons at the ortho and ortho’ positions of Fe(TPP) was found to be effective in enhancing the efficiency of CO2 electroreduction to CO and the catalyst durability (Scheme 7) [53,54]. The catalyst (FeTDMPP) operates at a lower overpotential (450 mV) compared with FeTPP (600 mV), while the substitution of a trimethylammonio group (Fe-p-TMA) not only increases the CO conversion efficiency and selectivity, but also catalyzes the reduction of CO2 to methane [55,67,68].
Heterogeneous catalytic materials based on solid-support or supramolecular porphyrin catalysts of Co, Cu, and Fe for the reduction of CO2 in aqueous solutions were explored. A catalytic electrode constructed from two-dimensional (2D) covalent organic frameworks (COFs) of cobalt tetrakis(4-aminophenyl)porphyrin (Co(TAP)) on porous conductive carbon fabric was found to demonstrate significantly improved (26-fold) catalytic activity for CO2-to-CO conversion at an overpotential of −0.55 V (FE of 90%, TON up to 290,000, and initial TOF of 9400 h−1) at pH 7 with respect to the molecular cobalt complex under the same condition [69]. A similar catalytic electrode was prepared by depositing a porous organic cage (Fe-PB), which bears six Fe(TPP) centers, onto a glassy carbon electrode coated with carbon nanotubes (CNTS/GCE) [70]. Both electrochemically active surface area (3.7 nmol EA-Fe∙cm−2 vs. 2.5 nmol EA-Fe∙cm−2) and mass transport were increased for Fe-PB/CNTs/GCE with respect to the Fe(TPP)/CNTs/GCE containing an equivalent amount of Fe(TPP), resulting in an enhanced catalytic current response, CO/H2 selectivity, and product turnover (TON = 55,250 after 24 h and TOF = 0.64 s−1 vs. TON = 32,770 after 24 h and TOF = 0.38 s−1) for controlled potential electrolysis at −0.63 V vs. RHE. Cu(II)-5,10,15,20-tetrakis-(2,6-dihydroxyphenyl)porphyrin) Cu(TDMPP) deposited on a commercial porous polytetrafluoroethylene-treated carbon fiber paper reduces CO2 to hydrocarbons (methane and ethane) at −0.976 V vs. RHE at 44% FE [71].
A number of 3d metal complexes (Co, Fe, and Ni) bearing tetradentate chelating ligands of N, C, and O donor atoms were reported to be active electrocatalysts for the selective reduction of CO2 to CO. The N-heterocyclic carbene–isoquinoline complexes [Ni(Prbimiq1)]2+ (Prbimiq1 = bis(3-(imidazolyl)isoquinolinyl)propane) (Scheme 8) were also reported to catalyze CO2-to-CO electroreduction, yielding CO at an optimal FE of 90% (overpotential = 840 mV) on a glassy carbon electrode; however, the catalytic efficiency was found to decrease significantly upon prolonged electrolysis [72]. Co-azacalix[4](2,6)pyridine catalysts (Scheme 8) containing methyl- and allyl- substituted macrocyclic aminopyridine ligands were reported to selectively produce (optimal FE = 98%) CO in a CO2-saturated DMF solution with 1.2 M trifluoroethanol on a glassy carbon electrode (680 mV overpotential) at a TON of 6.2, and the pendant NH moiety was suggested to stabilize the CoI–CO2 adduct [73].
The FeI catalyst containing 2,9-bis(2-hydroxyphenyl)-1,10-phenanthroline [FeI(dophen)]− yields HCOOH at 74% FE, as well as oxalate (7%) and CO (13%) as minor byproducts, upon electrolysis in dimethyl sulfoxide (DMSO) at −1.76 V vs. SCE on a carbon electrode (Scheme 9) [74]. An iron(III) complex of 6,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine (Fe(III)Cl(tbudhbpy)) was also reported to catalyze the reduction of CO2 to formate in the presence of an added proton source (PhOH; FE = 68%, TON = 2.7, t = 10 h), whereas, in the absence of acid, only CO is formed (FE = 1.1%, TON = 3, t = 15 h) [75].
Recently, Lau and Robert reported Co and Fe quaterpyridine complexes (Scheme 10), [M(qpy)(OH2)2]2+ (qpy = 2,2′:6′,2”:6”,2‴-quaterpyridine), as catalysts for CO2-to-CO electroreduction in acetonitrile with a selectivity of >95% in the presence of phenol at low overpotentials of 140 and 240 mV, respectively, and an impressive turnover frequency (TOF) of 3.3 × 104 s−1 was reported for the Fe catalyst [76].
Chardon-Noblat and Deronzier et al. reported fac-Mn(L)(CO)3Br complexes (L = 2,2′-bipyridine or 4,4′-dimethyl-2,2′-bipyridine) as an active electrocatalyst for CO2 reduction (Scheme 11) [77,78]. Controlled-potential electrolysis with Mn(L)(CO)3Br (L = 2,2′-bipyridine) at −1.40 vs. SCE (420 mV overpotential) in ACN with 5% H2O converts CO2 into CO quantitatively, while CO is also selectively obtained (TON = 34) with R = Me. Kubiak et al. later reported the activity of Mn(L)(CO)3Br bearing other substituents (R) on L in the presence of weak acids [79]. With R = t-butyl, CO is produced at quantitative FE (estimated TOF of 340 s−1) in controlled-potential electrolysis at −2.2 V vs. SCE with 1.4 M trifluoroethanol. When L = 6,6′-dimesityl-2,2′- bipyridine, CO is selectively produced at high FE in electrolysis and a TOF of 5 × 103 s−1 in acetonitrile also containing 1.4 M trifluoroethanol.
When pendent proton sources are introduced in proximity to the metal center on Mn(L)(CO)3Br, i.e., L = 4-phenyl-6-(1,3-dihydroxybenzen-2-yl)-2,2′-bipyridine (Scheme 11), the catalyst is active for CO2 reduction in acetonitrile at −1.8 V vs. SCE without added acids, producing both CO (70% FE) and HCOOH (22% FE) [80,81]. A reaction pathway involving the formation of Mn hydride via intramolecular proton transfer from the phenolic moiety was suggested. Interestingly, a similar Mn(L)(CO)3Br with a single pendent hydroxyphenyl substituent on the bpy ligand selectively produces CO at 76% FE (at 540 mV overpotential) in electrolysis in acetonitrile containing 5% H2O [82]. The reactivity of the similarly structured Mn(I)-tricarbonyl catalysts bearing two and three acidic hydroxyphenyl functions was also compared in the presence of added acids (H2O, 2,2,2-trifluoroethanol (TFE), and phenol), and the product (CO, HCOOH, and H2) distribution was dependent on the strength of the added acid. [81,83]. Recently, a series of fac-Mn(CO)3 catalysts bearing imidazolium-functionalized bipyridine, {Mn[bpyMe(lmMe)](CO)3Br}+ (Scheme 11), were found to demonstrate a superior reactivity (FE = approximately 70%) toward CO2 reduction in comparison to the dimesityl analogue (FE = 49.6%) at a mild potential (−1.44 V vs. SCE) in acetonitrile with 9.25 M H2O [84]. The imidazolium moiety is suggested to favor the formation of a local hydration shell which promotes more efficient protonation of the reaction intermediates [84].
An Mn-MeCN/MWCNT catalytic cathode ([Mn-MeCN] = [Mn{4,4′-di(1H-pyrrolyl- 3-propylcarbonate)-2,2′-bipyridine}(CO)3(MeCN)]+ and MWCNT = multi-walled carbon nanotube), prepared by loading the functionalized fac-Mn(CO)3 catalysts (Scheme 11) onto conductive MWCNTs, promotes CO2 reduction at a low overpotential of 100 mV in the presence of K+ to produce CO at a steady rate for 48 h at −0.39 V (vs. RHE) [85]. The activities toward electrochemical CO2 reduction of a similar catalyst bearing an amine-functionalized bpy ligand, fac-Mn(apbpy)(CO)3Br (apbpy = 4-(4-aminophenyl)-2,2′-bipyridine), were studied under both heterogeneous and homogeneous conditions [86,87]. A glassy carbon electrode (GCE) functionalized with the complex was found to produce CO electrocatalytically in aqueous acetonitrile at a TON 30 times higher than that in homogeneous condition [86]. When grafted electrochemically onto carbon cloth, the catalyst also reduces CO2 to syngas in aqueous solution (FE for CO and H2 = 60% and 40%, respectively) at −1.35 V and a TON of up to 33,200 in 10 h [87].
Multinuclear catalysts of first-row transition metals draw much attention, as a their multiple reaction centers are considered potentially more effective for mediating the simultaneous reduction of more than one CO2, thus leading to the formation of C-2 (or higher) products. Kubiak reported a number of multinuclear nickel and copper phosphine complexes as electrocatalysts for CO2 reduction (Scheme 12). The catalyst [Ni2(μ-CNMe)(CNMe)2(dppm)2] (dppm = 1,1-bis(diphenylphosphino)methane) operates at −0.87 V vs. SCE [88]. The similar dinuclear nickel complex [Ni2(μ-CNR)(CNR)2(μ-dppa)2] (dppa = bis(diphenylphosphino)amine; CNR = isocyanide ligand) also catalyzes CO2 reduction [89]. However, these catalysts suffer from carbonylation upon extended cycles of catalysis [88]. Trinuclear nickel clusters [Ni3(μ3-I)(μ3-CNR)(μ2-dppm)3] were also found to catalyze CO2 selective reduction to CO and CO3− at −1.08 to −1.18 V vs. SCE [90,91]. The binuclear copper complexes [Cu2(μ-PPh2bpy)(MeCN)2][PF6]2 (at −1.53 V vs. SCE) and [Cu2(μ-PPh2bpy)(py)2][PF6]2 (PPh2bpy = 3-diphenylphosphino-2,2′-dipyridyl) were also reported to be active electrocatalysts for CO2 reduction [92]. More recently, the [FeFe]-hydrogenase model, [(μ-bdt)Fe2(CO)6] (bdt = benzene-1,2-dithiolato), was found to demonstrate distinctive activity for electroreduction of CO2 in acetonitrile in the presence of CH3OH or H2O as the proton source at an estimated maximum TOF of 195 s−1 [93]. Controlled-potential electrolysis using the catalyst under optimized conditions produces HCOOH at a good Faradaic yield of 88% as the major product (selectivity ≈ 81%), together with a small amount of CO (selectivity ≈ 11%) and H2 (selectivity ≈ 8%) [93].
Despite the formation of C-2 or longer hydrocarbons being reported using electrode modified with nanocatalysts, C2 products are less common for the electroreduction of CO2 on molecular catalysts. Apart from the abovementioned Cu(TDMPP) supported on porous polytetrafluoroethylene-treated carbon fiber paper, which reduces CO2 to hydrocarbons (methane and ethane) [71], so far, only a dinuclear copper(I) complex, [Cu2(L)]2+ (HL = [N-(2-mercaptopropyl)-N,N-bis(2-pyridylmethyl)amine]), was reported to produce a tetranuclear copper(II) complex bearing two CO2-derived oxalate groups upon reaction with atmospheric CO2 [94]. Treatment of the oxalate-bridged copper(II) complex with a soluble lithium salt in acetonitrile quantitatively produces lithium oxalate, and a subsequent electrochemical reduction of the copper(II) complex regenerates the initial dinuclear copper(I) compound, which reacts again with CO2 and demonstrates six turnovers (producing 12 equivalents of oxalate) at an applied potential of −0.03 V vs. NHE in 7 h [94].
3. Functionalization of Carbon Dioxide
In parallel, chemical processes for using CO2 as a renewable, non-toxic, and cost-efficient feedstock for producing fine chemicals are emerging [95,96,97,98,99,100]. Confined by the kinetic inertness of CO2, widely applied CO2 functionalization processes include the industrial preparation of urea, salicylic acid, inorganic carbonates, cyclic/acyclic organic carbonates, and pigments, as well as application as an additive in methanol synthesis [95,96,97,98,99,100,101,102]. Therefore, novel and efficient catalytic processes for CO2 functionalization are highly valuable. Two major approaches for the functionalization of CO2, i.e., “horizontal” and “diagonal” (reductive) approaches, evolved, and they both require the use of high-energy co-reactants, catalysts, and often stringent reaction conditions to overcome the kinetic barrier (Scheme 13) [96,97,98,99,100,101,102,103,104,105].
3.1. Horizontal Functionalization of CO2
A number of metal-catalyzed reactions, which utilize CO2 as a single-carbon (C-1) feedstock, were reported, and they require usually high pressure and temperature to proceed. In these reactions, functionalization occurs “horizontally” via simple bond formation without changing the formal oxidation state of the CO2 carbon, typically giving products such as R–X–C(O)O–R’ (X = O, N, or C) with limited variety (Schemes 13 and 18). For example, porphine [106,107,108,109,110,111,112] and salen-type [113,114,115,116,117,118,119] complexes catalyze the coupling of CO2 with epoxide to cyclic [113,114] and polycarbonates [115,116,117,118,119].
The reactions of CO2 with amine-containing nucleophiles were studied (Scheme 14), mainly using precious-metal (Ag [120,121], Pd [122,123,124], and Ru [125,126] or organotin [127,128] (Sn)) catalysts, e.g., (a) oxazolidinones from aziridine [129], 1,2-aminoalcohols [127], and α-allenyl amines [124]; (b) benzoxazin-2-ones from o-alkynylanilines [120,121]; (c) carbamates from amine/allylic chloride [122,123], amine/alkyne [130], and N-substituted propargylamine [126]; and (d) urea or urethane from primary amine [128,131]. The carboxylation of carbon nucleophiles with CO2 was studied (Scheme 15), mainly with Cu [132,133], Ni [133,134,135,136,137,138,139], and Pd [140,141,142,143,144,145] catalysts, e.g., (a) substituted carboxylic acid from arylhalide [139,140], benzyl chloride [134], alkynes [135,136], allenes [122], and alkenes [132,146]; (b) ester from alkyne/allylic chloride [133], phenylpyridine [147], and aryl methane [148]; and (c) lactone from diene [142,149], 2-hydroxystyrene [144], allenes [138], and alkynes [137,138]. Similar CO2 functionalization was also performed using organocatalysts, e.g., (a) organic N-heterocyclic bases (DBU) for coupling with primary amino alcohol to oxazolidines [150,151,152] and carboxylation of cyclopentadiene [153]; (b) N-heterocyclic carbenes (iPrNHC) for coupling with epoxide, aziridine, or propagyl alcohol to yield heterocycles and carbonates [154,155,156]; and (c) organosalt or ionic liquid with aziridine or epoxide [157,158].
3.2. Electro- and Photocatalytic CO2 Functionalization
In contrast to chemical approaches, electro- and photocatalytic functionalizations of CO2, which usually proceed under mild conditions (room temperature and 1 atm), were much less investigated. In these reactions, CO2 carbon was mostly incorporated with substrates without changing its oxidation state. Duñach et al. reported the Ni-catalyzed electrochemical carboxylation of alkynes and diynes, as well as electro-coupling with epoxides and aziridines under 1 atm CO2, using Ni-L (L = bpy, cyclam, PMDTA) catalysts and an Mg sacrificial anode (Scheme 14 and Scheme 16) [159,160,161,162,163]. Notably, electrocatalytic formylation of dimethyl amine (DMA) to DMF was reported using Ru(bpy)2(CO)2 [164]. Visible-light-driven carboxylation of aryl halides [165] and alkenes [166] was carried out using Pd(OAc)2/PR3/Ir(ppy)2(dtbpy)(PF6) [165] and Rh(PPh3)3X or [Rh(PR3)2Cl]2/[Ru(bpy)3]2+ (R = Cy or Ar; X = OAc, Cl, or H) [166] with iPr2EtN as the sacrificial donor (Scheme 17). Ultraviolet (UV)-driven α-carboxylation of tertiary N-benzylpiperidines C5H10N(CH2Ph) and carboxylation of alkenes were achieved using p-terphenyl/trifluoroacetate [167] and diphenylxanthone/Cu(iPrNHC)Cl [168], respectively.
3.3. Diagonal (Reductive) CO2 Functionalization
While, in the above “horizontal” transformations, the CO2 carbon is functionalized with its formal oxidation state unchanged, the “vertical” reduction of CO2 very often results in a lower oxidation state and, thus, higher-energy products, such as CO, formic acid/formate, methanol, and methane, in which no new bonds, other than C–H, are formed to the CO2 carbon (Scheme 18) [19,23,24,25,26]. For CO2 functionalization processes to be more versatile and widely applied, the spectrum of products directly obtained from CO2 has to be broadened in terms of functionalities and energy. Thus, a “diagonal” reductive approach, where CO2 is reacted in a concerted manner with a nucleophilic functionalization reagent and a reducing agent, was proposed and explored [98,169,170].
Recently, reductive coupling of CO2 with amine-type co-reactants emerged as a novel approach for accessing products bearing deoxygenated carbons, e.g., N–C(O)H, originated from CO2 (Scheme 19). Examples of metal-catalyzed N–H formylation with CO2 are shown in Table 3, and the structures of selected catalysts are shown in Scheme 20. The formylation of an N–H bond of primary or secondary amines to yield formamide derivatives was reported using precious-metal (Ir, Pd, Pt, and Ru) [164,171,172,173,174,175,176] catalysts, e.g., (PPh3)3Ir(CO)Cl [167], PdCl2 [172], [Pt2(μ-dppm)3] (dppm = bis(diphenylphosphino)methane) [173,177], RuCl2(PMe3)4 [178] RuCl2(dppe) (dppe = bis(diphenylphosphino)ethane) [164,179], and Ru(PNP)(CO)(H)Cl (PNP = N,N-bis(2-(diphenylphosphinoethyl)methylamine) (Scheme 20), and H2 as the reductant at a usually moderate to high turnover number (TON). Similar N–H formylation was also explored using non-precious-metal catalysts, particularly with phosphine ligands, e.g., MX2/dmpe (M = Co, Cu, Fe, Ni, and Mn; X= Cl−, CH3CO2−, and acac−; dmpe = bis(dimethylphosphino)ethane) [180], Cu(PPh3)3Cl [141,150], M(BF4)2/PP3 (M = Fe and Co; PP3 = tris[2-(diphenylphosphino)ethyl]phosphine), and Fe(BF4)2/PAr3 (PAr3 = tris(2-(diphenylphosphino)phenyl)phosphine) [19,181,182,183]. A molybdenum silylphosphine hydride complex, [MoH3{Si(Ph)[Ph2PCH2CH2P(Ph)C6H4-o]2}], also catalyzes DMF production from DMA and CO2 [184]. The formylations of aniline (NH2Ar) and ammonia (NH3) were demonstrated using RuCl2(PMe3)4 [185,186], [Ru(triphos)(tmm)] [187], and IrCl(CO)(PPh3)3 [188] with H2 as the reductant. Amine–CO2 reductive coupling was also performed using hydrosilane (R3Si–H) or hydroborane (R2B–H) as the reductant on metal (Cu, Fe, Ni, and Rh) [189,190,191,192,193] catalysts such as [Ni(μ-H)(dippe)]2 (dippe = 1,2-bis(diisopropylphosphino)ethane) [192] and Cu(iPr-NHC)(OtBu) (iPrNHC) [193].
More recently, non-carbonyl or fully deoxygenated products, e.g., formamidines (RN = C(H)NR′2) and methylamines (R2NCH3) were obtained at elevated temperature and pressure (Scheme 21) from o-phenylenediamines with RuCl2(dppe)2 [194], as well as from primary and secondary amines with Ru(acac)3/triphos [195] or Ru(triphos)(tmm) [187], [RuII(dmso)4Cl2]/P(nBu)(Ad)2 [196], and Zn(iPrNHC)Cl [196] using H2 and PhSiH3 as reductants. Recently, reductive coupling of CO2 with amines and o-phenylenediamines was achieved using organo-base (TBD) [197,198], iPrNHC [199,200], and proazaphosphatrane (VBMe) [201] to produce formamides [196,199], formamidines [200], tertiary methylamines, and methylene diamines [198,201] using hydrosilane (PhSiH3) or hydroborane (9-BBN) as reductants (Scheme 22).
4. Summary and Outlook
While CO2 utilization remains a major scientific challenge, it is also a promising means for providing petroleum substitutes, thus achieving sustainable and carbon-neutral resource utilization, in face of the current rising atmospheric CO2 level. The direct reduction and the concerted reductive functionalization of CO2 enable us to assess a broad spectrum of products which are higher in energy and varied in functionality, in comparison with the “horizontal” pathways. However, the search of novel, economic, and environmentally friendly catalysts, which circumvent the current reaction bottlenecks, will be highly important in terms of the access to desirable products, as well as the enhancement of catalyst stability and energy efficiency. Much is to be done for molecular catalysts with respect to understanding how the coordination spheres will facilitate yielding the desired products of varied oxidation states on the CO2-derived carbon, particularly in obtaining C-2 or higher hydrocarbons [30,53,54,55,67,68,71,80,81,82,83,84,85,86,87,94,98,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193]. For the reductive transformations of CO2 to be sustainably driven by renewable energy sources, the exploration of direct photo-driven redox catalysis using systems of photosensitizer–catalyst combinations without precious metals [17,18,19,20] and the development of catalytic electrodes bearing the immobilized macromolecular or molecular catalysts [24,62,63,64,65,69,70,71,85,86,87] will be essential, such that the technology will be further advanced and materialized in the form of photoelectrochemical cells [31].
Author Contributions
Conceptualization, C-F.L.; Blibliographic research, C-F.L. and P-Y.H.; Writing-Original Draft Preparation, C-F.L. and P-Y.H.; Writing-Review & Editing, C-F.L.; Visualization, C-F.L and P-Y.H.
Funding
This research received no external funding. And APC was funded by the Education University of Hong Kong.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ghaib, K.; Nitz, K.; Ben-Fares, F.-Z. Chemical Methanation of CO2: A Review. ChemBioEng Rev. 2016, 3, 266–275. [Google Scholar] [CrossRef]
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent Advances in CO2 Capture and Utilization. ChemSusChem Chem. Sustain. Energy Mater. 2008, 1, 893–899. [Google Scholar] [CrossRef]
- Spinner, N.S.; Vega, J.A.; Mustain, W.E. Recent Progress in the Electrochemical Conversion and Utilization of CO2. Catal. Sci. Technol. 2012, 2, 19–28. [Google Scholar] [CrossRef]
- Wilcox, E.M.; Roberts, G.W.; Spivey, J.J. Direct Catalytic Formation of Acetic Acid from CO2 and Methane. Catal. Today 2003, 88, 83–90. [Google Scholar] [CrossRef]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A Short Review of Catalysis for CO2 Conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Bian, Z.; Das, S.; Wai, M.H.; Hongmanorom, P.; Kawi, S. A Review on Bimetallic Nickel-Based Catalysts for CO2 Reforming of Methane. ChemPhysChem 2017, 18, 3117–3134. [Google Scholar] [CrossRef]
- Buelens, L.C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-Dry Reforming of Methane Intensifies CO2 Utilization via Le Chatelier’s Principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef]
- Habisreutinger, S.N.; Schmidt-Mende, L.; Stolarczyk, J.K. Photocatalytic Reduction of CO2 on TiO2 and Other Semiconductors. Angew. Chem. Int. Ed. 2013, 52, 7372–7408. [Google Scholar] [CrossRef]
- Schreier, M.; Héroguel, F.; Steier, L.; Ahmad, S.; Luterbacher, J.S.; Mayer, M.T.; Luo, J.; Grätzel, M. Solar Conversion of CO2 to CO Using Earth-Abundant Electrocatalysts Prepared by Atomic Layer Modification of CuO. Nat. Energy 2017, 2, 17087. [Google Scholar] [CrossRef]
- Sorcar, S.; Hwang, Y.; Grimes, C.A.; In, S.-I. Highly Enhanced and Stable Activity of Defect-Induced Titania Nanoparticles for Solar Light-Driven CO2 Reduction into CH4. Mater. Today 2017, 20, 507–515. [Google Scholar] [CrossRef]
- Park, H.; Ou, H.-H.; Colussi, A.J.; Hoffmann, M.R. Artificial Photosynthesis of C1--C3 Hydrocarbons from Water and CO2 on Titanate Nanotubes Decorated with Nanoparticle Elemental Copper and CdS Quantum Dots. J. Phys. Chem. A 2015, 119, 4658–4666. [Google Scholar] [CrossRef] [PubMed]
- Sorcar, S.; Thompson, J.; Hwang, Y.; Park, Y.H.; Majima, T.; Grimes, C.A.; Durrant, J.R.; In, S.-I. High-Rate Solar-Light Photoconversion of CO2 to Fuel: Controllable Transformation from C1 to C2 Products. Energy Environ. Sci. 2018, 11, 3183–3193. [Google Scholar] [CrossRef]
- Li, N.; Wang, B.; Si, Y.; Xue, F.; Zhou, J.; Lu, Y.; Liu, M. Toward High-Value Hydrocarbon Generation by Photocatalytic Reduction of CO2 in Water Vapor. ACS Catal. 2019, 9, 5590–5602. [Google Scholar] [CrossRef]
- Thampi, K.R.; Lucarelli, L.; Kiwi, J. Characterization of a Ruthenium/Titania Catalyst for Selective Methanation at Room Temperature and Atmospheric Pressure. Langmuir 1991, 7, 2642–2648. [Google Scholar] [CrossRef]
- Yang, X.; Fugate, E.A.; Mueanngern, Y.; Baker, L.R. Photoelectrochemical CO2 Reduction to Acetate on Iron--Copper Oxide Catalysts. ACS Catal. 2016, 7, 177–180. [Google Scholar] [CrossRef]
- Jeon, H.S.; Kunze, S.; Scholten, F.; Roldan Cuenya, B. Prism-Shaped Cu Nanocatalysts for Electrochemical CO2 Reduction to Ethylene. ACS Catal. 2017, 8, 531–535. [Google Scholar] [CrossRef]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc. Chem. Res. 2009, 42, 1983–1994. [Google Scholar] [CrossRef]
- Windle, C.D.; Perutz, R.N. Advances in molecular photocatalytic and electrocatalytic CO2 reduction. Coord. Chem. Rev. 2012, 256, 2562–2570. [Google Scholar] [CrossRef]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 2016, 7, 70–88. [Google Scholar] [CrossRef]
- Hunt, A.J.; Sin, E.H.K.; Marriott, R.; Clark, J.H. Generation, capture, and utilization of industrial carbon dioxide. ChemSusChem Chem. Sustain. Energy Mater. 2010, 3, 306–322. [Google Scholar] [CrossRef] [PubMed]
- Langanke, J.; Wolf, A.; Hofmann, J.; Böhm, K.; Subhani, M.A.; Müller, T.E.; Leitner, W.; Gürtler, C. Carbon dioxide (CO2) as sustainable feedstock for polyurethane production. Green Chem. 2014, 16, 1865–1870. [Google Scholar] [CrossRef]
- Liu, Q.; Wu, L.; Jackstell, R.; Beller, M. Using carbon dioxide as a building block in organic synthesis. Nat. Commun. 2015, 6, 5933. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.D.; Grills, D.C.; Muckerman, J.T.; Polyansky, D.E.; Fujita, E. Toward more efficient photochemical CO2 reduction: Use of scCO2 or photogenerated hydrides. Coord. Chem. Rev. 2010, 254, 2472–2482. [Google Scholar] [CrossRef]
- Shen, J.; Kortlever, R.; Kas, R.; Birdja, Y.Y.; Diaz-Morales, O.; Kwon, Y.; Ledezma-Yanez, I.; Schouten, K.J.P.; Mul, G.; Koper, M.T.M. Electrocatalytic reduction of carbon dioxide to carbon monoxide and methane at an immobilized cobalt protoporphyrin. Nat. Commun. 2015, 6, 8177. [Google Scholar] [CrossRef]
- Liu, X.; Inagaki, S.; Gong, J. Heterogeneous molecular systems for photocatalytic CO2 reduction with water oxidation. Angew. Chem. Int. Ed. 2016, 55, 14924–14950. [Google Scholar] [CrossRef] [PubMed]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re (bipy)(CO)3Cl (bipy= 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Clark, M.L.; Cheung, P.L.; Lessio, M.; Carter, E.A.; Kubiak, C.P. Kinetic and Mechanistic Effects of Bipyridine (bpy) Substituent, Labile Ligand, and Brønsted Acid on Electrocatalytic CO2 Reduction by Re(bpy) Complexes. ACS Catal. 2018, 8, 2021–2029. [Google Scholar] [CrossRef]
- Bolinger, C.M.; Story, N.; Sullivan, B.P.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by 2, 2′-bipyridine complexes of rhodium and iridium. Inorg. Chem. 1988, 27, 4582–4587. [Google Scholar] [CrossRef]
- Sung, S.; Kumar, D.; Gil-Sepulcre, M.; Nippe, M. Electrocatalytic CO2 Reduction by Imidazolium-Functionalized Molecular Catalysts. J. Am. Chem. Soc. 2017, 139, 13993–13996. [Google Scholar] [CrossRef]
- Nakada, A.; Ishitani, O. Selective Electrocatalysis of a Water-Soluble Rhenium (I) Complex for CO2 Reduction Using Water as an Electron Donor. ACS Catal. 2017, 8, 354–363. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. The effect of pH on the formation of CO and HCOO−. Organometallics 1987, 6, 181–186. [Google Scholar] [CrossRef]
- Bruce, M.R.M.; Megehee, E.; Sullivan, B.P.; Thorp, H.; O’Toole, T.R.; Downard, A.; Meyer, T.J. Electrocatalytic reduction of carbon dioxide by associative activation. Organometallics 1988, 7, 238–240. [Google Scholar] [CrossRef]
- Machan, C.W.; Sampson, M.D.; Kubiak, C.P. A molecular ruthenium electrocatalyst for the reduction of carbon dioxide to CO and formate. J. Am. Chem. Soc. 2015, 137, 8564–8571. [Google Scholar] [CrossRef] [PubMed]
- Slater, S.; Wagenknecht, J.H. Electrochemical reduction of carbon dioxide catalyzed by Rh(diphos)2Cl. J. Am. Chem. Soc. 1984, 106, 5367–5368. [Google Scholar] [CrossRef]
- DuBois, D.L.; Miedaner, A.; Haltiwanger, R.C. Electrochemical reduction of CO2 catalyzed by [Pd(triphosphine)(solvent)](BF4)2 complexes: Synthetic and mechanistic studies. J. Am. Chem. Soc. 1991, 113, 8753–8764. [Google Scholar] [CrossRef]
- Raebiger, J.W.; Turner, J.W.; Noll, B.C.; Curtis, C.J.; Miedaner, A.; Cox, B.; DuBois, D.L. Electrochemical reduction of CO2 to CO catalyzed by a bimetallic palladium complex. Organometallics 2006, 25, 3345–3351. [Google Scholar] [CrossRef]
- Fisher, B.J.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361–7363. [Google Scholar] [CrossRef]
- Fujita, E. Photochemical carbon dioxide reduction with metal complexes. Coord. Chem. Rev. 1999, 185, 373–384. [Google Scholar] [CrossRef]
- Fujita, E.; Szalda, D.J.; Creutz, C.; Sutin, N. Carbon dioxide activation: Thermodynamics of carbon dioxide binding and the involvement of two cobalt centers in the reduction of carbon dioxide by a cobalt (I) macrocycle. J. Am. Chem. Soc. 1988, 110, 4870–4871. [Google Scholar] [CrossRef]
- Fujita, E.; Creutz, C.; Sutin, N.; Szalda, D.J. Carbon dioxide activation by cobalt (I) macrocycles: Factors affecting carbon dioxide and carbon monoxide binding. J. Am. Chem. Soc. 1991, 113, 343–353. [Google Scholar] [CrossRef]
- Creutz, C.; Schwarz, H.A.; Wishart, J.F.; Fujita, E.; Sutin, N. Thermodynamics and kinetics of carbon dioxide binding to two stereoisomers of a cobalt (I) macrocycle in aqueous solution. J. Am. Chem. Soc. 1991, 113, 3361–3371. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.P.; Ruppert, R.; Sauvage, J.P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Fujita, E.; Haff, J.; Sanzenbacher, R.; Elias, H. High Electrocatalytic Activity of RRSS-[NiIIHTIM](ClO4)2 and [NiIIDMC](ClO4)2 for Carbon Dioxide Reduction (HTIM= 2,3,9,10-Tetramethyl-1,4,8,11-tetraazacyclotetradecane, DMC = C-meso-5,12-Dimethyl-1,4,8,11-tetraazacyclotetradecane). Inorg. Chem. 1994, 33, 4627–4628. [Google Scholar] [CrossRef]
- Froehlich, J.D.; Kubiak, C.P. Homogeneous CO2 reduction by Ni(cyclam) at a glassy carbon electrode. Inorg. Chem. 2012, 51, 3932–3934. [Google Scholar] [CrossRef]
- Schneider, J.; Jia, H.; Kobiro, K.; Cabelli, D.E.; Muckerman, J.T.; Fujita, E. Nickel(II) macrocycles: Highly efficient electrocatalysts for the selective reduction of CO2 to CO. Energy Environ. Sci. 2012, 5, 9502–9510. [Google Scholar] [CrossRef]
- Collin, J.P.; Jouaiti, A.; Sauvage, J.P. Electrocatalytic properties of Ni(cyclam)2+ and Ni2(biscyclam)4+ with respect to CO2 and H2O reduction. Inorg. Chem. 1988, 27, 1986–1990. [Google Scholar] [CrossRef]
- Tinnemans, A.H.A.; Koster, T.P.M.; Thewissen, D.; Mackor, A. Tetraaza-macrocyclic cobalt(II) and nickel(II) complexes as electron-transfer agents in the photo(electro)chemical and electrochemical reduction of carbon dioxide. Recl. Trav. Chim. Pays Bas 1984, 103, 288–295. [Google Scholar] [CrossRef]
- Che, C.-M.; Mak, S.-T.; Lee, W.-O.; Fung, K.-W.; Mak, T.C.W. Electrochemical studies of nickel(II) and cobalt(II) complexes of tetra-azamacrocycles bearing a pyridine functional group and X-ray structures of [NiII(L3)Cl]ClO4 and [NiII(L3)][ClO4]2·H2O {L3 = meso-2,3,7,11,12-pentamethyl-3,7,11,17-tetra-azabicyclo[11.3.1]heptadeca-1,13,15-triene}. J. Chem. Soc. Dalt. Trans. 1988, 2153–2159. [Google Scholar]
- Chen, L.; Guo, Z.; Wei, X.-G.; Gallenkamp, C.; Bonin, J.; Anxolabéhère-Mallart, E.; Lau, K.-C.; Lau, T.-C.; Robert, M. Molecular catalysis of the electrochemical and photochemical reduction of CO2 with Earth-abundant metal complexes. Selective production of CO vs. HCOOH by switching of the metal center. J. Am. Chem. Soc. 2015, 137, 10918–10921. [Google Scholar] [CrossRef] [PubMed]
- Savéant, J.-M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. A local proton source enhances CO2 electroreduction to CO by a molecular Fe catalyst. Science 2012, 338, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide. Chem. Soc. Rev. 2013, 42, 2423–2436. [Google Scholar] [CrossRef] [PubMed]
- Costentin, C.; Robert, M.; Savéant, J.-M. Current issues in molecular catalysis illustrated by iron porphyrins as catalysts of the CO2-to-CO electrochemical conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef] [PubMed]
- Hammouche, M.; Lexa, D.; Momenteau, M.; Saveant, J.M. Chemical catalysis of electrochemical reactions. Homogeneous catalysis of the electrochemical reduction of carbon dioxide by iron(“0”) porphyrins. Role of the addition of magnesium cations. J. Am. Chem. Soc. 1991, 113, 8455–8466. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins: Synergystic effect of weak Brönsted acids. J. Am. Chem. Soc. 1996, 118, 1769–1776. [Google Scholar] [CrossRef]
- Bhugun, I.; Lexa, D.; Savéant, J.-M. Catalysis of the electrochemical reduction of carbon dioxide by iron(0) porphyrins. Synergistic effect of Lewis acid cations. J. Phys. Chem. 1996, 100, 19981–19985. [Google Scholar] [CrossRef]
- Costentin, C.; Drouet, S.; Robert, M.; Savéant, J.-M. Turnover numbers, turnover frequencies, and overpotential in molecular catalysis of electrochemical reactions. Cyclic voltammetry and preparative-scale electrolysis. J. Am. Chem. Soc. 2012, 134, 11235–11242. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Yanagida, S.; Brunschwig, B.S.; Fujita, E. Mechanistic and kinetic studies of cobalt macrocycles in a photochemical CO2 reduction system: Evidence of Co-CO2 adducts as intermediates. J. Am. Chem. Soc. 1995, 117, 6708–6716. [Google Scholar] [CrossRef]
- Grodkowski, J.; Neta, P.; Fujita, E.; Mahammed, A.; Simkhovich, L.; Gross, Z. Reduction of cobalt and iron corroles and catalyzed reduction of CO2. J. Phys. Chem. A 2002, 106, 4772–4778. [Google Scholar] [CrossRef]
- Meshitsuka, S.; Ichikawa, M.; Tamaru, K. Electrocatalysis by metal phthalocyanines in the reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1974, 158–159. [Google Scholar] [CrossRef]
- Furuya, N.; Matsui, K. Electroreduction of carbon dioxide on gas-diffusion electrodes modified by metal phthalocyanines. J. Electroanal. Chem. Interfacial Electrochem. 1989, 271, 181–191. [Google Scholar] [CrossRef]
- Atoguchi, T.; Aramata, A.; Kazusaka, A.; Enyo, M. Cobalt(II)--tetraphenylporphyrin--pyridine complex fixed on a glassy carbon electrode and its prominent catalytic activity for reduction of carbon dioxide. J. Chem. Soc. Chem. Commun. 1991, 156–157. [Google Scholar] [CrossRef]
- Sonoyama, N.; Kirii, M.; Sakata, T. Electrochemical reduction of CO2 at metal-porphyrin supported gas diffusion electrodes under high pressure CO2. Electrochem. Commun. 1999, 1, 213–216. [Google Scholar] [CrossRef]
- Aoi, S.; Mase, K.; Ohkubo, K.; Fukuzumi, S. Selective electrochemical reduction of CO2 to CO with a cobalt chlorin complex adsorbed on multi-walled carbon nanotubes in water. Chem. Commun. 2015, 51, 10226–10228. [Google Scholar] [CrossRef]
- Bonin, J.; Chaussemier, M.; Robert, M.; Routier, M. Homogeneous photocatalytic reduction of CO2 to CO using iron(0) porphyrin catalysts: Mechanism and intrinsic limitations. ChemCatChem 2014, 6, 3200–3207. [Google Scholar] [CrossRef]
- Rao, H.; Schmidt, L.C.; Bonin, J.; Robert, M. Visible-light-driven methane formation from CO2 with a molecular iron catalyst. Nature 2017, 548, 74. [Google Scholar] [CrossRef]
- Lin, S.; Diercks, C.S.; Zhang, Y.-B.; Kornienko, N.; Nichols, E.M.; Zhao, Y.; Paris, A.R.; Kim, D.; Yang, P.; Yaghi, O.M.; et al. Covalent Organic Frameworks Comprising Cobalt Porphyrins for Catalytic CO2 Reduction in Water. Science 2015, 349, 1208–1213. [Google Scholar] [CrossRef]
- Smith, P.T.; Benke, B.P.; Cao, Z.; Kim, Y.; Nichols, E.M.; Kim, K.; Chang, C.J. Iron Porphyrins Embedded into a Supramolecular Porous Organic Cage for Electrochemical CO2 Reduction in Water. Angew. Chem. Int. Ed. 2018, 57, 9684–9688. [Google Scholar] [CrossRef]
- Weng, Z.; Jiang, J.; Wu, Y.; Wu, Z.; Guo, X.; Materna, K.L.; Liu, W.; Batista, V.S.; Brudvig, G.W.; Wang, H. Electrochemical CO2 Reduction to Hydrocarbons on a Heterogeneous Molecular Cu Catalyst in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 8076–8079. [Google Scholar] [CrossRef] [PubMed]
- Thoi, V.S.; Kornienko, N.; Margarit, C.G.; Yang, P.; Chang, C.J. Visible-light photoredox catalysis: Selective reduction of carbon dioxide to carbon monoxide by a nickel N-heterocyclic carbene-isoquinoline complex. J. Am. Chem. Soc. 2013, 135, 14413–14424. [Google Scholar] [CrossRef] [PubMed]
- Chapovetsky, A.; Do, T.H.; Haiges, R.; Takase, M.K.; Marinescu, S.C. Proton-assisted reduction of CO2 by cobalt aminopyridine macrocycles. J. Am. Chem. Soc. 2016, 138, 5765–5768. [Google Scholar] [CrossRef] [PubMed]
- Pun, S.-N.; Chung, W.-H.; Lam, K.-M.; Guo, P.; Chan, P.-H.; Wong, K.-Y.; Che, C.-M.; Chen, T.-Y.; Peng, S.-M. Iron(I) complexes of 2,9-bis(2-hydroxyphenyl)-1,10-phenanthroline (H2dophen) as electrocatalysts for carbon dioxide reduction. X-ray crystal structures of [Fe(dophen)Cl]2·2HCON(CH3)2 and [Fe (dophen)(N-MeIm)2]ClO4 (N-MeIm = 1-methylimidazole). J. Chem. Soc. Dalton Trans. 2002, 575–583. [Google Scholar] [CrossRef]
- Nichols, A.W.; Chatterjee, S.; Sabat, M.; Machan, C.W. Electrocatalytic Reduction of CO2 to Formate by an Iron Schiff Base Complex. Inorg. Chem. 2018, 57, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Cometto, C.; Chen, L.; Lo, P.-K.; Guo, Z.; Lau, K.-C.; Anxolabéhère-Mallart, E.; Fave, C.; Lau, T.-C.; Robert, M. Highly Selective Molecular Catalysts for the CO2-to-CO Electrochemical Conversion at Very Low Overpotential. Contrasting Fe vs. Co Quaterpyridine Complexes upon Mechanistic Studies. ACS Catal. 2018, 8, 3411–3417. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef] [PubMed]
- Bourrez, M.; Orio, M.; Molton, F.; Vezin, H.; Duboc, C.; Deronzier, A.; Chardon-Noblat, S. Pulsed-EPR Evidence of a Manganese(II) Hydroxycarbonyl Intermediate in the Electrocatalytic Reduction of Carbon Dioxide by a Manganese Bipyridyl Derivative. Angew. Chem. Int. Ed. 2014, 53, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Sampson, M.D.; Kubiak, C.P. Manganese electrocatalysts with bulky bipyridine ligands: Utilizing Lewis acids to promote carbon dioxide reduction at low overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a substitute for rhenium in CO2 reduction catalysts: The importance of acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Franco, F.; Cometto, C.; Vallana, F.F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A Local Proton Source in a [Mn(bpy-R)(CO)3Br]-Type Redox Catalyst Enables CO2 Reduction Even in the Absence of Brønsted Acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, J.; Shaw, T.W.; Schaefer III, H.F.; Bocarsly, A.B. Design of a catalytic active site for electrochemical CO2 reduction with Mn(I)-tricarbonyl species. Inorg. Chem. 2015, 54, 5285–5294. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3Br] Complexes. Chem. Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.; Li, X.; Wolf, L.M.; Meeder, J.R.; Bhuvanesh, N.S.; Grice, K.A.; Panetier, J.A.; Nippe, M. Synergistic Effects of Imidazolium-Functionalization on Fac-Mn(CO)3 Bipyridine Catalyst Platforms for Electrocatalytic Carbon Dioxide Reduction. J. Am. Chem. Soc. 2019, 141, 6569–6582. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Saita, K.; Sekizawa, K.; Maeda, S.; Morikawa, T. Low-Energy Electrocatalytic CO2 Reduction in Water over Mn-Complex Catalyst Electrode Aided by a Nanocarbon Support and K+ Cations. ACS Catal. 2018, 8, 4452–4458. [Google Scholar] [CrossRef]
- Sun, C.; Rotundo, L.; Garino, C.; Nencini, L.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrochemical CO2 Reduction at Glassy Carbon Electrodes Functionalized by MnI and ReI Organometallic Complexes. ChemPhysChem 2017, 18, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Filippi, J.; Gobetto, R.; Miller, H.A.; Rocca, R.; Nervi, C.; Vizza, F. Electrochemical CO2 Reduction in Water at Carbon Cloth Electrodes Functionalized with a Fac-Mn (apbpy)(CO)3Br Complex. Chem. Commun. 2019, 55, 775–777. [Google Scholar] [CrossRef] [PubMed]
- Simón-Manso, E.; Kubiak, C.P. Dinuclear nickel complexes as catalysts for electrochemical reduction of carbon dioxide. Organometallics 2005, 24, 96–102. [Google Scholar] [CrossRef]
- DeLaet, D.L.; DelRosario, R.; Fanwick, P.E.; Kubiak, C.P. Carbon dioxide chemistry and electrochemistry of a binuclear “Cradle” complex of Ni, Ni2(µ-CNMe)(CNMe)2(PPh2CH2PPh2)2. J. Am. Chem. Soc. 1987, 109, 754–758. [Google Scholar] [CrossRef]
- Ratliff, K.S.; Lentz, R.E.; Kubiak, C.P. Carbon dioxide chemistry of the trinuclear complex [Ni3 (µ3-CNMe)( µ3-I)(dppm)3][PF6]. Electrocatalytic reduction of carbon dioxide. Organometallics 1992, 11, 1986–1988. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; Ferrence, G.M.; Washington, J.; Henderson, J.I.; Rosenhein, L.; Heise, J.D.; Fanwick, P.E.; Kubiak, C.P. A Class of Halide-Supported Trinuclear Nickel Clusters [Ni3(µ3-L)(µ3-X)(µ2-dppm)3]n+ (L = I−, Br−, CO, CNR.; X = I−, Br−; n = 0, 1; dppm= Ph2PCH2PPh2): Novel Physical Properties and the Fermi Resonance of Symmetric µ3-η1 Bound Isocyanide Ligands. J. Am. Chem. Soc. 1996, 118, 2198–2207. [Google Scholar] [CrossRef]
- Haines, R.J.; Wittrig, R.E.; Kubiak, C.P. Electrocatalytic Reduction of Carbon Dioxide by the Binuclear Copper Complex [Cu2(6-(diphenylphosphino-2,2′-bipyridyl)2(MeCN)2][PF6]2. Inorg. Chem. 1994, 33, 4723–4728. [Google Scholar] [CrossRef]
- Cheng, M.; Yu, Y.; Zhou, X.; Luo, Y.; Wang, M. Chemical Versatility of [FeFe]-Hydrogenase Models: Distinctive Activity of [μ-C6H4-1,2-(κ2-S)2][Fe2(CO)6] for Electrocatalytic CO2 Reduction. ACS Catal. 2018, 9, 768–774. [Google Scholar] [CrossRef]
- Angamuthu, R.; Byers, P.; Lutz, M.; Spek, A.L.; Bouwman, E. Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex. Science 2010, 327, 313–315. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Maeda, C.; Miyazaki, Y.; Ema, T. Recent progress in catalytic conversions of carbon dioxide. Catal. Sci. Technol. 2014, 4, 1482–1497. [Google Scholar] [CrossRef] [Green Version]
- Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Reductive functionalization of CO2 with amines: An entry to formamide, formamidine and methylamine derivatives. Green Chem. 2015, 17, 157–168. [Google Scholar] [CrossRef]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Borjesson, M.; Moragas, T.; Gallego, D.; Martin, R. Metal-catalyzed carboxylation of organic (pseudo)halides with CO2. ACS Catal. 2016, 6, 6739–6749. [Google Scholar] [CrossRef]
- Huang, K.; Sun, C.-L.; Shi, Z.-J. Transition-metal-catalyzed C-C bond formation through the fixation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 2435–2452. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365–2387. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Kleij, A.W. Myth or reality? Fixation of carbon dioxide into complex organic matter under mild conditions. ChemSusChem 2011, 4, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Köhler, B.; Kuckshinrichs, W.; Leitner, W.; Markewitz, P.; Müller, T.E. Chemical technologies for exploiting and recycling carbon dioxide into the value chain. ChemSusChem 2011, 4, 1216–1240. [Google Scholar] [CrossRef] [PubMed]
- Juliá-Hernández, F.; Gaydou, M.; Serrano, E.; van Gemmeren, M.; Martin, R. Ni- and Fe-catalyzed carboxylation of unsaturated hydrocarbons with CO2. In Ni-and Fe-Based Cross-Coupling Reactions; Springer: Dodrecht, The Netherlands, 2017; pp. 91–128.CO2. In Ni-and Fe-Based Cross-Coupling Reactions; Springer: Dodrecht, The Netherlands, 2017; pp. 91–128. [Google Scholar]
- Aida, T.; Inoue, S. Activation of carbon dioxide with aluminum porphyrin and reaction with epoxide. Studies on (tetraphenylporphinato) aluminum alkoxide having a long oxyalkylene chain as the alkoxide group. J. Am. Chem. Soc. 1983, 105, 1304–1309. [Google Scholar] [CrossRef]
- Kruper, W.J.; Dellar, D.D. Catalytic formation of cyclic carbonates from epoxides and CO2 with chromium metalloporphyrinates. J. Org. Chem. 1995, 60, 725–727. [Google Scholar] [CrossRef]
- Paddock, R.L.; Hiyama, Y.; McKay, J.M.; Nguyen, S.T. Co(III) porphyrin/DMAP: An efficient catalyst system for the synthesis of cyclic carbonates from CO2 and epoxides. Tetrahedron Lett. 2004, 45, 2023–2026. [Google Scholar] [CrossRef]
- Jin, L.; Jing, H.; Chang, T.; Bu, X.; Wang, L.; Liu, Z. Metal porphyrin/phenyltrimethylammonium tribromide: High efficient catalysts for coupling reaction of CO2 and epoxides. J. Mol. Catal. A Chem. 2007, 261, 262–266. [Google Scholar] [CrossRef]
- Bai, D.; Wang, Q.; Song, Y.; Li, B.; Jing, H. Synthesis of cyclic carbonate from epoxide and CO2 catalyzed by magnetic nanoparticle-supported porphyrin. Catal. Commun. 2011, 12, 684–688. [Google Scholar] [CrossRef]
- Ahmadi, F.; Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Khosropour, A.R. Highly efficient chemical fixation of carbon dioxide catalyzed by high-valent tetraphenylporphyrinatotin(IV) triflate. Inorg. Chem. Commun. 2011, 14, 1489–1493. [Google Scholar] [CrossRef]
- Ema, T.; Miyazaki, Y.; Koyama, S.; Yano, Y.; Sakai, T. A bifunctional catalyst for carbon dioxide fixation: Cooperative double activation of epoxides for the synthesis of cyclic carbonates. Chem. Commun. 2012, 48, 4489–4491. [Google Scholar] [CrossRef] [PubMed]
- Melendez, J.; North, M.; Villuendas, P. One-component catalysts for cyclic carbonate synthesis. Chem. Commun. 2009, 2577–2579. [Google Scholar] [CrossRef] [PubMed]
- Escárcega-Bobadilla, M.V.; Martinez Belmonte, M.; Martin, E.; Escudero-Adán, E.C.; Kleij, A.W. A Recyclable Trinuclear Bifunctional Catalyst Derived from a Tetraoxo Bis-Zn(salphen) Metalloligand. Chem. Eur. J. 2013, 19, 2641–2648. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Nakamura, M.; Nozaki, K. Alternating copolymerization of cyclohexene oxide with carbon dioxide catalyzed by (salalen)CrCl complexes. Macromolecules 2009, 42, 6972–6980. [Google Scholar] [CrossRef]
- Wu, G.-P.; Wei, S.-H.; Ren, W.-M.; Lu, X.-B.; Xu, T.-Q.; Darensbourg, D.J. Perfectly alternating copolymerization of CO2 and epichlorohydrin using cobalt(III)-based catalyst systems. J. Am. Chem. Soc. 2011, 133, 15191–15199. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Niu, Y. Bifunctional cobalt Salen complex: A highly selective catalyst for the coupling of CO2 and epoxides under mild conditions. Appl. Organomet. Chem. 2011, 25, 424–428. [Google Scholar] [CrossRef]
- Vagin, S.I.; Reichardt, R.; Klaus, S.; Rieger, B. Conformationally flexible dimeric salphen complexes for bifunctional catalysis. J. Am. Chem. Soc. 2010, 132, 14367–14369. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Hashimoto, S.; Nozaki, K. Bimetallic mechanism operating in the copolymerization of propylene oxide with carbon dioxide catalyzed by cobalt-salen complexes. Chem. Sci. 2010, 1, 369–373. [Google Scholar] [CrossRef]
- Ishida, T.; Kikuchi, S.; Yamada, T. Efficient preparation of 4-hydroxyquinolin-2(1H)-one derivatives with silver-catalyzed carbon dioxide incorporation and intramolecular rearrangement. Org. Lett. 2013, 15, 3710–3713. [Google Scholar] [CrossRef]
- Ishida, T.; Kikuchi, S.; Tsubo, T.; Yamada, T. Silver-catalyzed incorporation of carbon dioxide into o-alkynylaniline derivatives. Org. Lett. 2013, 15, 848–851. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous hydrogenation of carbon dioxide. Chem. Rev. 1995, 95, 259–272. [Google Scholar] [CrossRef]
- McGhee, W.D.; Riley, D.P.; Christ, M.E.; Christ, K.M. Palladium-catalyzed generation of O-allylic urethanes and carbonates from amines/alcohols, carbon dioxide, and allylic chlorides. Organometallics 1993, 12, 1429–1433. [Google Scholar] [CrossRef]
- Kayaki, Y.; Mori, N.; Ikariya, T. Palladium-catalyzed carboxylative cyclization of α-allenyl amines in dense carbon dioxide. Tetrahedron Lett. 2009, 50, 6491–6493. [Google Scholar] [CrossRef]
- Sasaki, Y.; Dixneuf, P.H. A novel catlytic synthesis of vinyl carboamtes from carbon dioxide, diethylamine, and alkynes in the presence of Ru3(CO)12. J. Chem. Soc. Chem. Commun. 1986, 790–791. [Google Scholar] [CrossRef]
- Mitsudo, T.; Hori, Y.; Yamakawa, Y.; Watanabe, Y. Ruthenium catalyzed selective synthesis of enol carbamates by fixation of carbon dioxide. Tetrahedron Lett. 1987, 28, 4417–4418. [Google Scholar] [CrossRef]
- Tominaga, K.; Sasaki, Y. Synthesis of 2-oxazolidinones from CO2 and 1,2-aminoalcohols catalyzed by n-Bu2SnO. Synlett 2002, 2002, 307–309. [Google Scholar] [CrossRef]
- Shi, F.; Deng, Y.; SiMa, T.; Peng, J.; Gu, Y.; Qiao, B. Alternatives to phosgene and carbon monoxide: Synthesis of symmetric urea derivatives with carbon dioxide in ionic liquids. Angew. Chem. 2003, 115, 3379–3382. [Google Scholar] [CrossRef]
- Kawanami, H.; Matsumoto, H.; Ikushima, Y. Effective scCO2-ionic liquid reaction system based on symmetric aliphatic ammonium salts for the rapid CO2 fixation with aziridine to 2-oxazolidinone. Chem. Lett. 2004, 34, 60–61. [Google Scholar] [CrossRef]
- Mahe, R.; Sasaki, Y.; Bruneau, C.; Dixneuf, P.H. Catalytic synthesis of vinyl carbamates from carbon dioxide and alkynes with ruthenium complexes. J. Org. Chem. 1989, 54, 1518–1523. [Google Scholar] [CrossRef]
- Abla, M.; Choi, J.-C.; Sakakura, T. Halogen-free process for the conversion of carbon dioxide to urethanes by homogeneous catalysis. Chem. Commun. 2001, 2238–2239. [Google Scholar] [CrossRef]
- Ohmiya, H.; Tanabe, M.; Sawamura, M. Copper-catalyzed carboxylation of alkylboranes with carbon dioxide: Formal reductive carboxylation of terminal alkenes. Org. Lett. 2011, 13, 1086–1088. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Z.; Li, W.-J.; Zhang, X.; Zhou, H.; Lu, X.-B. Cu(I)-catalyzed carboxylative coupling of terminal alkynes, allylic chlorides, and CO2. Org. Lett. 2010, 12, 4748–4751. [Google Scholar] [CrossRef] [PubMed]
- Leon, T.; Correa, A.; Martin, R. Ni-catalyzed direct carboxylation of benzyl halides with CO2. J. Am. Chem. Soc. 2013, 135, 1221–1224. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.R.; Santos, R.H.A.; Gambardella, M.T.P.; DeMacedo, L.G.M.; Rodrigues-Filho, U.P.; Launay, J.-C.; Franco, D.W. Activation of carbon dioxide by bicyclic amidines. J. Org. Chem. 2004, 69, 8005–8011. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Nakagawa, S.; Koizumi, T.; Hirayama, K.; Yamamoto, Y. Nickel-mediated regio- and chemoselective carboxylation of alkynes in the presence of carbon dioxide. J. Org. Chem. 1999, 64, 3975–3978. [Google Scholar] [CrossRef]
- Inoue, Y.; Itoh, Y.; Kazama, H.; Hashimoto, H. Reaction of dialkyl-substituted alkynes with carbon dioxide catalyzed by nickel(0) complexes. Incorporation of carbon dioxide in alkyne dimers and novel cyclotrimerization of the alkynes. Bull. Chem. Soc. Jpn. 1980, 53, 3329–3333. [Google Scholar] [CrossRef]
- Tsuda, T.; Maruta, K.; Kitaike, Y. Nickel(0)-catalyzed alternating copolymerization of carbon dioxide with diynes to poly(2-pyrones). J. Am. Chem. Soc. 1992, 114, 1498–1499. [Google Scholar] [CrossRef]
- Fujihara, T.; Nogi, K.; Xu, T.; Terao, J.; Tsuji, Y. Nickel-catalyzed carboxylation of aryl and vinyl chlorides employing carbon dioxide. J. Am. Chem. Soc. 2012, 134, 9106–9109. [Google Scholar] [CrossRef] [PubMed]
- Correa, A.; Martin, R. Palladium-catalyzed direct carboxylation of aryl bromides with carbon dioxide. J. Am. Chem. Soc. 2009, 131, 15974–15975. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Iwasawa, N. Hydrocarboxylation of allenes with CO2 catalyzed by silyl pincer-type palladium complex. J. Am. Chem. Soc. 2008, 130, 15254–15255. [Google Scholar] [CrossRef] [PubMed]
- Behr, A.; Brehme, V.A. Homogeneous and heterogeneous catalyzed three-step synthesis of 2-ethylheptanoic acid from carbon dioxide, butadiene and hydrogen. J. Mol. Catal. A Chem. 2002, 187, 69–80. [Google Scholar] [CrossRef]
- Feng, X.; Sun, A.; Zhang, S.; Yu, X.; Bao, M. Palladium-catalyzed carboxylative coupling of benzyl chlorides with allyltributylstannane: Remarkable effect of Palladium nanoparticles. Org. Lett. 2012, 15, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Sasano, K.; Takaya, J.; Iwasawa, N. Palladium(II)-catalyzed direct carboxylation of alkenyl C-H bonds with CO2. J. Am. Chem. Soc. 2013, 135, 10954–10957. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, T.; Yamamoto, T.; Saegusa, T. Palladium-catalyzed cycloaddition of carbon dioxide with methoxyallene. J. Organomet. Chem. 1992, 429, C46–C48. [Google Scholar] [CrossRef]
- Greenhalgh, M.D.; Thomas, S.P. Iron-catalyzed, highly regioselective synthesis of α-aryl carboxylic acids from styrene derivatives and CO2. J. Am. Chem. Soc. 2012, 134, 11900–11903. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, H.; Takaya, J.; Iwasawa, N. Rhodium(I)-catalyzed direct carboxylation of arenes with CO2 via chelation-assisted C-H bond activation. J. Am. Chem. Soc. 2010, 133, 1251–1253. [Google Scholar] [CrossRef]
- Mita, T.; Michigami, K.; Sato, Y. Sequential Protocol for C(sp3)-H Carboxylation with CO2: Transition-Metal-Catalyzed Benzylic C-H Silylation and Fluoride-Mediated Carboxylation. Org. Lett. 2012, 14, 3462–3465. [Google Scholar] [CrossRef]
- Dai, Y.; Feng, X.; Wang, B.; He, R.; Bao, M. Preparation and application of air-stable P,N-bidentate ligands for the selective synthesis of δ-lactone via the palladium-catalyzed telomerization of 1,3-butadiene with carbon dioxide. J. Organomet. Chem. 2012, 696, 4309–4314. [Google Scholar] [CrossRef]
- Saylik, D.; Horvath, M.J.; Elmes, P.S.; Jackson, W.R.; Lovel, C.G.; Moody, K. Preparation of Isocyanates from Primary Amines and Carbon Dioxide Using Mitsunobu Chemistry1. J. Org. Chem. 1999, 64, 3940–3946. [Google Scholar] [CrossRef]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1981, 1–28. [Google Scholar] [CrossRef]
- Dinsmore, C.J.; Mercer, S.P. Carboxylation and Mitsunobu reaction of amines to give carbamates: Retention vs. inversion of configuration is substituent-dependent. Org. Lett. 2004, 6, 2885–2888. [Google Scholar] [CrossRef] [PubMed]
- Haruki, E.; Hara, T.; Inoue, H. Syntheses of Tricyclo (5.2. 1.02, 6) deca-3, 8-diene-1, 8-dicarboxylic Acid and Its Derivatives from Cyclopentadiene and Carbon Dioxide. Chem. Express. 1990, 5, 493. [Google Scholar]
- Ashley, A.E.; Thompson, A.L.; O’Hare, D. Non-metal-mediated homogeneous hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [Google Scholar] [CrossRef] [PubMed]
- Kayaki, Y.; Yamamoto, M.; Ikariya, T. N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. 2009, 121, 4258–4261. [Google Scholar] [CrossRef]
- Ueno, A.; Kayaki, Y.; Ikariya, T. Cycloaddition of tertiary aziridines and carbon dioxide using a recyclable organocatalyst, 1,3-di-tert-butylimidazolium-2-carboxylate: Straightforward access to 3-substituted 2-oxazolidones. Green Chem. 2013, 15, 425–430. [Google Scholar] [CrossRef]
- He, Q.; O’Brien, J.W.; Kitselman, K.A.; Tompkins, L.E.; Curtis, G.C.T.; Kerton, F.M. Synthesis of cyclic carbonates from CO2 and epoxides using ionic liquids and related catalysts including choline chloride-metal halide mixtures. Catal. Sci. Technol. 2014, 4, 1513–1528. [Google Scholar] [CrossRef]
- Calo, V.; Nacci, A.; Monopoli, A.; Fanizzi, A. Cyclic carbonate formation from carbon dioxide and oxiranes in tetrabutylammonium halides as solvents and catalysts. Org. Lett. 2002, 4, 2561–2563. [Google Scholar] [CrossRef]
- Derien, S.; Clinet, J.C.; Dunach, E.; Perichon, J. Activation of carbon dioxide: Nickel-catalyzed electrochemical carboxylation of diynes. J. Org. Chem. 1993, 58, 2578–2588. [Google Scholar] [CrossRef]
- Tascedda, P.; Dunach, E. Electrosynthesis of cyclic carbamates from aziridines and carbon dioxide. Chem. Commun. 2000, 449–450. [Google Scholar] [CrossRef]
- Derien, S.; Dunach, E.; Perichon, J. From stoichiometry to catalysis: Electroreductive coupling of alkynes and carbon dioxide with nickel-bipyridine complexes. Magnesium ions as the key for catalysis. J. Am. Chem. Soc. 1991, 113, 8447–8454. [Google Scholar] [CrossRef]
- Tascedda, P.; Dunãch, E. Novel electrochemical reactivity of Ni(cyclam)Br2: Catalytic carbon dioxide incorporation into epoxides. J. Chem. Soc. Chem. Commun. 1995, 43–44. [Google Scholar] [CrossRef]
- Dérien, S.; Clinet, J.-C.; Duñach, E.; Périchon, J. First example of direct carbon dioxide incorporation into 1,3-diynes: A highly regio-and stereo-selective nickel-catalysed electrochemical reaction. J. Chem. Soc. Chem. Commun. 1991, 549–550. [Google Scholar] [CrossRef]
- Kröcher, O.; Köppel, R.A.; Baiker, A. Highly active ruthenium complexes with bidentate phosphine ligands for the solvent-free catalytic synthesis of N,N-dimethylformamide and methyl formate. Chem. Commun. 1997, 453–454. [Google Scholar] [CrossRef]
- Shimomaki, K.; Murata, K.; Martin, R.; Iwasawa, N. Visible-light-driven carboxylation of aryl halides by the combined use of palladium and photoredox catalysts. J. Am. Chem. Soc. 2017, 139, 9467–9470. [Google Scholar] [CrossRef] [PubMed]
- Murata, K.; Numasawa, N.; Shimomaki, K.; Takaya, J.; Iwasawa, N. Construction of a visible light-driven hydrocarboxylation cycle of alkenes by the combined use of Rh(I) and photoredox catalysts. Chem. Commun. 2017, 53, 3098–3101. [Google Scholar] [CrossRef] [PubMed]
- Seo, H.; Katcher, M.H.; Jamison, T.F. Photoredox activation of carbon dioxide for amino acid synthesis in continuous flow. Nat. Chem. 2017, 9, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Ishida, N.; Masuda, Y.; Uemoto, S.; Murakami, M. A Light/Ketone/Copper System for Carboxylation of Allylic C-H Bonds of Alkenes with CO2. Chem. Eur. J. 2016, 22, 6524–6527. [Google Scholar] [CrossRef]
- DasNeves Gomes, C.; Jacquet, O.; Villiers, C.; Thuéry, P.; Ephritikhine, M.; Cantat, T. A diagonal approach to chemical recycling of carbon dioxide: Organocatalytic transformation for the reductive functionalization of CO2. Angew. Chem. Int. Ed. 2012, 51, 187–190. [Google Scholar] [CrossRef]
- Li, Y.; Cui, X.; Dong, K.; Junge, K.; Beller, M. Utilization of CO2 as a C1 Building Block for Catalytic Methylation Reactions. ACS Catal. 2017, 7, 1077–1086. [Google Scholar] [CrossRef]
- Haynes, P.; Slaugh, L.H.; Kohnle, J.F. Formamides from carbon dioxide, amines and hydrogen in the presence of metal complexes. Tetrahedron Lett. 1970, 11, 365–368. [Google Scholar] [CrossRef]
- Kudo, K.; Phala, H.; Sugita, N.; Takezaki, Y. Synthesis of dimethyl formamide from carbon dioxide, hydrogen and dimethyl amine catalyzed by palladium (II) chloride. Chem. Lett. 1977, 6, 1495–1496. [Google Scholar] [CrossRef]
- Schreiner, S.; Yu, J.Y.; Vaska, L. Reversible homogeneous catalysis of carbon dioxide hydrogenation/reduction at room temperature and low pressures. J. Chem. Soc. Chem. Commun. 1988, 602–603. [Google Scholar] [CrossRef]
- Morimoto, Y.; Fujiwara, Y.; Taniguchi, H.; Hori, Y.; Nagano, Y. PdCl2(MeCN)2-catalyzed carbonylation of diethylamine with carbon dioxide: Selective synthesis of tetraethylurea and diethylformamide. Tetrahedron Lett. 1986, 27, 1809–1810. [Google Scholar] [CrossRef]
- Süss-Fink, G.; Langenbahn, M.; Jenke, T. Rutheniumcluster als Katalysatoren für die Carbonylierung von cyclischen Aminen. J. Organomet. Chem. 1989, 368, 103–109. [Google Scholar] [CrossRef]
- Zhang, L.; Han, Z.; Zhao, X.; Wang, Z.; Ding, K. Highly Efficient Ruthenium-Catalyzed N-Formylation of Amines with H2 and CO2. Angew. Chem. 2015, 127, 6284–6287. [Google Scholar] [CrossRef]
- Schreiner, S.; Yu, J.Y.; Vaska, L. Carbon dioxide reduction via homogeneous catalytic synthesis and hydrogenation of N,N-dimethylformamide. Inorg. Chim. Acta 1988, 147, 139–141. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Catalytic production of dimethylformamide from supercritical carbon dioxide. J. Am. Chem. Soc. 1994, 116, 8851–8852. [Google Scholar] [CrossRef]
- Liu, F.; Abrams, M.B.; Baker, R.T.; Tumas, W. Phase-separable catalysis using room temperature ionic liquids and supercritical carbon dioxide. Chem. Commun. 2001, 433–434. [Google Scholar] [CrossRef] [Green Version]
- Affan, M.A.; Jessop, P.G. Catalytic Formylation of Primary and Secondary Amines with CO2 and H2 Using Abundant-Metal Catalysts. Inorg. Chem. 2017, 56, 7301–7305. [Google Scholar] [CrossRef]
- Federsel, C.; Boddien, A.; Jackstell, R.; Jennerjahn, R.; Dyson, P.J.; Scopelliti, R.; Laurenczy, G.; Beller, M. A Well-Defined Iron Catalyst for the Reduction of Bicarbonates and Carbon Dioxide to Formates, Alkyl Formates, and Formamides. Angew. Chem. Int. Ed. 2010, 49, 9777–9780. [Google Scholar] [CrossRef]
- Federsel, C.; Ziebart, C.; Jackstell, R.; Baumann, W.; Beller, M. Catalytic Hydrogenation of Carbon Dioxide and Bicarbonates with a Well-Defined Cobalt Dihydrogen Complex. Chem. Eur. J. 2012, 18, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Ziebart, C.; Federsel, C.; Anbarasan, P.; Jackstell, R.; Baumann, W.; Spannenberg, A.; Beller, M. Well-Defined Iron Catalyst for Improved Hydrogenation of Carbon Dioxide and Bicarbonate. J. Am. Chem. Soc. 2012, 134, 20701–20704. [Google Scholar] [CrossRef] [PubMed]
- Minato, M.; Zhou, D.-Y.; Sumiura, K.; Hirabayashi, R.; Yamaguchi, Y.; Ito, T. Reactions of quadruply chelated silyl– and germyl–molybdenum hydrido complexes with carboxylic acids and carbon dioxide: A first example of carbon dioxide fixation utilizing the trans effect of a silyl ligand. Chem. Commun. 2001, 2654–2655. [Google Scholar] [CrossRef]
- Jessop, P.G.; Hsiao, Y.; Ikariya, T.; Noyori, R. Homogeneous Catalysis in Supercritical Fluids: Hydrogenation of Supercritical Carbon Dioxide to Formic Acid, Alkyl Formates, and Formamides. J. Am. Chem. Soc. 1996, 118, 344–355. [Google Scholar] [CrossRef]
- Munshi, P.; Heldebrant, D.J.; McKoon, E.P.; Kelly, P.A.; Tai, C.-C.; Jessop, P.G. Formanilide and carbanilide from aniline and carbon dioxide. Tetrahedron Lett. 2003, 44, 2725–2727. [Google Scholar] [CrossRef]
- Beydoun, K.; vomStein, T.; Klankermayer, J.; Leitner, W. Ruthenium-Catalyzed Direct Methylation of Primary and Secondary Aromatic Amines Using Carbon Dioxide and Molecular Hydrogen. Angew. Chem. 2013, 125, 9733–9736. [Google Scholar] [CrossRef] [Green Version]
- Vaska, L.; Schreiner, S.; Felty, R.A.; Yu, J.Y. Catalytic Reduction of Carbon Dioxide to Methane and Other Species via Formamide Intermediation: Synthesis and Hydrogenation of HC(O)NH2 in the Presence of (Ir(Cl)CO)(Ph3P)2. J. Mol. Catal. 1988, 52, L11–L16. [Google Scholar] [CrossRef]
- Motokura, K.; Takahashi, N.; Kashiwame, D.; Yamaguchi, S.; Miyaji, A.; Baba, T. Copper-diphosphine complex catalysts for N-formylation of amines under 1 atm of carbon dioxide with polymethylhydrosiloxane. Catal. Sci. Technol. 2013, 3, 2392–2396. [Google Scholar] [CrossRef]
- Frogneux, X.; Jacquet, O.; Cantat, T. Iron-catalyzed hydrosilylation of CO2: CO2 conversion to formamides and methylamines. Catal. Sci. Technol. 2014, 4, 1529–1533. [Google Scholar] [CrossRef]
- Itagaki, S.; Yamaguchi, K.; Mizuno, N. Catalytic synthesis of silyl formates with 1 atm of CO2 and their utilization for synthesis of formyl compounds and formic acid. J. Mol. Catal. A Chem. 2013, 366, 347–352. [Google Scholar] [CrossRef]
- González-Sebastián, L.; Flores-Alamo, M.; García, J.J. Nickel-catalyzed hydrosilylation of CO2 in the presence of Et3B for the synthesis of formic acid and related formates. Organometallics 2013, 32, 7186–7194. [Google Scholar] [CrossRef]
- Shintani, R.; Nozaki, K. Copper-catalyzed hydroboration of carbon dioxide. Organometallics 2013, 32, 2459–2462. [Google Scholar] [CrossRef]
- Yu, B.; Zhang, H.; Zhao, Y.; Chen, S.; Xu, J.; Huang, C.; Liu, Z. Cyclization of o-phenylenediamines by CO2 in the presence of H2 for the synthesis of benzimidazoles. Green Chem. 2013, 15, 95–99. [Google Scholar] [CrossRef]
- Li, Y.; Sorribes, I.; Yan, T.; Junge, K.; Beller, M. Selective methylation of amines with carbon dioxide and H2. Angew. Chem. 2013, 125, 12378–12382. [Google Scholar] [CrossRef]
- Jacquet, O.; Frogneux, X.; Gomes, C.D.N.; Cantat, T. CO2 as a C1-building block for the catalytic methylation of amines. Chem. Sci. 2013, 4, 2127–2131. [Google Scholar] [CrossRef]
- Li, Y.; Fang, X.; Junge, K.; Beller, M. A general catalytic methylation of amines using carbon dioxide. Angew. Chem. 2013, 125, 9747–9750. [Google Scholar] [CrossRef]
- Frogneux, X.; Blondiaux, E.; Thuéry, P.; Cantat, T. Bridging amines with CO2: Organocatalyzed reduction of CO2 to aminals. ACS Catal. 2015, 5, 3983–3987. [Google Scholar] [CrossRef]
- Jacquet, O.; DasNeves Gomes, C.; Ephritikhine, M.; Cantat, T. Recycling of carbon and silicon wastes: Room temperature formylation of N-H bonds using carbon dioxide and polymethylhydrosiloxane. J. Am. Chem. Soc. 2012, 134, 2934–2937. [Google Scholar] [CrossRef]
- Jacquet, O.; DasNeves Gomes, C.; Ephritikhine, M.; Cantat, T. Complete catalytic deoxygenation of CO2 into formamidine derivatives. ChemCatChem 2013, 5, 117–120. [Google Scholar] [CrossRef]
- Blondiaux, E.; Pouessel, J.; Cantat, T. Carbon Dioxide Reduction to Methylamines under Metal-Free Conditions. Angew. Chem. Int. Ed. 2014, 53, 12186–12190. [Google Scholar] [CrossRef]
Scheme 1.
Structures of {Re[(bpy)Me(ImMe)](CO)3Cl}+ and [Re(CH2OH)–OH2]+ [30,31]; bpy—2,2′-bipyridine.







Scheme 8.
Structures of Ni N-heterocyclic carbene–isoquinoline [Ni(Prbimiq1)]2+ and Co-azacalix[4](2,6)pyridine complexes [71,72]; Prbimiq1—bis(3-(imidazolyl)isoquinolinyl)propane.

Scheme 9.
Structures of [FeI(dophen)]− and (Fe(III)Cl(tbudhbpy) [74,75]; dophen—2,9-bis(2-hydroxyphenyl)-1,10-phenanthroline; tbudhbpy—,6′-di(3,5-di-tert-butyl-2-hydroxybenzene)-2,2′-bipyridine.

Scheme 10.
Structures of [CoII(qpy)(OH2)2]2+ and [FeII(qpy)(OH2)2]2+ [76]; qpy—2,2′:6′,2”:6”,2‴-quaterpyridine.
Scheme 10.
Structures of [CoII(qpy)(OH2)2]2+ and [FeII(qpy)(OH2)2]2+ [76]; qpy—2,2′:6′,2”:6”,2‴-quaterpyridine.

Scheme 11.
Structures of Mn(L)(CO)3Br and its derivatives with various pendant substituents [77,78,79,80,81,82,83,84,85,86,87]; L—2,2′-bipyridine or 4,4′-dimethyl-2,2′-bipyridine.


Scheme 13.
Schematic illustration of thermodynamic barrier for CO2 functionalization [96,97,98,99,100,101,102,103,104,105].

Scheme 14.
Examples of metal-catalyzed CO2 functionalization with amine-containing nucleophiles [120,121,122,123,124,125,126,127,128,129,130,131].

Scheme 15.
Examples of metal-catalyzed carboxylation of carbon nucleophiles with CO2 [132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158].

Scheme 16.
Examples of electrocatalytic coupling of CO2 with various carbon nucleophiles [161,162,163].

Scheme 17.
Examples of photocatalytic carboxylation of carbon nucleophiles with CO2 [164,165,166,167,168].

Scheme 18.
Horizontal and diagonal (reductive) approaches for CO2 functionalization [19,23,24,25,26].


Scheme 20.
Transition-metal catalysts for reductive coupling of CO2 and amines [189,190,191,192,193].

Scheme 21.
Examples of non-carbonyl products formed via reductive coupling of amines with CO2 [194,195,196].



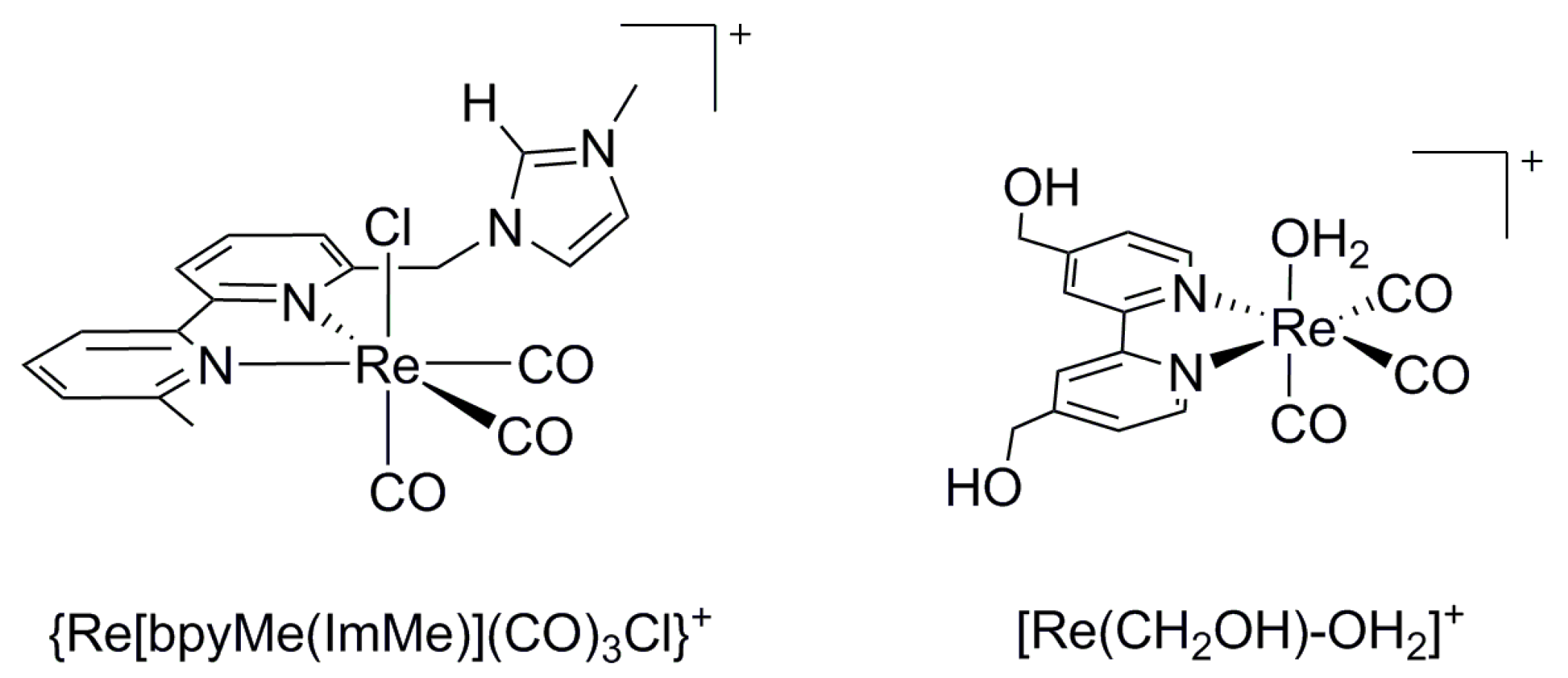{kind=link}
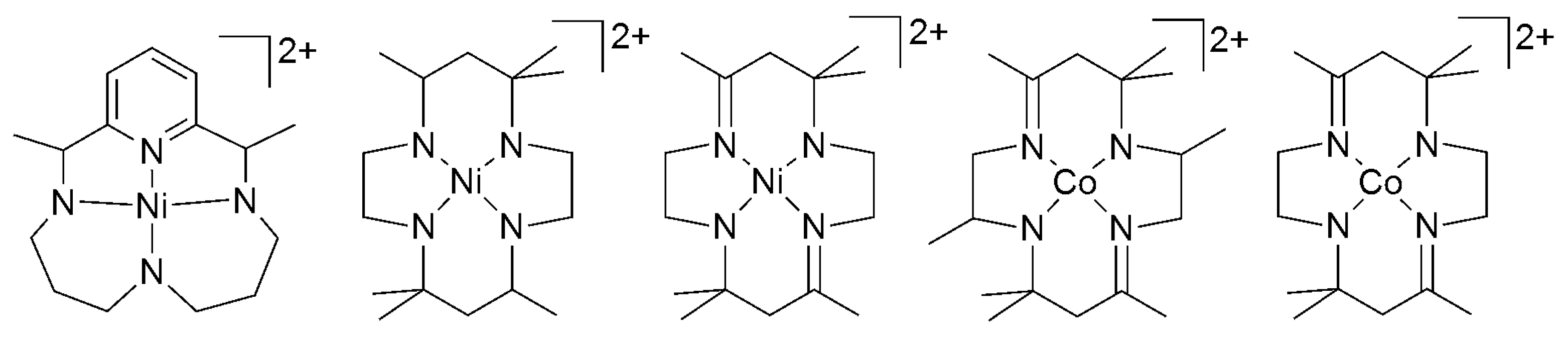{kind=link}
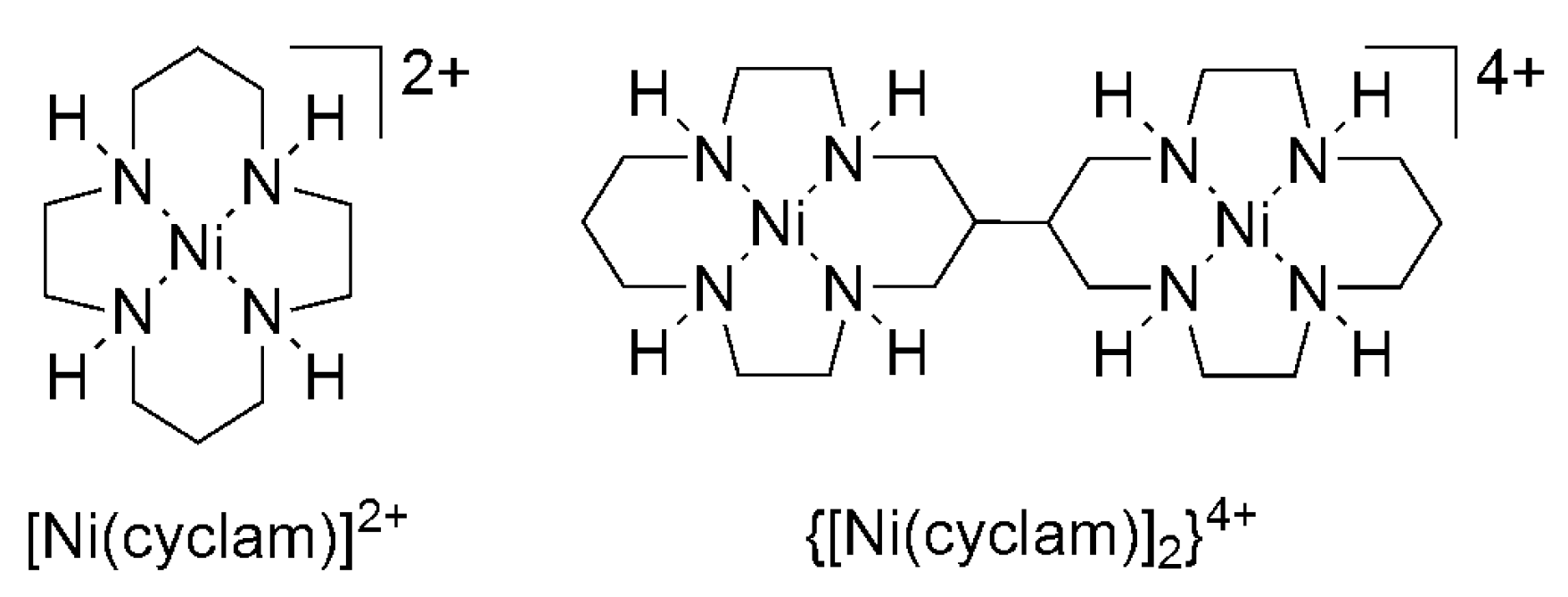{kind=link}
{kind=link}
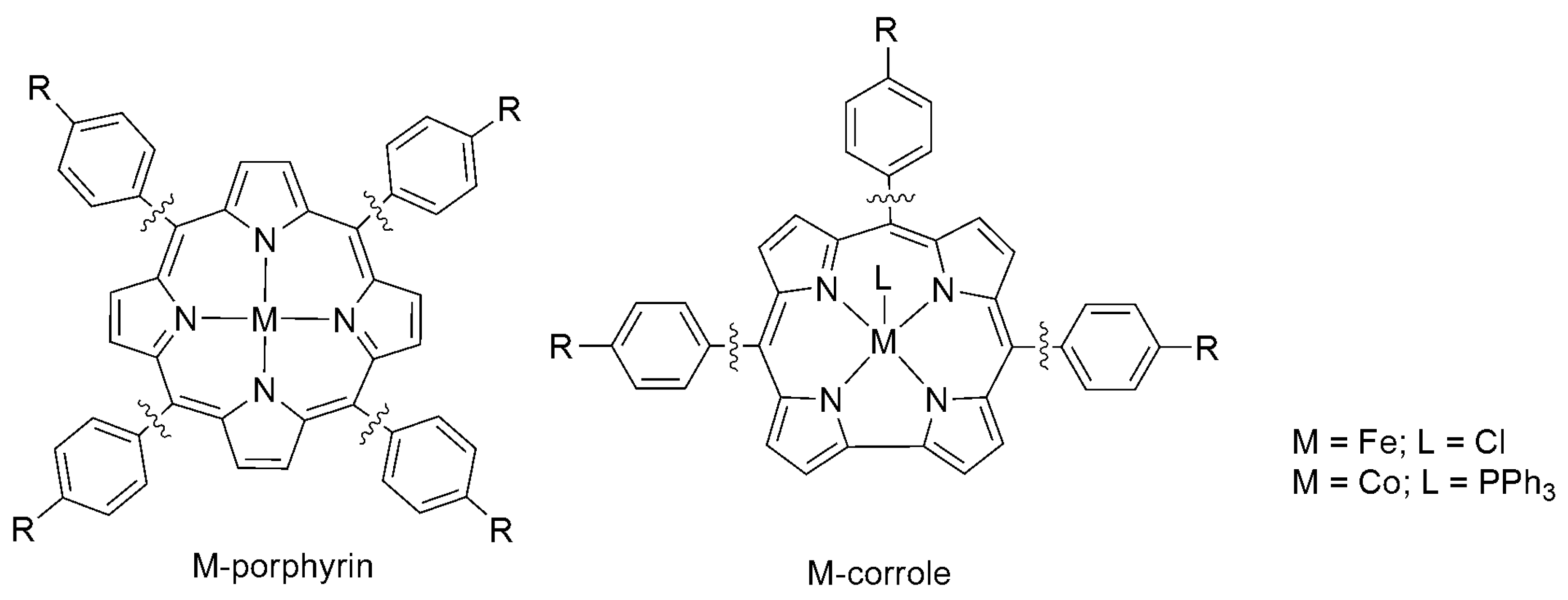{kind=link}
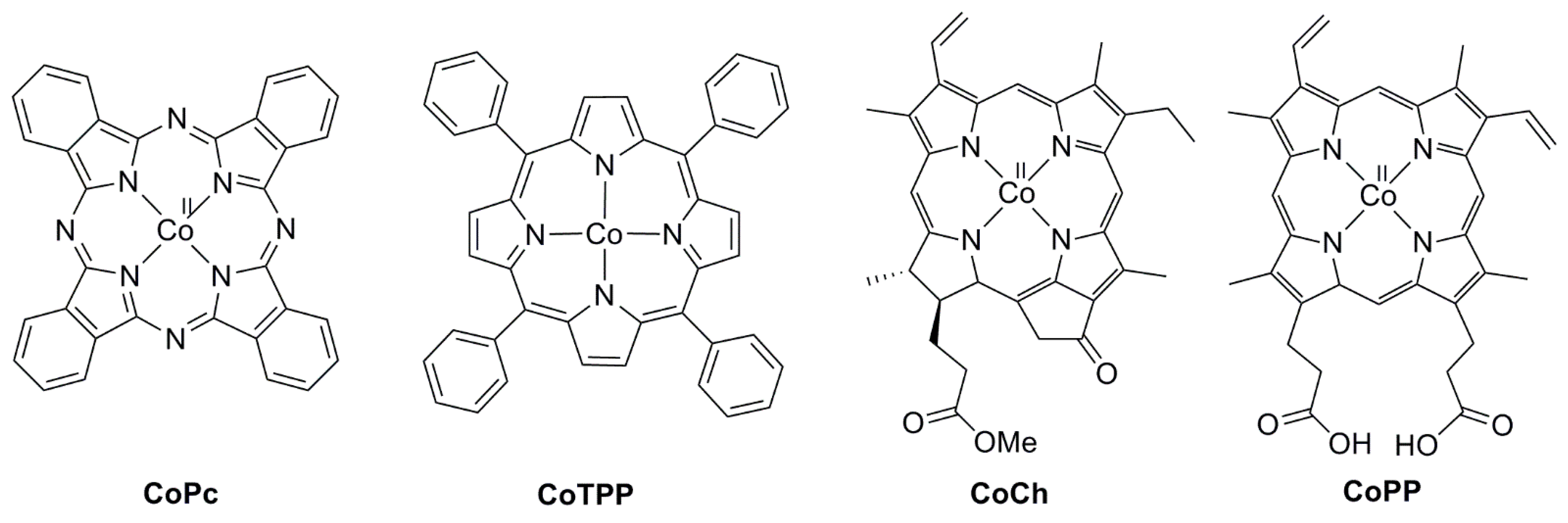{kind=link}
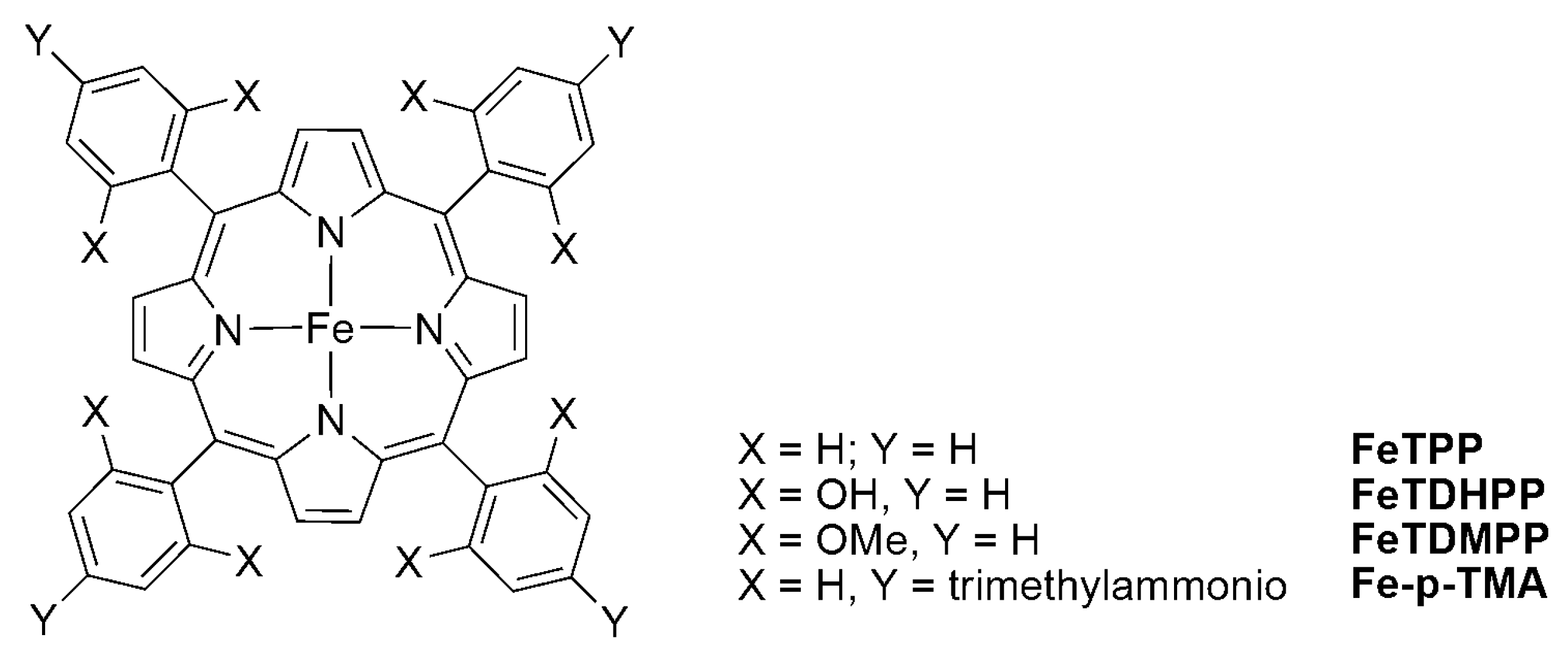{kind=link}
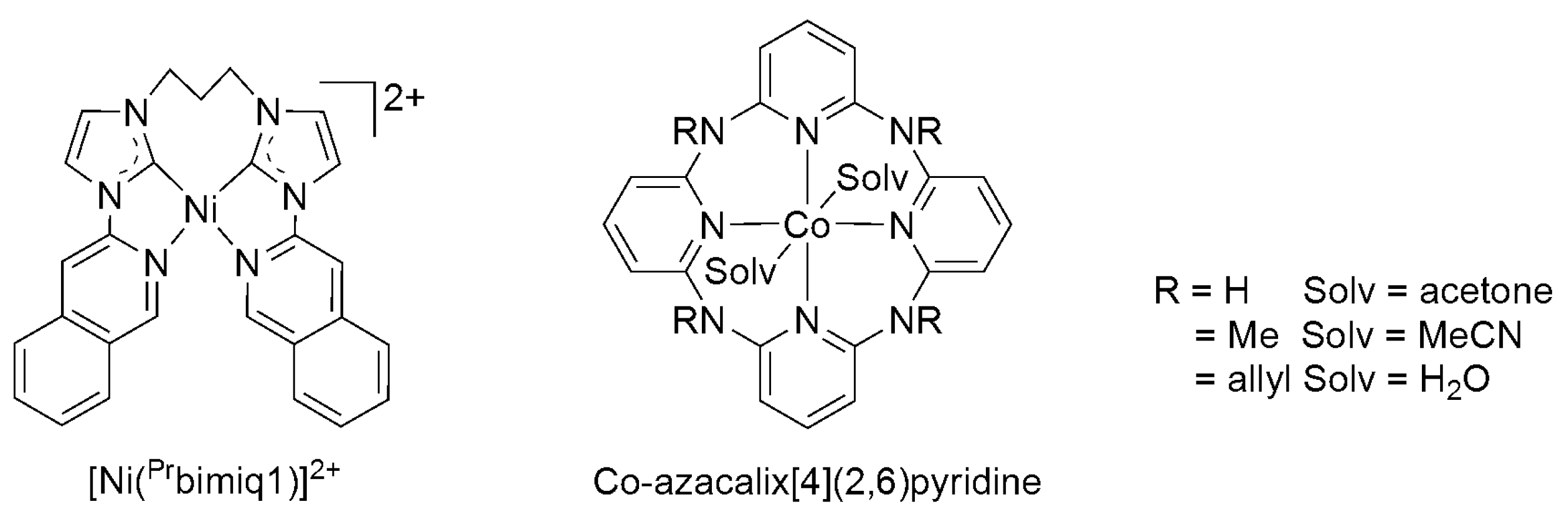{kind=link}
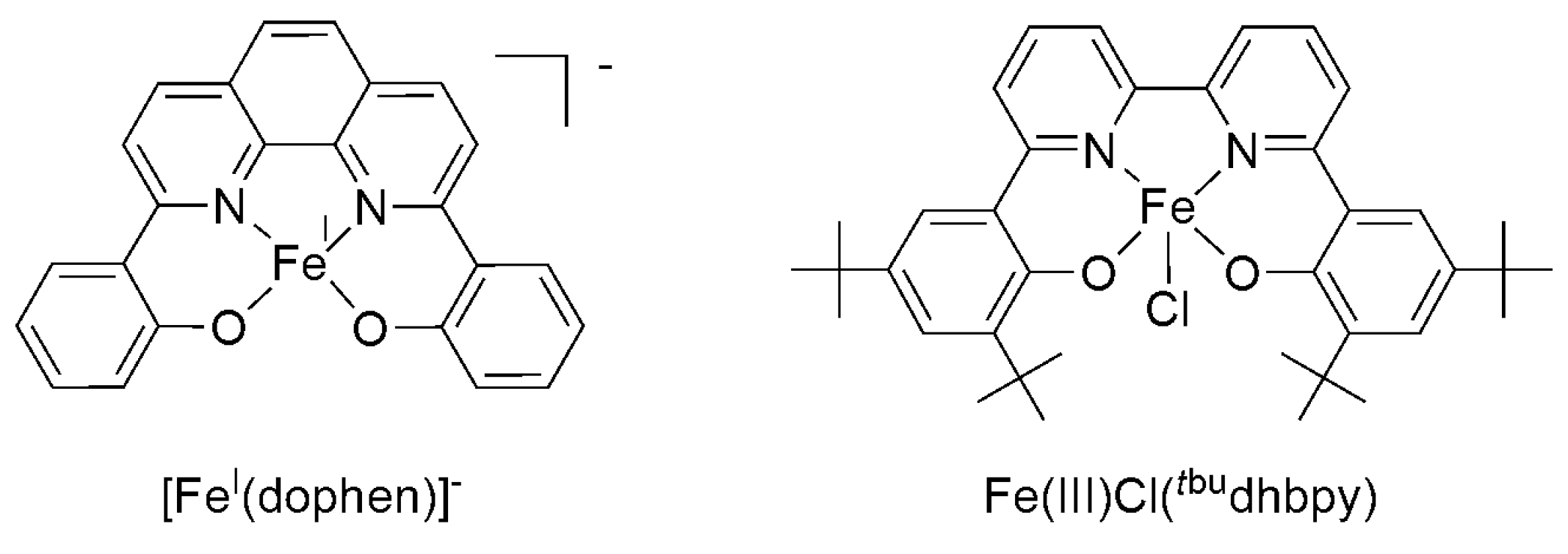{kind=link}
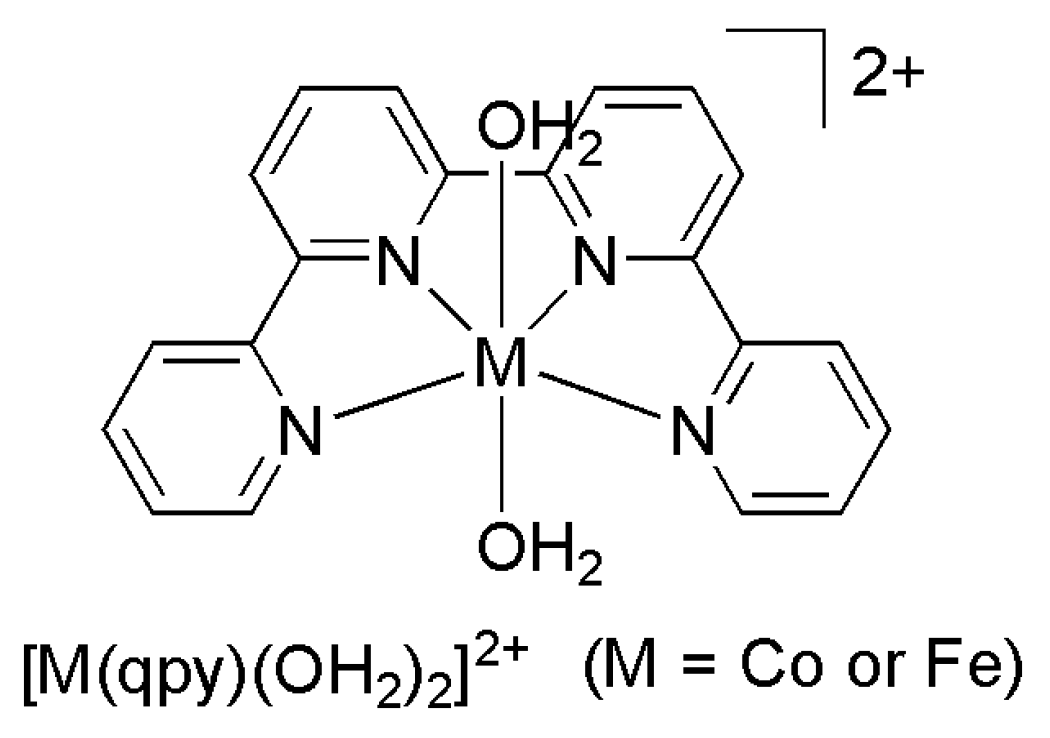{kind=link}
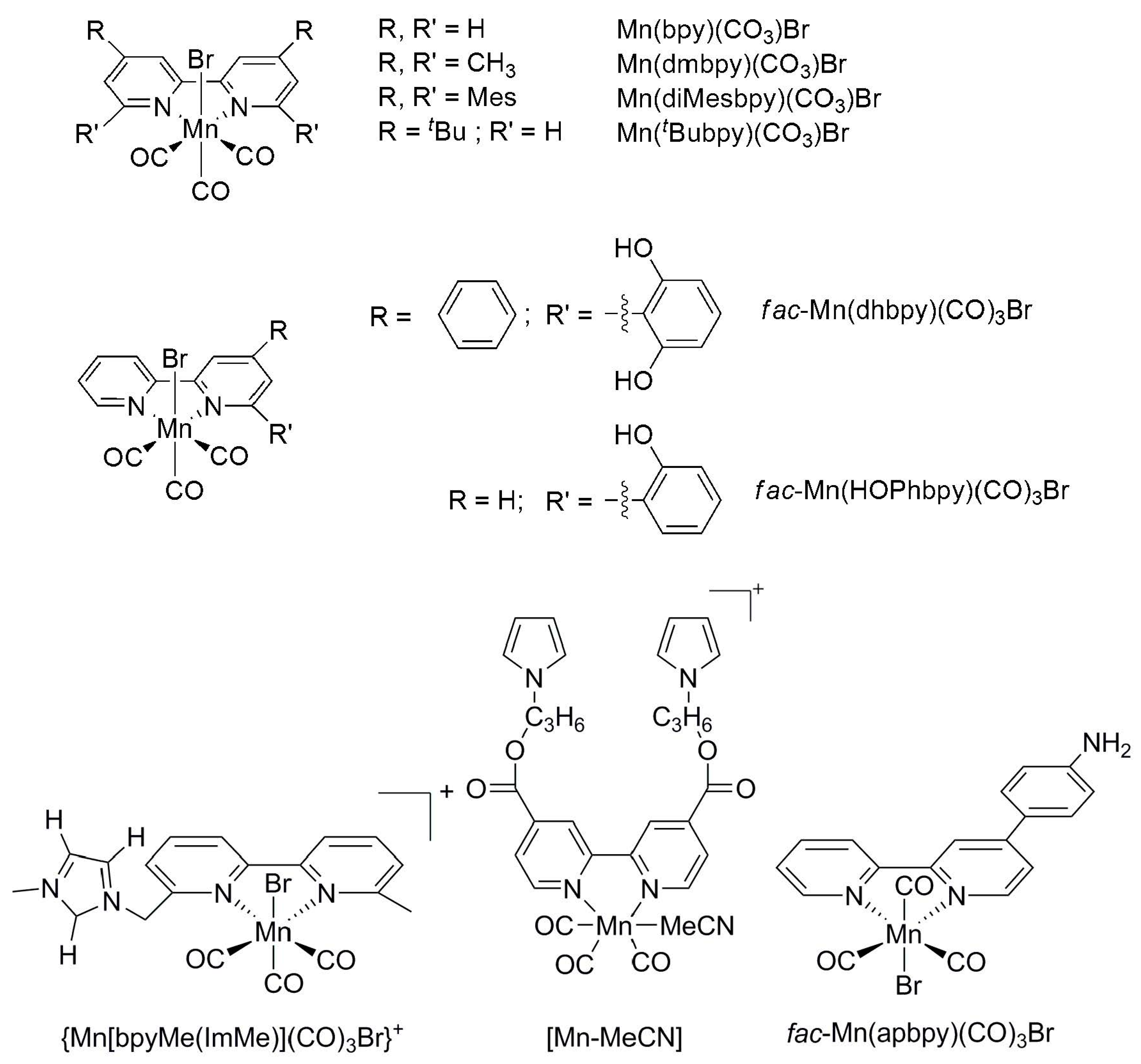{kind=link}
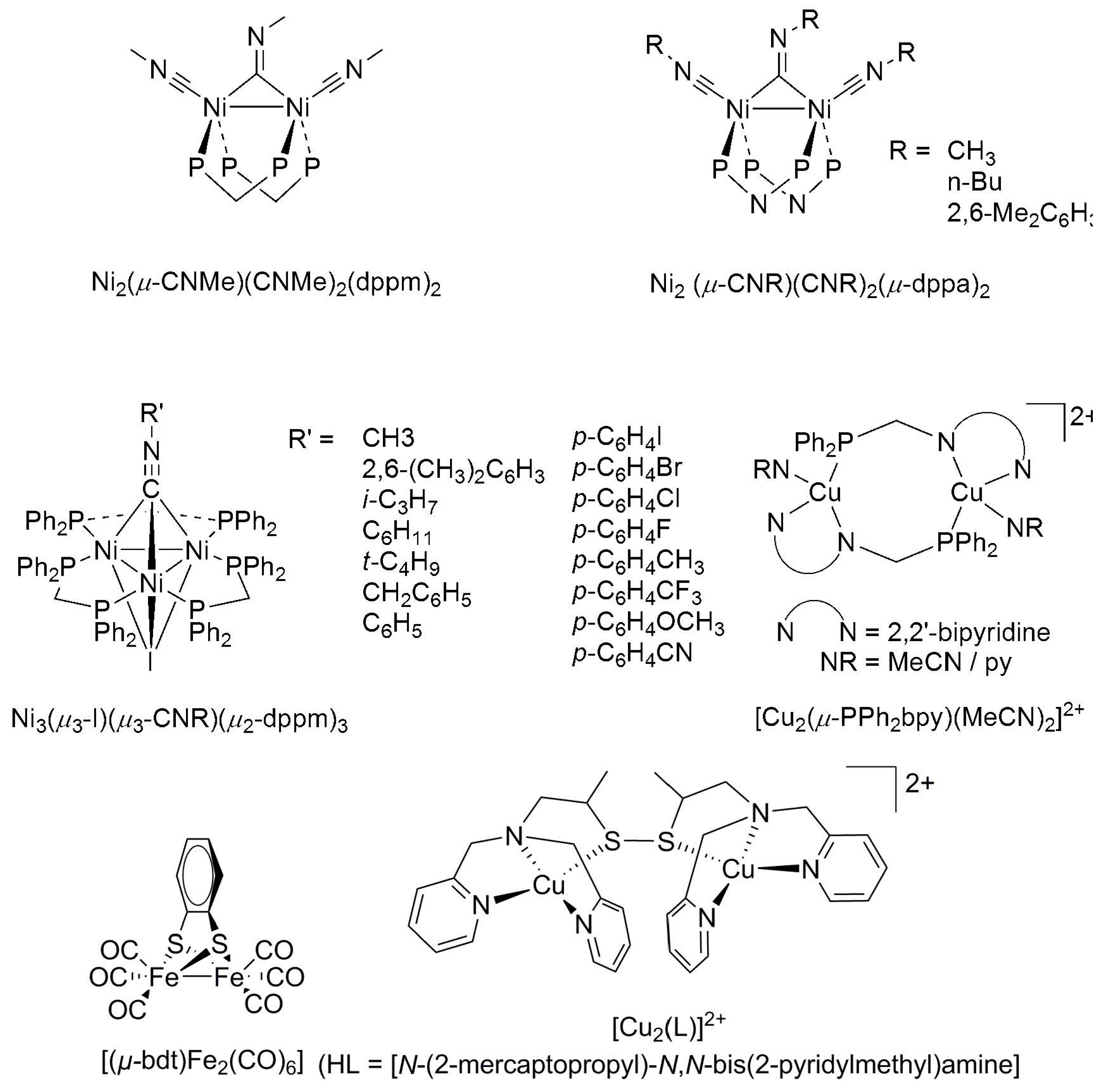{kind=link}
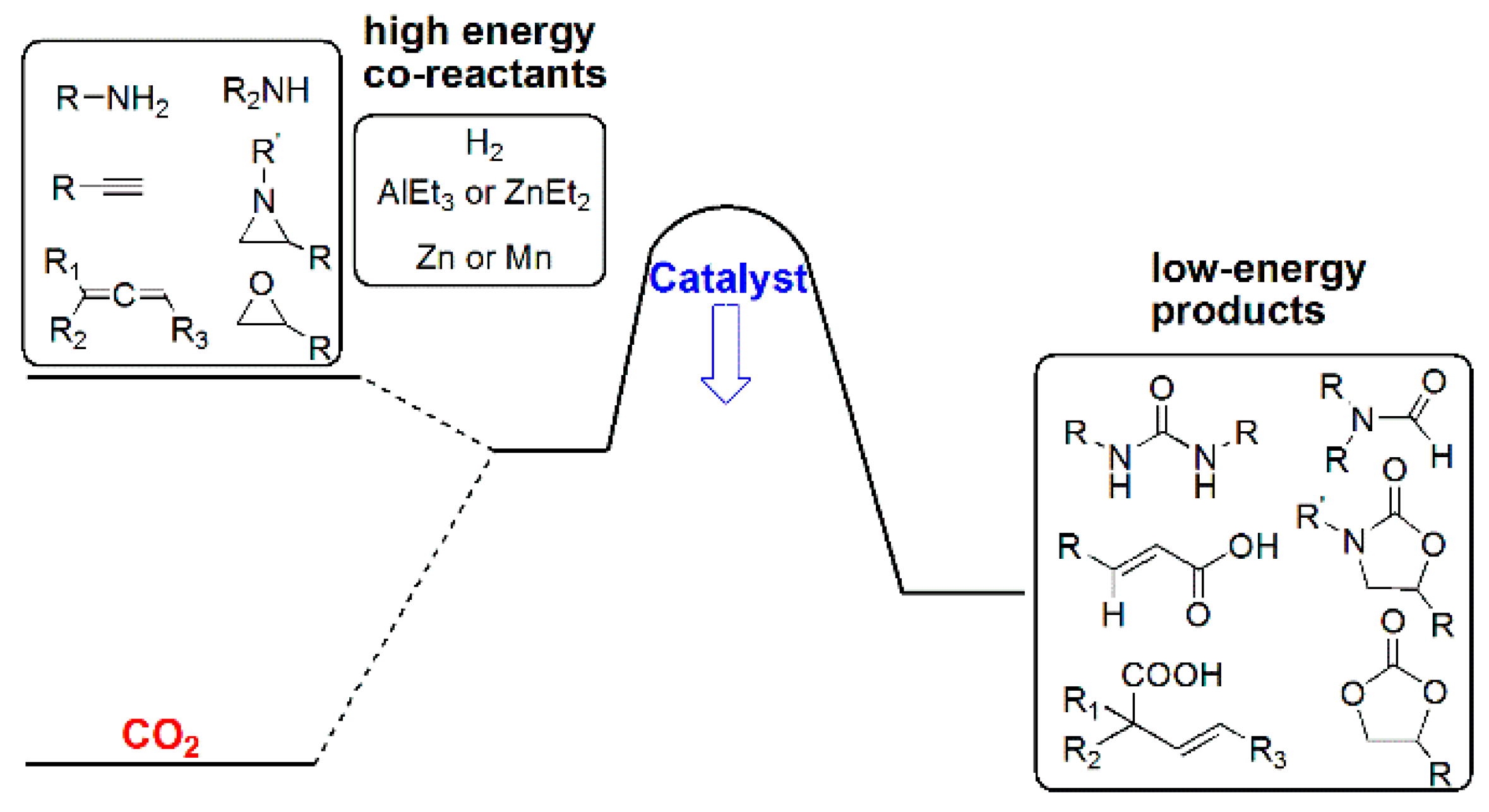{kind=link}
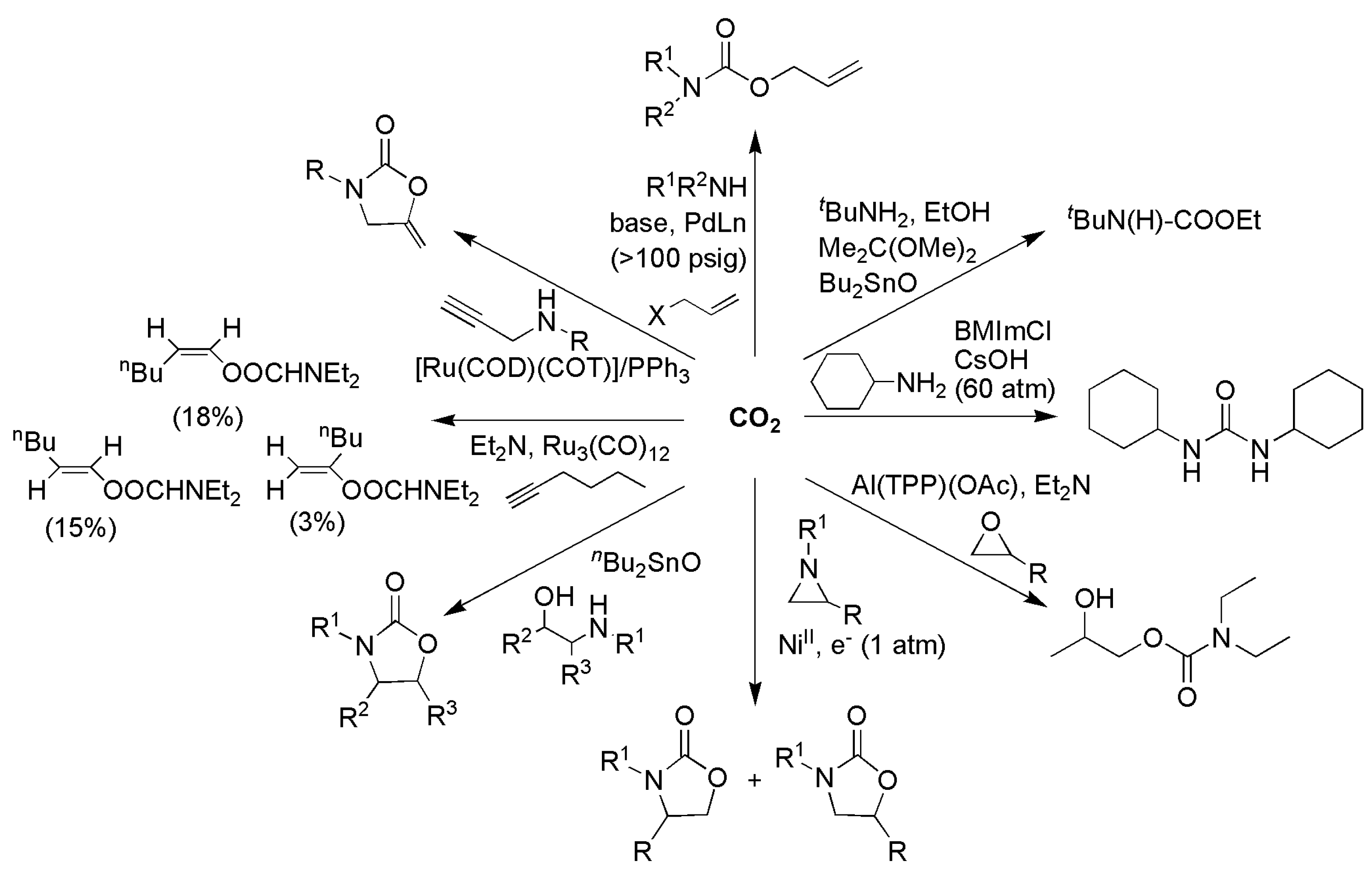{kind=link}
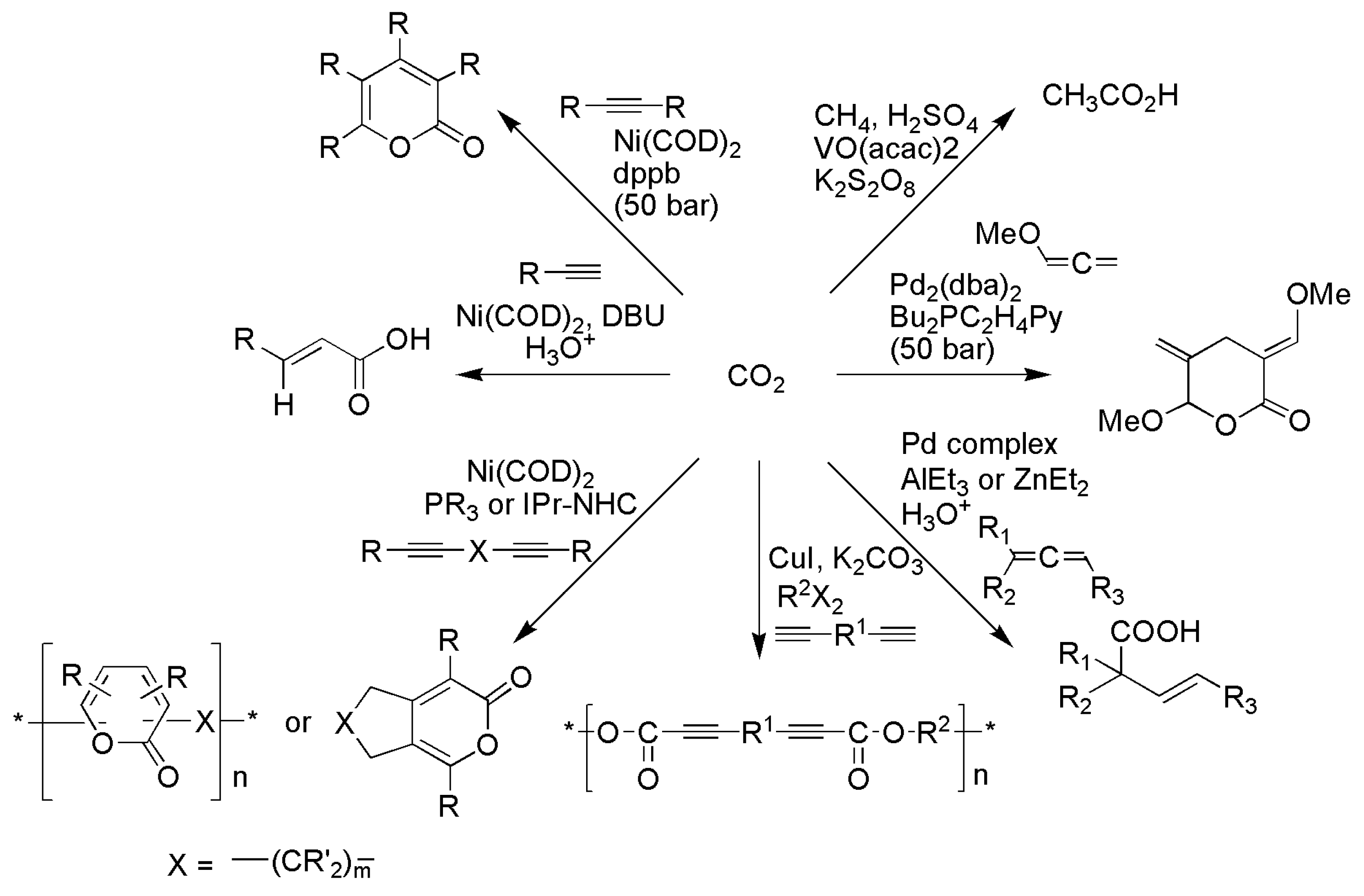{kind=link}
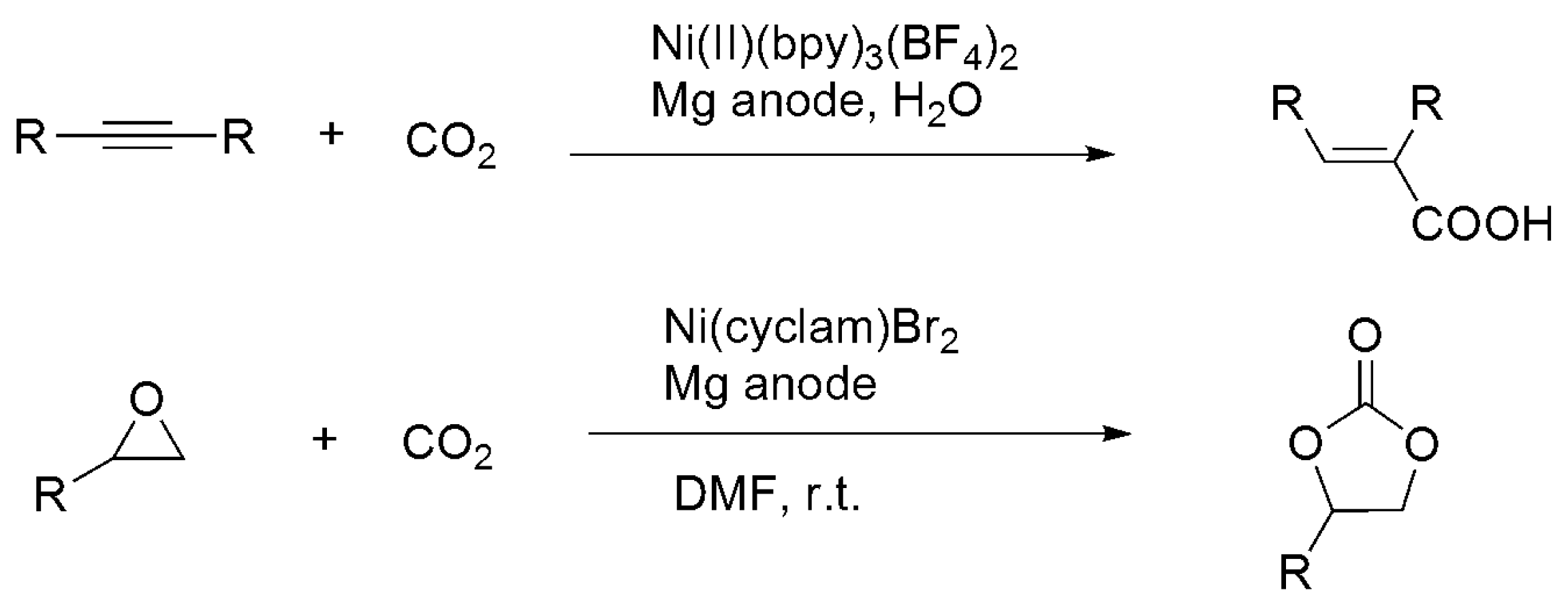{kind=link}
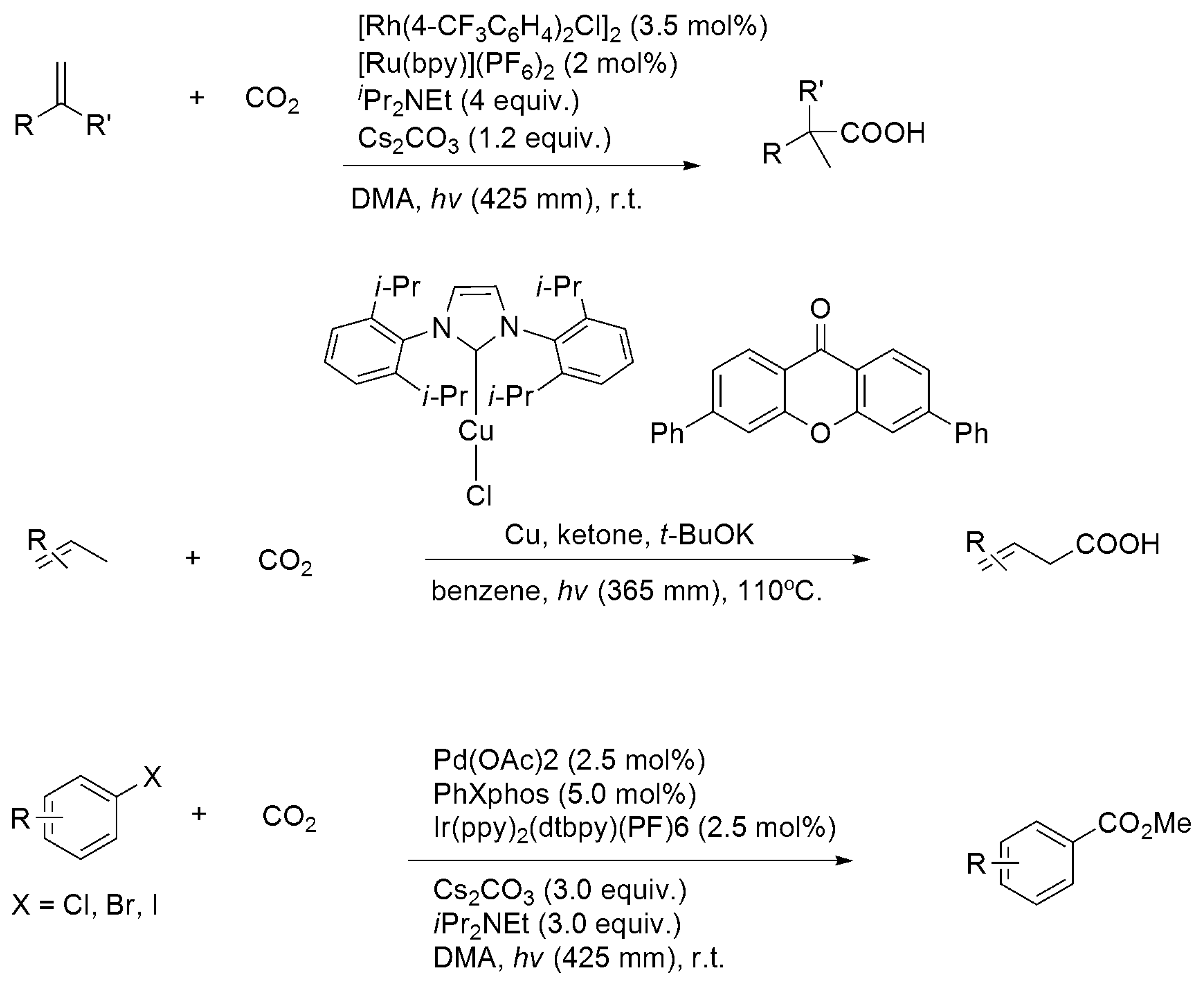{kind=link}
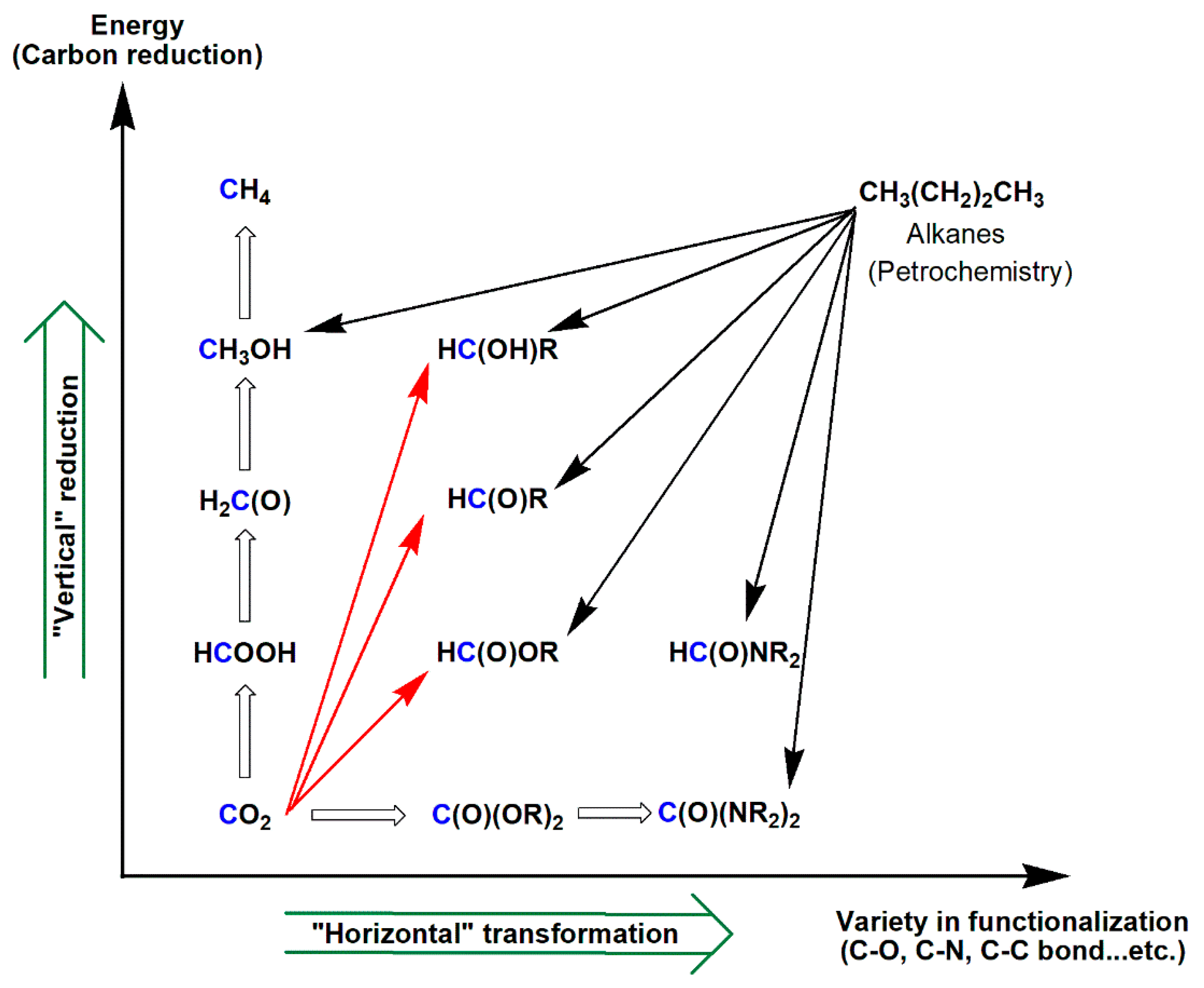{kind=link}
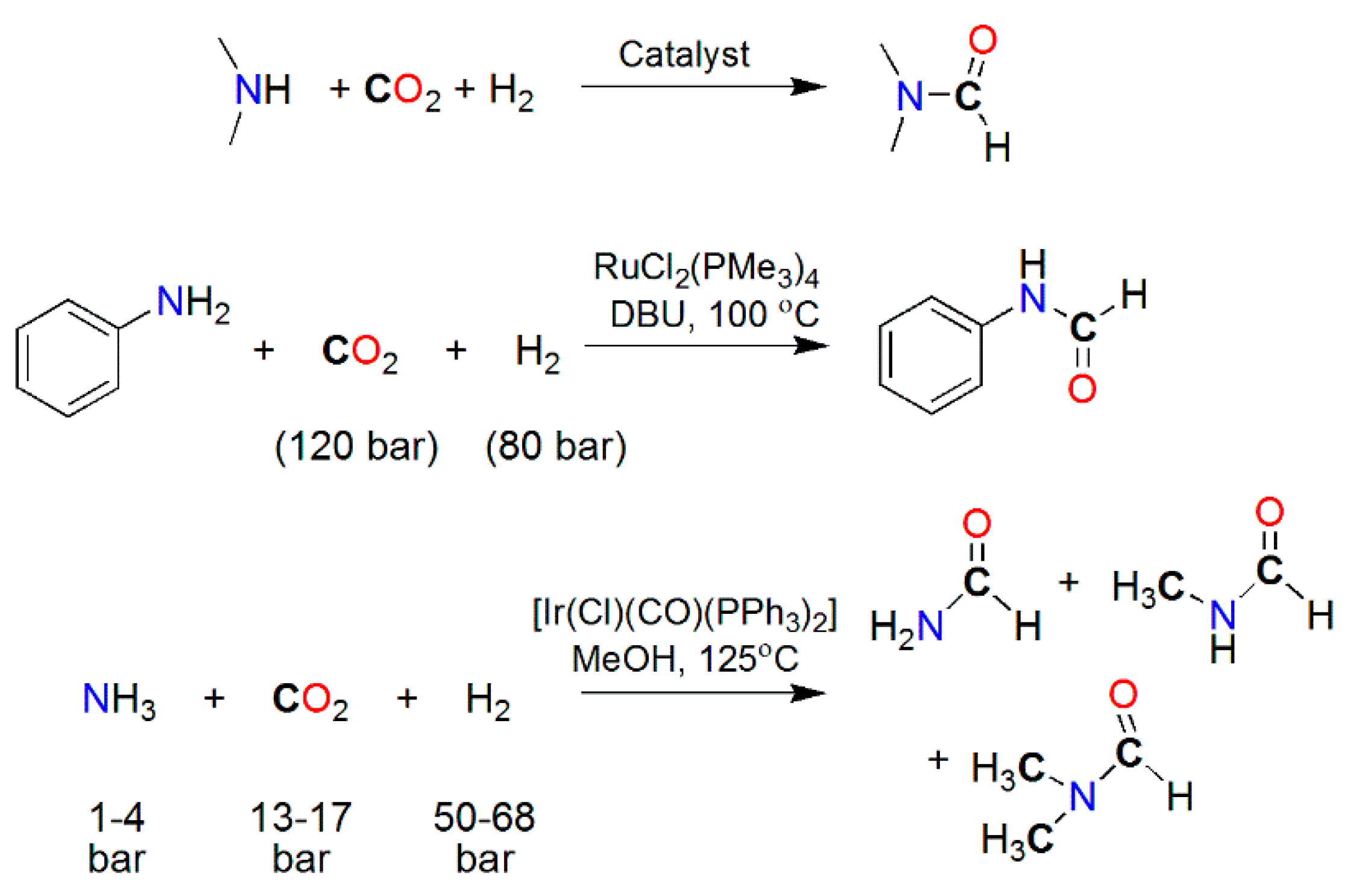{kind=link}
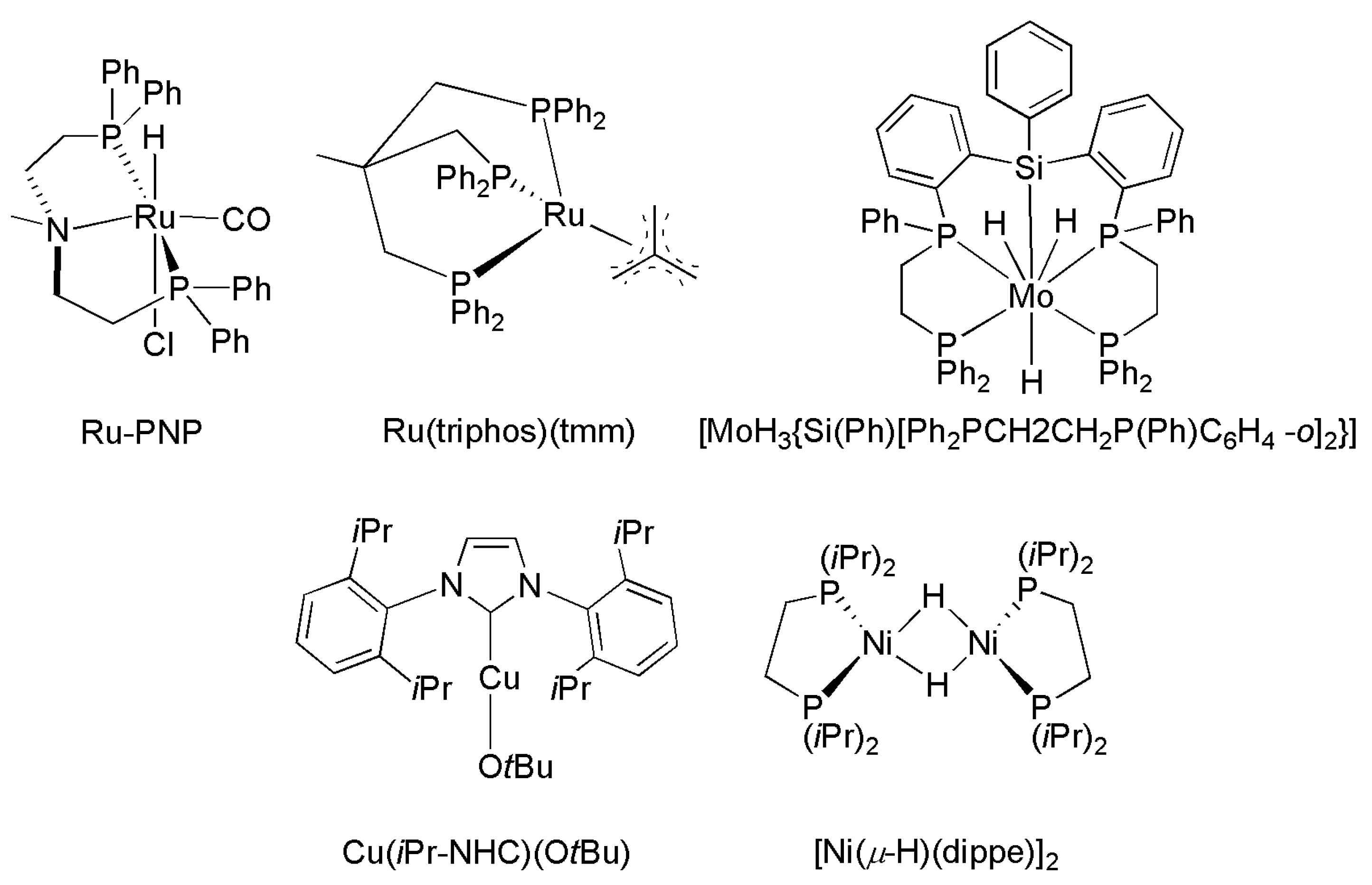{kind=link}
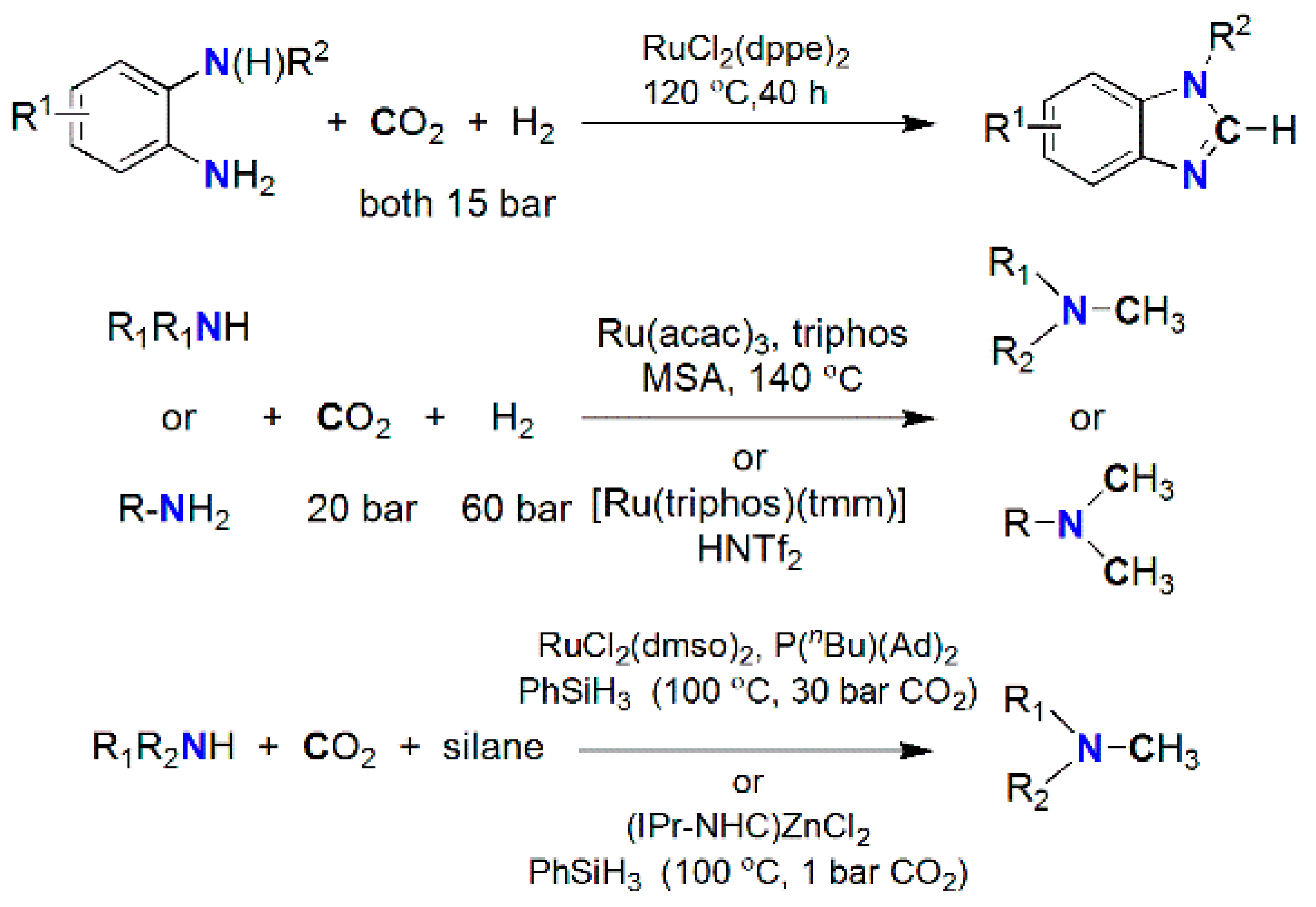{kind=link}
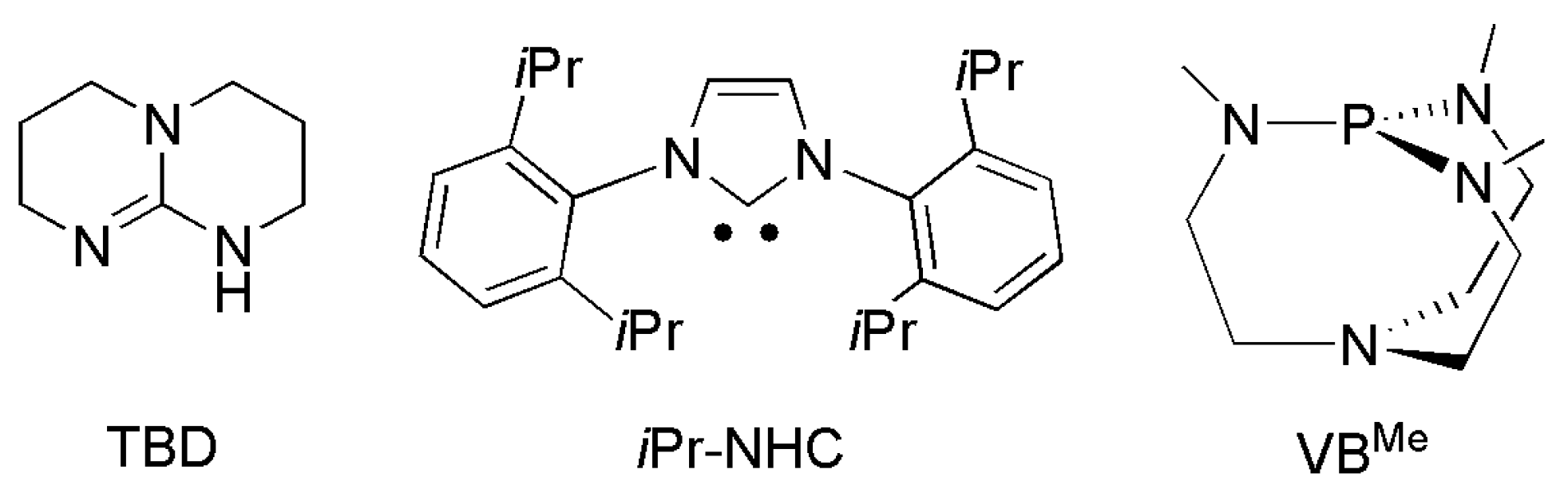{kind=link}
Table 1.
4d and 5d metal 2,2′-bipyridine (bpy) catalysts for two-electron CO2 electroreduction. SCE—saturated calomel electrode; TON—turnover number; DMF—dimethylformamide.
Table 1.
4d and 5d metal 2,2′-bipyridine (bpy) catalysts for two-electron CO2 electroreduction. SCE—saturated calomel electrode; TON—turnover number; DMF—dimethylformamide.
| Catalyst | E1/2 vs. SCE | TON | Solvent | Faradaic Efficiency | Reference | ||
|---|---|---|---|---|---|---|---|
| CO | HCOO− | H2 | |||||
| Re(bpy)(CO)3Cl | −1.49 V | 300 | - b | - a | DMF/H2O | 98% | [27] |
| fac-Re(4,4′-OCH3-bpy)(CO)3X | −2.32 V c | 3.9 | - b | - b | MeCN | 59% | [28] |
| cis-[Rh(bpy)2OTf2]+ | −1.55 V | - a | 6.8–12.3 | 8.5–28.5 | MeCN | 64% | [29] |
| [Ru(bpy)2(CO)Cl]+ | −1.50 V | 10.7–25.5 | 7.8–10.1 | 2.1–21.7 | DMF/H2O | - b | [29] |
| [Ru(bpy)2(CO)2]2+ | −1.50 V | 8.8–26.2 | 18.2–19.9 | 0.2–19.2 | DMF/H2O | - b | [29] |
| cis-[Os(bpy)2(CO)H]+ | −1.50 V | 5.5 a | - a | - b | MeCN | 90% | [30] |
| cis-[Os(bpy)2(CO)H]+ | −1.50 V | - b | 1.8 | - b | MeCN/H2O | 25% | [30] |
| trans-Cl-Ru(mesbpy)(CO)2Cl2 | −2.2 V c | 5.2 | - b | - b | MeCN | 95% | [31] |
a Not detected; b not mentioned; c vs. Fc/Fc+.
Table 2.
Polyphosphine metal catalysts for two-electron CO2 electroreduction. dppe—1,2-bis(diphenylphosphino)ethane; triphos—bis(diphenylphosphinoethyl)phenylphosphine.
Table 2.
Polyphosphine metal catalysts for two-electron CO2 electroreduction. dppe—1,2-bis(diphenylphosphino)ethane; triphos—bis(diphenylphosphinoethyl)phenylphosphine.
| Catalyst | TON | Solvent | Faradaic Efficiency | Reference | ||
|---|---|---|---|---|---|---|
| CO | HCOO− | H2 | ||||
| Rh(dppe)2Cl | - | 1.58 | - | MeCN | 42% | [35] |
| [Pd(etpC)(DMF)](BF4)2 | 130 | - | 154 | DMF | 85% | [36] |
| {m-(triphos)2- [Pd(CH3CN)]2}(BF4)4} | 190 | - | - | DMF | 80% | [37] |
- Not mentioned.
Table 3.
A comparison of metal-catalyzed N–H formylation of selected primary and secondary amines with CO2. dppm—bis(diphenylphosphino)methane; dppe—bis(diphenylphosphino)ethane; PNP—N,N-bis(2-(diphenylphosphinoethyl)methylamine; dmpe—bis(dimethylphosphino)ethane; PP3—tris[2-(diphenylphosphino)ethyl]phosphine; PAr3—tris(2-(diphenylphosphino)phenyl)phosphine; dippe—1,2-bis(diisopropylphosphino)ethane.
Table 3.
A comparison of metal-catalyzed N–H formylation of selected primary and secondary amines with CO2. dppm—bis(diphenylphosphino)methane; dppe—bis(diphenylphosphino)ethane; PNP—N,N-bis(2-(diphenylphosphinoethyl)methylamine; dmpe—bis(dimethylphosphino)ethane; PP3—tris[2-(diphenylphosphino)ethyl]phosphine; PAr3—tris(2-(diphenylphosphino)phenyl)phosphine; dippe—1,2-bis(diisopropylphosphino)ethane.
| Catalyst | Amine | P CO2/H2 (T) Bar (°C) | Reductant | TON/Yield (%) | Reference |
|---|---|---|---|---|---|
| IrCl(CO)(PPh3)2 | Me2NH | 27/27 (125) | H2 | 1200 | [188] |
| PdCl2 | Me2NH | 40/80 (170) | H2 | 34 | [172] |
| [Pt2(μ-dppm)3] | Me2NH | 12/94 (75) | H2 | 1460 | [173,177] |
| RuCl2(dppe)2 | Me2NH | 130/85 (100) | H2 | 74,000 | [164] |
| RuCl2(PMe3)4 | Me2NH | 130/80 (100) | H2 | 370,000 | [178] |
| Ru(PNP)(CO)(H)Cl | Morpholine | 35/35 a (120) | H2 | 1,940,000 | [176] |
| NiX2/dmpe(X = CH2COO− or acac) | Morpholine | 100 b (100–135) | H2 | 18,000 | [180] |
| (PPh3)3CuCl | Me2NH | 27/27 (125) | H2 | 900 | [171] |
| [MoH3{Si(Ph)[Ph2PCH2CH2P(Ph)C6H4-o]2}] | Me2NH | 30/20 a (110) | H2 | 115 | [184] |
| Fe(BF4)2⋅6H2O/PP3 | Me2NH | 30/60 | H2 | 727 | [181] |
| Co(BF4)2⋅6H2O/PP3 | Me2NH | 30/60 | H2 | 1308 | [182] |
| Fe(BF4)2⋅6H2O/PAr3 | Me2NH | 30/60 | H2 | 5104 | [183] |
| Rh2(OAc)4/K2CO3 | PhCH2NH2 | 1 a (50) | PhMe2SiH | 41% | [191] |
| piperidine | 43% | ||||
| PhNH2 | 34% | ||||
| [Ni(μ-H)(dippe)]2/BEt3 | PhCH2NH2 | 1 a (80) | Et3SiH | 85% | [192] |
| (PhCH2)2NH | 47% | ||||
| piperidine | 52% | ||||
| Cu(OAc)2⋅H2O/Bz(PR2)2 | Piperidine | 1 a (80) | PMHS | 11,700/94% | [189] |
| Fe(acac)2/P(C2H4PPh2)3 | (Ph)(Me)NH | 1 (RT) | PhSiH3 | 95% | [190] |
| Cu(iPrNHC)(OtBu) | (Ph)(Me)NH | 1 a (35/65) | H-Bpin | 81% | [193] |
| Ph(CH2)2NH2 | 98% | ||||
| (PhCH2)2NH | 90% |
a Unit = atm; b total pressure.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Leung, C.-F.; Ho, P.-Y. Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock. Catalysts 2019, 9, 760. https://doi.org/10.3390/catal9090760
AMA Style
Leung C-F, Ho P-Y. Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock. Catalysts. 2019; 9(9):760. https://doi.org/10.3390/catal9090760
Chicago/Turabian StyleLeung, Chi-Fai, and Pui-Yu Ho. 2019. "Molecular Catalysis for Utilizing CO2 in Fuel Electro-Generation and in Chemical Feedstock" Catalysts 9, no. 9: 760. https://doi.org/10.3390/catal9090760
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.